
Abstract
Besides its well-known role as energy storage tissue, adipose tissue is a biologically active tissue that can also be considered as an endocrine organ, as it is able to secrete adipokines. These bioactive factors, similar in structure to cytokines, are involved in several physiological and pathological conditions, such as glucose homeostasis, angiogenesis, blood pressure regulation, control of food intake, and also inflammation and bone homeostasis via endocrine, paracrine, and autocrine mechanisms. Given their pleiotropic functions, the role of adipokines has been evaluated in chronic rheumatic osteoarticular inflammatory diseases, particularly focusing on their effects on inflammatory and immune response and on bone alterations. Indeed, these diseases are characterized by different bone complications, such as local and systemic bone loss and new bone formation. The aim of this review is to summarize the role of adipokines in rheumatoid arthritis, ankylosing spondylitis, psoriatic arthritis, osteoarthritis, and osteoporosis, especially considering their role in the pathogenesis of bone complications typical of these conditions.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Adipose tissue is recognized to perform several functions; besides its role in energetic homeostasis and energy storage, it is involved in thermogenesis, it is responsible for structural support of joints, gastrointestinal tract and skin and it can be considered as an endocrine organ as it is able to produce several biological mediators known as adipokines that are involved in blood pressure regulation, glucose homeostasis, angiogenesis, control of food intake, and regulation of inflammation and immune system via endocrine, paracrine, and autocrine mechanisms [1,2,3]. Adipokines have both pro-inflammatory and anti-inflammatory properties and provide a link between systemic metabolism and immune response, both at a local and systemic level. In healthy individuals with normal metabolic status, there is a balance between pro- and anti-inflammatory adipokines. In fasting conditions, the production of anti-inflammatory adipokines within the white adipose tissue increases, while the pro-inflammatory adipokine levels decline, resulting in suppressed immune function. On the contrary, obesity is characterized by a shift towards proinflammatory adipokines: this pro-inflammatory status contributes to the chronic “low-grade inflammatory state” associated with obesity [2].
Given their pleiotropic functions, increasing evidence leads to consider adipokines as significant component of the complex network of soluble mediators involved in the pathophysiology of chronic inflammatory and immune mediated disease, such as rheumatic diseases [4, 5]. Indeed, although mainly released by adipocytes, adipokines are also synthesized by osteoblasts, chondrocytes, synoviocytes, and immune cells in the joint microenvironment [4].
For instance, resistin is mainly produced by circulating monocytes and macrophages in humans [3, 4], and resistin expression was observed in rheumatoid arthritis (RA) joints in synovial stromal cells and infiltrating immune cells, such as macrophages and B cells [4]. Another adipokine, visfatin, is primarily found in liver, bone marrow, and muscle tissue, and it is also secreted by macrophages [3]. Moreover, although mainly produced by white adipose tissue cells, the adipokine leptin gene expression has been found in placenta trophoblasts and amnion cells, in fetal mouse bone and cartilage, in rat gastric epithelial cells [5, 6]. Furthermore, chondrocytes and synovial fibroblasts from RA and osteoarthritis (OA) patients express the adipokine chemerin [4]. Also, another adipokine, vaspin, is expressed in tissues other than adipose tissue, like skeletal muscle, and OA cartilage and synovium [4].
The main chronic inflammatory joint diseases are characterized by an important bone involvement, with drastic impact on quality of life. In RA, systemic and local bone loss prevail, leading to the typical joint erosions and juxta-articular and systemic osteoporosis (OP). Conversely, in OA and spondyloarthritis (SpA), local new bone formation is observed, with subsequent formation of osteophytes and syndesmophytes [7]. OP is a systemic bone disease characterized by impairment of bone mass and microarchitecture, leading to fragility fractures; it is most frequently observed in postmenopausal women and in older people in general. Aging is associated with a low-grade inflammatory state distinguished by overexpression of proinflammatory cytokines, such as IL-1, IL-6, and TNFα, which could lead to the increased bone resorption observed in OP. Indeed, these cytokines are potent stimulators of bone resorption and inhibitors of bone formation [8].
In the latest years, the close relationship between adipose tissue and bone tissue has become clearer. Bone mass positively correlates with fat mass, and epidemiological studies showed that low body weight is a major risk factor for fractures and low bone mineral density [9]. A possible explanation for the positive association between adipose tissue and bone mass could be found in the skeleton response to mechanical loading due to body weight; nevertheless, in the last few years, several authors investigated the effect of adipokines in bone metabolism and bone remodeling as mediators of the fat-bone crosstalk [10].
The aim of this review is to summarize the role of adipokines in rheumatoid arthritis, ankylosing spondylitis, psoriatic arthritis, osteoarthritis, and osteoporosis, especially considering their role in the pathogenesis of bone complications typical of these conditions.
Adipokines and Inflammation
Adipokines affect several biological functions: their actions impact not only on metabolism but also on different features of the immune system. Therefore, these mediators may embody a junction between lipid metabolism, inflammation and autoimmunity. Adipokines might play a part in creating a chronic inflammatory state, stimulating the production of pro-inflammatory cytokines by different immune cells, like macrophages. Nevertheless, the results of adipokines’ action on inflammation and immune system can be considerably different and are often conflicting (Fig. 1).

Effect of adipokines on immune cells
Adiponectin is a glycoprotein mainly secreted not only by white adipose tissue but also by skeletal muscle, bone cells, lymphocytes, and liver. Adiponectin is the most represented circulating adipokine. Adiponectin circulates in blood in multiple isoforms whose biological relevance is not fully known: low molecular weight (LMW) trimer, medium molecular weight (MMW) hexamer and high molecular weight (HMW) multimer. In addition, a smaller form called globular adiponectin (gAd) has been described [11]. The HMW isoform is thought to be the most biologically active. Adiponectin acts through different signaling pathways, such as AMPK and PPAR, after binding to its two receptors: AdipoR1, expressed ubiquitously but mainly in the skeletal muscle, and AdipoR2, predominantly described in the liver [11]. The pleiotropic actions of adiponectin in regard to insulin sensitivity, inflammation, and angiogenesis may actually be due to the existence of different isoforms with different receptor binding affinity in different target tissues [11, 12]. Several evidences have shown that adiponectin has a protective anti-inflammatory effect, especially in atherosclerosis, obesity, and type 2 diabetes [2, 12]; however, recent studies suggested that it could play an opposite role in other conditions, such as autoimmune diseases [12]. As a modulator of the immune system, adiponectin has both anti- and pro-inflammatory activity. Adiponectin suppresses the pro-inflammatory M1 macrophage activation, while it promotes the anti-inflammatory M2 macrophage activation and proliferation [12]. Moreover, it induces the production of anti-inflammatory factors in these cells and also in dendritic cells [13]. Further, this adipokine modulates the inflammatory response by inhibiting eosinophil and endothelial cell chemotactic response [14, 15], suppressing macrophage and nuclear factor (NF)-kB signaling, stimulating monocyte and macrophage apoptosis, and inhibiting their phagocytic activity [16]. Moreover, transcription of adiponectin is suppressed by inflammatory cytokines such as TNFα and IL-6 [17]. However, HWW adiponectin and gAd have been shown to induce TNFα in human monocytes, while mutated adiponectin that cannot form HMW could not lead to enhanced TNFα production [12]. Moreover, gAd appears to be a powerful inducer of pro-inflammatory cytokines in macrophages; indeed, macrophages exposed to gAd showed enhanced TNFα and IL-6 secretion in a dose-dependent manner [18]. Furthermore, adiponectin has been found to inhibit human neutrophil phagocytosis of Escherichia coli, both in whole blood and in isolated cells [19]; these data further support the existence of an immune modulatory role for this adipokine.
Leptin is a 16-kDa non-glycosylated cytokine-like hormone encoded by the LEP gene, and its action is mediated through the leptin receptor (LEPR), which belongs to the class I cytokine receptor superfamily. At least six isoforms of LEPR are known, which differ by the length of the cytoplasmic domain, but only the long isoform seems to be able of transducing the leptin signal. LEPR usually signals through the Janus kinase (JAK) and signal transducer and activator of transcription (STAT) signaling pathway, but it is also able of signaling via alternative pathways including ERK1/2, p38 MAPK, JNK, PKC, SHP2/GRB2 and PI3K/Akt pathways [5, 20]. Leptin is mainly produced by adipocytes and, in physiological conditions, circulating levels positively correlate with white adipose tissue mass and body mass index. This hormone could be considered an energy homeostasis regulating factor that balances food intake and energy consumption by inducing anorexigenic factors and by suppressing orexigenic factors in the hypothalamus. Several other factors are involved in leptin synthesis, such as energy status, sex hormones (increased by female sex hormones estrogen and progesterone, suppressed by testosterone), and various inflammatory mediators [5, 20]. Moreover, other than adipose tissue, leptin is produced to a lesser extent by skeletal muscle, intestine, gastric epithelium, placenta, mammary glands, brain, joint tissues, and bone [5] and is involved in several physiological processes, like vascular function, insulin secretion, bone metabolism, reproduction, inflammation, and immune response [5, 20, 21]. For instance, leptin and its receptor are expressed in cardiomyocytes and it can regulate cardiomyocyte contractility, hypertrophy, apoptosis, and metabolism [22]. This adipokine is also therefore associated with cardiovascular diseases, as shown by the association of elevated serum leptin levels and myocardial infarction and stroke; moreover, leptin is involved in atherosclerosis, since its positive association with inflammatory mediators involved in the pathogenesis of atherosclerosis C-reactive protein (CRP) and IL-6, the leptin-induced hypertrophy of vascular smooth muscle cells and the production of matrix metalloproteinase (MMP)-2, involved in plaque rupture. Furthermore, leptin enhances platelet aggregation and increases the production of several factors involved in the development of atherosclerosis, such as lipoprotein lipase, caveolin-1, and plasminogen activator inhibitor-1 [23]. Leptin has been suggested as the linking factor between adipose tissue, energy storage, and reproductive function: it seems to have an important role in the balance of the hypothalamic-pituitary-gonadal axis, since low levels of leptin have been associated with hypogonadotropic hypogonadism and hypogonadism in type 1 diabetes, whereas hyperleptinemia may be involved in hypogonadism associated with obesity, polycystic ovarian syndrome, and type 2 diabetes [24]. Additionally, leptin inhibits insulin synthesis and secretion from pancreatic β-cells, while insulin induces leptin secretion from adipose tissue. The alterations of this adipo-insular feedback are involved in the development of hyperinsulinemia and type 2 diabetes mellitus [25]. Accordingly, LEPR is present on the surface of blood vessel cells, cardiomyocytes, and innate and adaptive immune system cells, such as natural killer (NK) cells, granulocytes, monocytes, macrophages, dendritic cells, and B and T cells [21]. The mechanisms that control the immune-metabolic actions of leptin and its relationship with the immune system have been deeply investigated. Leptin secretion from white adipose tissue is regulated by the inflammatory status: acute inflammation and pro-inflammatory mediators, such as TNFα, IL-1, IL-6 and positively regulate leptin release from adipose tissue and plasma leptin levels [26]. Leptin is considered a pro-inflammatory adipokine involved in the “low-grade inflammatory state” observed in overweight and obese people [20] and is involved in the modulation of both innate and adaptive immunity. Leptin promotes activity and proliferation of NK cells; it activates chemotaxis and cytokine secretion of granulocytes (neutrophils, basophils, and eosinophils) and monocytes, and it is able to delay their apoptosis [20]; moreover, leptin promotes activation and proliferation of monocytes as well as the production of proinflammatory cytokines, such as IL-1, TNFα, and IL-6, in peripheral blood monocytes and resident tissue macrophages in mice and humans [2, 20]. Further, this adipokine induces the phagocytic activity of neutrophils and monocytes/macrophages [27, 28]. Likewise, leptin acts as an activator, chemoattractant, and survival factor for dendritic cells, with a possible involvement in their maturation and migration [20, 29, 30]. Leptin is also able to deeply affect the adaptative immune system, as it is able to enhance proliferation of naïve T and B cells whereas it decreases Treg cell proliferation while increasing Th17 proliferation [20].
Resistin is a cysteine-rich adipokine which in humans is predominantly secreted by macrophages. For this reason, it has been hypothesized that this mediator could affect the inflammatory response, albeit it exhibits pro- or anti-inflammatory actions in different tissues [4]. Studies have shown that resistin is able to enhance endothelial permeability and induce adhesion molecule expression and pro-inflammatory cytokine production in monocytes [31,32,33]. Additionally, this adipokine can downregulate the expression of reactive oxygen species (ROS) in macrophages and neutrophils, thus indirectly delaying apoptotic mechanisms in these cells [34].
Omentin is a glycoprotein first described in Crohn’s disease patients, but it is also found in plasma from healthy donors [4]. This adipokine has well-defined anti-atherogenic properties in obese patients and is negatively associated with metabolic syndrome [4]. Similar to adiponectin, omentin modulates macrophages activity by promoting the anti-inflammatory M2 phenotype activation and proliferation [35] and inhibits the expression of pro-inflammatory cytokines in these cells [36]. Given these data, omentin role in osteoarticular inflammatory diseases, such as psoriatic arthritis (PsA) and OA, has been investigated, as described in the following paragraphs.
Vaspin is an adipokine associated with several metabolic alterations, including insulin resistance, metabolic syndrome, atherosclerosis, and cardiovascular disease. Vaspin is not expressed only in adipose tissue, but also in skin, stomach, and skeletal muscle [4]. Furthermore, this adipokine plays a role in the regulation of the inflammation process. Vaspin modulates macrophage apoptosis [37] and has been reported to downregulate the expression of pro-inflammatory factors in monocytes/macrophages, neutrophils, and dendritic cells [38].
Visfatin is also called pre-B-cell colony-enhancing factor since it promotes B cell precursor differentiation (in synergy with IL-7). It is mainly produced by adipose tissue but also by other tissues such as the liver, bone marrow, and muscle tissue. As described for other adipokines, visfatin expression is induced by inflammatory factors such as TNFα, IL-1β, and IL-6. Visfatin in itself is considered a primarily pro-inflammatory adipokine [4]. Indeed, it promotes the chemotaxis of monocytes [39] and delays apoptosis of macrophages and neutrophils [40, 41], as well as stimulating their phagocytic activity [39, 42].
Other adipokines, like chemerin and apelin, are involved in the regulation of immune response. For instance, chemerin can modulate the migration of monocytes/macrophages and dendritic cells [43, 44], as well as inducing the phagocytic function of neutrophils and/or monocytes/macrophages [45], like leptin and visfatin. Conversely, alongside adiponectin, apelin suppresses this process; moreover, it inhibits chemotaxis of macrophages and the expression of pro-inflammatory mediators in these cells [46].
Adipokines and RA
RA is a chronic autoimmune inflammatory disease that primarily affects synovial joints, characterized by autoantibody production, cartilage and bone destruction, and systemic findings [17, 47]. The majority of RA patients present autoantibodies, including rheumatoid factors (RF) and anti-citrullinated protein antibodies (ACPA) [48]. Bone involvement in RA is characterized by both local and systemic bone loss. The first is represented by joint erosions (the radiological hallmark of the disease) and juxta-articular osteoporosis; the latter is exemplified by systemic osteoporosis [48]. Bone erosions occur early in RA patients and are predictive of a more severe course of disease with increased disability and mortality [48, 49]. The pathogenesis of bone loss in RA is multifactorial and involve glucocorticoid treatment, reduced mobility, sex hormone deficiency, disease activity and inflammatory status [50]. Indeed, both local and systemic bone complications are due to the loss of balance between osteoblast and osteoclast activity and the chronic inflammatory state typical of the disease expressed by pro-inflammatory cytokines TNFα, IL-1β, and IL-6, upregulates the RANK/RANKL pathway, leading to increased osteoclast activity, prolonged osteoclast lifespan, and bone resorption. The enhanced bone resorption is accompanied by impaired bone formation on account of the upregulation of WNT signaling inhibitors preventing osteoblast differentiation, such as Dickkopf-related protein 1 (Dkk-1), frizzled-related protein-1 and sclerostin. Indeed, Dkk-1 is upregulated in the synovial tissue by pro-inflammatory cytokines like TNFα and is associated with radiographic progression in early RA patients [48, 51]. In this regard, adipokines can be considered another factor coupling inflammation and bone loss in RA.
Many studies confirmed that adipokine secretion is altered in RA patients and presents a relationship with inflammatory status [3, 52, 53].
Although the well-known protective anti-inflammatory effect of adiponectin in the pathogenesis of obesity, diabetes mellitus, and atherosclerosis, several studies suggest that this adipokine plays a pro-inflammatory role in the pathogenesis of RA [3]. RA patients have higher serum and synovial levels of adiponectin than non-RA controls [4, 12, 21, 54]. However, the serum adiponectin levels did not correlate with RA disease activity based on Disease Activity Score of 28 joints (DAS28) and CRP levels [17, 55]. This discrepancy may be explained by the action of adiponectin on distinct cells and by the analysis of specific isoforms. In particular, the HMW isoform and the gAd serum levels were found higher in RA patients than in healthy subjects [12]. HMW adiponectin enhances the release of pro-inflammatory cytokines IL-6 and IL-8 in RA synovial fibroblasts [12, 56]; moreover, the secretion of inflammatory factors TGFβ, IL-6, IL-1Ra, PGE2, IL-8, and vascular endothelial growth factor (VEGF) by adipose mesenchymal stem cells derived from infrapatellar fat pad of RA patients was considerably increased by HMW adiponectin [57]. Moreover, adiponectin is involved in another hallmark of RA, neovascularization. Interestingly, adiponectin induces the expression of VEGF in cultured synovial fibroblasts isolated from RA patients and in osteoblasts [58]. Furthermore, authors investigated also the effect of adiponectin on endocan, an endothelial dysfunction biochemical marker also known as endothelial cell-specific molecule-1 (ESM-1), secreted by vascular endothelial cells and involved in cell proliferation, migration, and neovascularization [59, 60]. Endocan levels were increased in RA synovial tissues and adiponectin has been shown to stimulate its expression in RA synovial fibroblasts. Therefore, authors hypothesized that adiponectin-induced endocan expression in RA synovial cells may be involved in cell migration and invasion, together with angiogenesis in the pannus of arthritic joints [60]. Given these evidences, adiponectin seems to play a crucial role in the angiogenesis of RA.
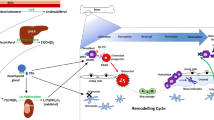
Several studies investigated the relationship between adiponectin and bone alterations occurring in RA (Fig. 2).

Effect of adipokines on bone cells leading to enhanced bone erosions in RA
Adiponectin was found to stimulate the expression of MMP in RA patients [58]. Matrix metalloproteinases are enzymes able of degrading all components of extracellular matrix; in particular, MMP-1, MMP-3, MMP-9, and MMP-13 expression is upregulated in RA synovial fibroblasts and plays a critical role in the pathogenesis of structural joint alterations in RA [61]. It has been shown that adiponectin is able to increase the secretion of MMP-1 and MMP-13 in osteoblasts [58] and adiponectin stimulation led to decreased mineralization and increased resorptive activity of osteoclasts [62]. Moreover, immunohistochemistry studies performed on RA bone tissue showed that adiponectin is expressed in osteoblasts and osteoclasts [62]. In a study conducted on a mice model of collagen-induced arthritis, adiponectin injection led to an earlier onset of arthritis, an accelerated joint damage progression, more severe synovial hyperplasia, bone erosion, and osteoporosis, compared to controls. In particular, this result was due to adiponectin-dependent enhancing of Th17 cell response and upregulation of RANKL/OPG ratio [63]. Qian et al. suggested that adiponectin mediates bone erosion in RA patients by enhancing the production of osteopontin, an important protein for osteoclast differentiation and activation. The authors found a positive association between adiponectin expression and osteopontin serum levels in RA patients; moreover, osteopontin expression was significantly increased in a dose-dependent manner in RA synovial fibroblasts in response to various dosages of adiponectin. The upregulation of osteopontin led to enhanced osteoclast precursor migration and osteoclastogenesis, thus contributing to bone resorption and subsequent joint destruction [64].
Recent clinical studies have suggested that adiponectin levels might correlate with the radiographic progression of RA; therefore, it may be involved in the development of bone erosions typical of RA. Different cross-sectional studies showed a positive association between adiponectin levels in RA patients and joint damage, in terms of erosions and joint space narrowing [65]. Giles et al. [66] conducted a prospective study evaluating the association of serum adipokine levels with progression of radiographic joint damage in RA patients, assessed by the van der Heijde modification of the Sharp method (SHS) [67]. The authors observed a positive association between adiponectin levels and yearly rate of SHS progression; notably, this associations remained significant also after adjusting for pertinent confounders. A cohort study conducted by Klein-Wieringa et al. [65] showed that baseline levels of adiponectin could predict the radiographic progression over a period of 4 years in patients with early RA; interestingly, this positive correlation was independent of ACPA that are associated with erosive disease. Meyer et al. [68] also suggested that adiponectin could be used as a predictor of radiographic progression and disease activity in early RA as their cohort study showed an association between serum adiponectin level and both erosive and narrowing sub-scores of radiographic disease progression. In conclusion, serum adiponectin levels in RA patients could be a good biomarker to evaluate the early stages of radiographic disease progression.
Similarly, leptin is involved in the pathophysiology of different rheumatic diseases, including RA. In several studies, leptin concentrations are higher in RA patients than in controls [3, 69]; further, DAS28 and CRP levels, expressing disease activity, are positively correlated with leptin levels [69, 70]. Meta-analysis confirmed these reports [70], showing higher serum leptin levels in RA patients with high disease activity [55]. Additionally, a correlation between ratio serum/synovial fluid leptin level and RA disease duration and activity parameters has been described [20].
Studies conducted on animal models of RA support the hypothesis of leptin involvement in the pathogenesis of RA. Compared with control mice, leptin-deficient mice presented a less severe antigen-induced arthritis, reduced levels of IL-1β and TNF in knee synovium and an impaired antigen-specific T cell proliferative response with lower IFN-γ and higher IL-10 production, which indicates a shift toward Th2 cell response [1]. Accordingly, in a preclinical model of collagen-induced arthritis in mice, articular injection of leptin exacerbated the severity of arthritis, through increased Th17 response [1].
Furthermore, in human and murine chondrocytes, leptin also increases vascular cell adhesion molecule 1 (VCAM-1) expression, a cell adhesion molecule, involved in leukocyte recruitment and extravasation from circulating blood to inflamed joints during RA [54].
Taken together, these data strongly suggest that leptin may play a key pro-inflammatory role in RA.
The relationship between leptin and radiographic damage in RA patients is controversial. It has been reported that synovial fluid leptin levels were significantly lower than in paired plasma in patients with non-erosive RA, but not in patients with erosive RA; this observation led to hypothesize that local reduction of leptin in the joint cavity could have a protective effect against erosions [71]. Accordingly, Olama et al. [69] found that serum leptin and the leptin synovial/serum ratio were higher in RA patients with erosive disease compared to patient without erosions. The presumed protective role of leptin against bone destruction might be due to leptin-induced IL-1 receptor antagonist expression and chondrocyte proliferation [69].
Conversely, leptin is known to increase Th17 proliferation [18], a Tcell subset, with a potential osteoclastogenic effect in the arthritic joint [72]. Th17 cells express RANKL and TNF, thus directly inducing osteoclastogenesis [72]. Moreover, IL-17, produced by Th17 cells, stimulates fibroblasts and osteoblasts to produce RANKL, therefore inducing osteoclast development [73]. Additionally, IL-17 promotes the inflammatory joint status by increasing the production of other several pro-inflammatory citokines, such as TNFα, IL-6, and IL-1, thus further inducing the inflammation mediated bone loss [74]. In this regard, RA patients are characterized by higher levels of peripheral Th17 frequencies and Th17-related cytokines, but also by higher levels of Th17 and IL-17 in synovial fluid compared to non-RA synovial fluid. Furthermore, Th17 levels correlated with disease activity in different studies [74], thus linking this Tcell subset, and therefore indirectly leptin, with RA prognosis and joint destruction.
However, other studies failed to demonstrate a correlation between leptin and bone erosions in RA [71]. The conflicting results could be explained by different study design and parameters used for adjustment; therefore, further research is needed in order to fully understand the contribution of leptin to RA pathogenesis.
The role of resistin in the pathogenesis of RA has been investigated in several studies. Resistin can be found in blood plasma or serum, synovial fluid, and synovial tissue of RA patients; moreover, it is expressed by synovial fibroblasts and infiltrating immune cells in joints affected by RA [4]. Higher synovial resistin levels correlate with RA disease activity, IL-6, and joint damage. Concerning serum resistin levels, most studies failed to find a difference between RA patients and controls, although there is evidence that they positively correlate with DAS28 and inflammatory markers, such as CRP or TNFα [3, 4, 75, 76]. Conversely, other authors have reported no association between circulating resistin levels and the aforementioned markers [3, 4, 75]. Despite these conflicting results, data collected from animal studies strongly suggest the hypothesis of a resistin involvement in RA pathogenesis. In fact, intra-articular resistin injection in healthy mice knee induced leukocyte infiltration and hyperplasia of the synovia, leading to a joint inflammation similar to human arthritis, and increased the expression of several pro-inflammatory cytokines [77]. Moreover, resistin has been considered a key player in RA angiogenesis, through the upregulation of VEGF expression and endothelial progenitor cell infiltration in a collagen-induced arthritis mice model [78]. Additionally, the authors observed greater bone erosion in this animal model. The pro-inflammatory action of resistin in RA patients was observed in most studies, suggesting that this adipokine could be a good marker to assess disease progression [3]. It has also been suggested that resistin could be considered as a prognostic marker in RA as it can predict radiographic progression of joint damage. In fact, resistin serum levels have been found to significantly correlate to Larsen score and therefore joint destruction [76].
Since its known role as a pro-inflammatory adipokine, several studies focused on the role of visfatin in rheumatic diseases. Circulating levels of visfatin were found to be higher in RA patients than in healthy controls, and visfatin levels positively correlated with CRP and RA disease activity assessed with DAS28 [3]. Moreover, both animal and clinical studies provided evidence of a role played by this adipokine in the pathogenesis of RA-associated bone loss. In a study conducted by Li et al. [79] on a murine collagen-induced arthritis model, visfatin-deficient mice presented reduced inflammatory bone destruction and disease progression; further, the authors observed that visfatin expression was required for osteoclastogenesis. These results are supported by previous findings by Busso et al. [80]. Visfatin was also connected to bone erosions in clinical studies. Mirfeizi et al. found [81] that visfatin serum levels were significantly higher in patients with radiographic joint damage dependently to disease duration. In a previous study, visfatin levels in RA patients were associated with radiographic progression, but the authors speculated that this association could be in fact mediated by ACPA, since visfatin levels also correlated with ACPA levels [65].
Similarly, chemerin and vaspin could be involved in RA pathogenesis. Indeed, chemerin and its receptor are expressed by chondrocytes and synovial fibroblasts from RA and OA patients and this adipokine induced the expression of pro-inflammatory cytokines and MMPs in these cells [4]. Moreover, chemerin plasma levels correlated with disease activity in RA patients [82].
Likewise, synovial fluid vaspin levels were found higher in RA patients compared to OA patients and showed a tendency to correlate with disease activity assessed by DAS28 [83]. Moreover, serum vaspin concentration was associated with the development of clinically manifest arthritis after follow-up [84].
Adipokines and SpA
The growing evidence indicating a role of adipokines in osteogenesis and bone metabolism has encouraged researchers to investigate the function of these factors in regard to SpA. Differently from RA bone involvement, SpA is distinguished by the co-existence of both destructive and productive bone lesions, due to alteration of bone remodeling in the affected joints [85]. Among the SpA family, the majority of studies evaluating the relationship with adipokines focused on ankylosing spondylitis (AS). AS is a chronic inflammatory disease that primarily affects the axial skeleton, i.e. spine and sacroiliac joint. Pathologically, AS is characterized by a three-stage process, resulting in a paradoxical coexistence of bone destruction and formation. The first stage is the inflammation driven mainly by TNFα, which causes erosions in cartilage and bone; these lesions are then filled in by fibrous tissue, which is later ossified in the last stage leading to abnormal bone outgrowth. New bone formation is actually a hallmark of the disease and is represented by syndesmophytes, bone bridges between the vertebral bodies and subsequent complete ankylosis of the spine [51, 86]. Undoubtedly, these productive bone lesions typical of AS deeply impact quality of life in the long-term, since they irreversibly constrain mobility of the spine [87].
Psoriatic arthritis (PsA) belongs to the SpA and is typically associated with psoriasis of the skin or nails. The dual bone involvement characteristic of PsA consists of bone erosions and new bone formation, typically at the edges of the cartilage joint at the insertion of the enthesis. Several evidences suggest that the osteo-proliferative processes prevalently occur by endochondral ossification and that several systemic factors, including adipokines, as well as local factors, angiogenesis and mechanical stress are involved [88].
Many studies found no statistical differences in serum adiponectin levels between AS patients and healthy control group [86, 87, 89, 90]. Further, several studies evaluating the radiographic changes using the modified Stoke Ankylosing Spondylitis Spine Score (mSASSS) [86, 87, 91] and the syndesmophyte formation/progression score [87] showed no correlation between circulating adiponectin and radiographic progression of disease and syndesmophyte formation [86, 87]. Conversely, Derdemezis et al. [92] found that serum adiponectin levels were significantly higher in AS patients compared with controls. These conflicting results may be explained by different measurement methods, but also by further evidences suggesting that factors like age and sex could influence serum adiponectin levels in AS patients. Yang et al. [89] observed that in the subgroup of patients aged over 40 years, serum adiponectin levels were higher than in controls. Further, Hartl et al. [93] found higher HMW adiponectin levels in female patients with AS compared to male patients. This result is consistent with data on gender-dependent differences in adipokine levels in the general population and with previous studies on AS patients [87], and it is ascribed to the inhibitory effect of androgens [87]. Interestingly, the authors observed also that serum HMW adiponectin levels were inversely associated with radiographic spinal progression (defined as mSASSS worsening by ≥ 2 points after 2 years, and/or syndesmophyte formation/progression after 2 years) in AS patients. The gender-dependent differences in adiponectin levels gain further importance since female patients in the study by Hartl et al. also had significantly lower mSASSS than men [93], once again accordingly to previous literature [94, 95]. The authors consequently speculated that HMW adiponectin might be a biomarker linking gender with radiographic spinal progression in AS patients and that adiponectin might play a natural protective role against radiographic spinal progression in the disease, explaining the generally slower axial new bone formation in female patients.
Adiponectin seems to have a role in PsA pathogenesis also. Xue et al. [96] found significantly lower levels of adiponectin in PsA patients compared to healthy and psoriasis controls. Moreover, the authors observed a negative correlation between adiponectin and peripheral blood osteoclast precursors in PsA patients. These findings support the evidence of adiponectin inhibition of osteoclast formation, in particular in the early stages of TNF/RANKL-induced osteoclastogenesis [10].
Studies on leptin role in patients with AS led to conflicting results. Higher levels of leptin in AS compared to healthy subjects and a correlation of leptin levels with disease activity, CRP and IL-6 levels in male patients have been described [26]. Different studies showed a significant correlation between leptin levels and Bath Ankylosing Spondylitis Metrology Index (BASMI) [97]. In contrast, other studies found lower leptin levels in AS patients than healthy controls [26, 98]. At the same time, no differences were found in leptin levels between AS patients and controls nor in AS patients during anti-TNFα therapy by Derdemesiz et al. [92]. A possible explanation for these ambiguous results could be found in the local action of leptin at inflammatory sites in AS (sacroiliac joints and spine, entheseal structures); in other words, leptin could exert its action in AS locally rather than systemically. In this regard, several studies found an interesting relation between leptin and syndesmophytes, a hallmark of this disease. Kim et al. [99] found that serum leptin levels were higher in male AS patients with syndesmophytes compared to those without, and correlated with bone specific alkaline phosphatase, a bone formation marker, suggesting a potential role of leptin in new bone formation in AS. Similar results were found by Gonzalez-Lopez et al. [90]. Moreover, in a study by Park et al. [86], leptin levels increased in AS patients in 2-year follow-up and this increase was associated with radiographic progression, assessed as mSASSS worsening by ≥ 2 points after 2 years. The authors speculated that the connection between leptin and new bone formation in AS might be found in the so-called fatty Romanus lesion, the focal fat infiltration visible on magnetic resonance imaging of the spine in AS patients, since it has been demonstrated that it is followed by new syndesmophyte formation after 2 years [99]. Thus, considering leptin involvement in bone metabolism described above, it has been suggested that leptin may upregulate osteoblast proliferation acting via a paracrine mechanism in the focal fat lesions in the spine, therefore contributing to new bone formation and radiographic progression in AS [86].
In addition to the aforementioned role of adiponectin in PsA, Xue et al. [96] found increased leptin and omentin levels in PsA patients compared with healthy controls; further, leptin and omentin were positively correlated with osteoclast precursor number. Moreover, leptin positively correlated also with PsA disease activity index. The authors hypothesized that the chronic inflammation status typical of the disease and leptin production might mutually enhance each other, leading to increased bone remodeling. Conversely, chemerin levels were significantly lower in PsA patients compared to healthy controls. Also, vaspin systemic levels were found higher in PsA patients compared to healthy controls, even though there was no significant correlation with disease activity, measured using the disease activity in PsA (DAPSA), Bath Ankylosing Spondylitis Disease Activity Index (BASDAI), Bath Ankylosing Spondylitis Functional Index (BASFI), Visual Analogue Scale (VAS), and Minimal Disease Activity (MDA) [100].
In AS patients, higher resistin serum levels compared to healthy controls have been reported [87]. Moreover, resistin serum levels correlated with CRP levels but not with radiographic progression assessed by mSASSS worsening of ≥ 2 units in 2 years and syndesmophyte formation/progression. Alongside leptin, Park et al. [86] investigated the association between resistin and AS: they found significantly higher baseline serum resistin levels in AS patients than in healthy subjects, and they observed a significant increase in resistin levels associated with radiographic progression. Therefore, the authors speculated that baseline resistin levels may be predictive of new bone formation in AS patients and that both leptin and resistin may be used as biomarkers of clinical outcome of AS.
Visfatin serum levels have been suggested as an inflammation-independent predictor of radiographic progression in AS patients. Indeed, in a study conducted by Syrbe et al. [87], they were higher in AS patients compared to healthy controls and in AS patients with subsequent radiographic spinal progression assessed by syndesmophyte formation/progression, compared to those without; visfatin levels, however, did not correlate with acute-phase reactant levels.
Adipokines and OA
OA is a multifactorial chronic disease considered one of the main causes of disability, in which mechanical, genetic, biochemical, and metabolic factors are involved. The hallmark of the disease are joint cartilage degradation and typical changes of adjacent subchondral bone with consequent osteophyte formation and subchondral bone sclerosis [101]. OA-affected joints could also present synovial inflammation, with synovial fibroblast and macrophage proliferation, alongside immune cell infiltration and consequent production of pro-inflammatory cytokines [4]. Adipokine role in OA has been deeply investigated in recent years.
Adiponectin serum levels are higher in OA patients than in healthy controls [102]. In addition, leptin serum and synovial fluid levels were found to be increased in OA patients and associated with pain and disease severity [103]. This adipokine could actually be involved in both OA inflammation and bone complications since it increases the expression of pro-inflammatory cytokines, such as IL-1, and MMPs in OA chondrocytes and animal models [4]. Accordingly, leptin has been found to enhance the production of IL-6 and IL-8 in T cells from OA patients [104]; these results supported the findings of a previous study conducted on leptin-deficient mice in which no increase in knee OA incidence compared to controls was observed [105]. Leptin has also been suggested to play a role in OA subchondral bone complications: its production was increased in OA osteoblasts compared to normal and was associated with higher levels of alkaline phosphatase, osteocalcin, collagen type I, and TGF-β [106, 107].
Growing body of evidence support the role of synovial fluid resistin in OA pathogenesis, reporting its involvement in pro-inflammatory response and cartilage catabolic activity. Resistin synovial fluid levels were indeed found higher in OA patients compared to healthy subjects [108] and were positively correlated with pro-inflammatory cytokines and MMPs expression in synovial fluid in OA-affected joints [109, 110]. Furthermore, resistin induced increased expression of pro-inflammatory cytokines and MMPs in human OA chondrocytes [111, 112].
Several studies described chemerin involvement in OA. This adipokine levels were found higher in synovial fluid of OA patients compared to controls and correlated with CRP serum levels and disease severity [113,114,115]. Accordingly, in a rat OA model, stimulation with chemerin and IL-1 combined administration lead to increased expression of MMPs, therefore aggravating the disease [116].
In addition, visfatin has been reported to increase expression of MMPs in OA chondrocytes [117] suggesting a catabolic function and therefore an important role in the pathophysiology of OA; in addition, its circulating levels were found increased in OA patients compared to healthy controls [118].
Lastly, serum vaspin levels were found lower in OA patients compared to healthy controls [119], while omentin synovial fluid levels in OA patients negatively correlated with symptomatic [120] and radiographic severity [121].
Adipokines and OP
OP is a multifactorial systemic bone disorder characterized by bone loss and micro-architectural deterioration of bone tissue, leading to increased fragility fracture risk, thus contributing to disability and mortality. The impaired bone mass is due to an imbalance in bone remodeling phases between osteoblastic bone formation and osteoclastic bone resorption processes, leading to increased bone loss. OP is most frequently observed in post-menopausal women and in the elderly population [8]. Postmenopausal osteoporosis is the most common type of osteoporosis and is mainly due to the menopause-related estrogen deficiency that promotes osteoclastogenesis and bone resorption [122]. Moreover, estrogen deficiency induces the low-grade inflammatory state typical of aging which in itself promotes bone resorption [122]. Indeed, pro-inflammatory cytokines, such as IL-6 and TNFα, enhance osteoclastogenesis and inhibit bone formation [8, 122, 123]. The close relationship between bone tissue and bone marrow adipose tissue (MAT), which secretes various adipokines, has led researchers to raise the hypothesis of MAT involvement in the pathogenesis of OP. Actually, some studies show an inverse relationship between bone mass and MAT in postmenopausal women and in animal models [124,125,126,127]. The adipokines produced by MAT can influence bone loss directly and indirectly via their pro-inflammatory activity. In the bone marrow of patients affected by osteoporosis, adiponectin and leptin levels were lower than in non-osteoporotic subjects [128]; conversely, adiponectin serum levels were inversely correlated with bone density in other studies [10]. These conflicting results may be explained by the multiple activity mechanisms of adiponectin in bone: local paracrine effects of adiponectin secreted from bone marrow adipocytes, endocrine effects of the adiponectin secreted by white adipose tissue and released into the blood stream [10]. In conclusion, although the understanding of the relationship between MAT and bone metabolism is still at the beginning stage and therefore further studies are needed, it can be speculated that the crosstalk between inflammation and MAT expansion creates a vicious circle in which adipokines may represent the pathogenic cornerstone, overall contributing to osteoporotic bone alterations.
Conclusions
Chronic rheumatic inflammatory diseases are characterized by important bone complications with severe ramifications on quality of life of these patients. In recent years, the importance of adipokines in the pathogenesis of rheumatic disease has been largely investigated; however, given the pleiotropic actions of these molecules and the complex pathophysiology of these diseases, their role is not fully understood yet. Further efforts are required to exhaustively unveil the crosstalk between adipose tissue, bone and immune system and therefore provide new knowledge for future innovative therapies for rheumatic diseases.
References
Francisco V, Pino J, Campos-Cabaleiro V, et al. Obesity, fat mass and immune system: role for leptin. Front Physiol. 2018;9:640.
Mancuso P. The role of adipokines in chronic inflammation. ImmunoTargets Ther. 2016;5:47–56.
Fatel EC de S, Rosa FT, Simão ANC, Dichi I. Adipokines in rheumatoid arthritis. Adv Rheumatol (London, England). 2018;58(1):25.
Carrión M, Frommer KW, Pérez-García S, Müller-Ladner U, Gomariz RP, Neumann E. The adipokine network in rheumatic joint diseases. Int J Mol Sci. 2019;20(17):4091.
Francisco V, Pino J, Gonzalez-Gay MA, et al. Adipokines and inflammation: is it a question of weight? Br J Pharmacol. 2018;175(10):1569–79.
Fantuzzi G, Faggioni R. Leptin in the regulation of immunity, inflammation, and hematopoiesis. J Leukoc Biol. 2000;68(4):437–46.
Neumann E, Junker S, Schett G, Frommer K, Müller-Ladner U. Adipokines in bone disease. Nat Rev Rheumatol. 2016;12(5):296–302.
Pietschmann P, Mechtcheriakova D, Meshcheryakova A, Ellinger UFI. Immunology of osteoporosis: a mini-review. Gerontology. 2016;62:128–37.
Reid IR. Fat and bone. Arch Biochem Biophys. 2010;503(1):20–7.
Naot D, Musson DS, Cornish J. The activity of adiponectin in bone. Calcif Tissue Int. 2017;100(5):486–99.
Pal China S, Sanyal S, Chattopadhyay N. Adiponectin signaling and its role in bone metabolism. Cytokine. 2018;112:116–31.
Choi HM, Doss HM, Kim KS. Multifaceted physiological roles of adiponectin in inflammation and diseases. Int J Mol Sci. 2020;21(4):1219.
Wolf AM, Wolf D, Rumpold H, Enrich B, Tilg H. Adiponectin induces the anti-inflammatory cytokines IL-10 and IL-1RA in human leukocytes. Biochem Biophys Res Commun. 2004;323(2):630–5.
Bråkenhielm E, Veitonmäki N, Cao R, et al. Adiponectin-induced antiangiogenesis and antitumor activity involve caspase-mediated endothelial cell apoptosis. Proc Natl Acad Sci U S A. 2004;101(8):2476–81.
Yamamoto R, Ueki S, Moritoki Y, et al. Adiponectin attenuates human eosinophil adhesion and chemotaxis: implications in allergic inflammation. J Asthma. 2013;50(8):828–35.
Yokota T, Oritani K, Takahashi I, et al. Adiponectin, a new member of the family of soluble defense collagens, negatively regulates the growth of myelomonocytic progenitors and the functions of macrophages. Blood. 2000;96(5):1723–32.
Lee YH, Bae SC. Circulating adiponectin and visfatin levels in rheumatoid arthritis and their correlation with disease activity: a meta-analysis. Int J Rheum Dis. 2018;21(3):664–72.
Tsatsanis C, Zacharioudaki V, Androulidaki A. Adiponectin induces TNF-alpha and IL-6 in macrophages and promotes tolerance to itself and other pro-inflammatory stimuli. Biochem Biophys Res Commun. 2005;335(4):1254–63.
Rossi A, Lord J. Adiponectin inhibits neutrophil phagocytosis of Escherichia coli by inhibition of PKB and ERK 1/2 MAPK signalling and Mac-1 activation. PLoS One. 2013;8(7):e69108.
Abella V, Scotece M, Conde J, et al. Leptin in the interplay of inflammation, metabolism and immune system disorders. Nat Rev Rheumatol. 2017;13(2):100–9.
Francisco V, Ruiz-Fernández C, Pino J, et al. Adipokines: Linking metabolic syndrome, the immune system, and arthritic diseases. Biochem Pharmacol. 2019;165:196–206.
Feijóo-Bandín S, Portolés M, Roselló-Lletí E, et al. 20 years of leptin: Role of leptin in cardiomyocyte physiology and physiopathology. Life Sci. 2015;140:10–8.
Scotece M, Conde J, Gómez R, et al. Role of adipokines in atherosclerosis: interferences with cardiovascular complications in rheumatic diseases. Mediators Inflamm. 2012;2012:125458.
Chou SH, Mantzoros C. 20 years of leptin: role of leptin in human reproductive disorders. J Endocrinol. 2014;223(1):T49-62.
Amitani M, Asakawa A, Amitani H, et al. The role of leptin in the control of insulin-glucose axis. Front Neurosci. 2013;7:51.
Navarini L, Margiotta DPE, Vadacca M, Afeltra A. Leptin in autoimmune mechanisms of systemic rheumatic diseases. Cancer Lett. 2018;423:139–46.
Mancuso P, Canetti C, Gottschalk A, Tithof PK, Peters-Golden M. Leptin augments alveolar macrophage leukotriene synthesis by increasing phospholipase activity and enhancing group IVC iPLA2 (cPLA2gamma) protein expression. Am J Physiol Lung Cell Mol Physiol. 2004;287(3):L497-502.
Caldefie-Chezet F, Poulin A, Tridon A, Sion B, Vasson MP. Leptin: a potential regulator of polymorphonuclear neutrophil bactericidal action? J Leukoc Biol. 2001;69(3):414–8.
Mattioli B, Straface E, Matarrese P, et al. Leptin as an immunological adjuvant: enhanced migratory and CD8+ T cell stimulatory capacity of human dendritic cells exposed to leptin. FASEB J Off Publ Fed Am Soc Exp Biol. 2008;22(6):2012–22.
Mattioli B, Straface E, Quaranta MG, Giordani L, Viora M. Leptin promotes differentiation and survival of human dendritic cells and licenses them for Th1 priming. J Immunol. 2005;174(11):6820–8.
Jamaluddin MS, Yan S, Lü J, Liang Z, Yao Q, Chen C. Resistin increases monolayer permeability of human coronary artery endothelial cells. PLoS One. 2013;8(12):e84576.
Cho Y, Lee SE, Lee HC, et al. Adipokine resistin is a key player to modulate monocytes, endothelial cells, and smooth muscle cells, leading to progression of atherosclerosis in rabbit carotid artery. J Am Coll Cardiol. 2011;57(1):99–109.
Silswal N, Singh AK, Aruna B, Mukhopadhyay S, Ghosh S, Ehtesham NZ. Human resistin stimulates the pro-inflammatory cytokines TNF-alpha and IL-12 in macrophages by NF-kappaB-dependent pathway. Biochem Biophys Res Commun. 2005;334(4):1092–101.
Chao WC, Yen CL, Wu YH, et al. Increased resistin may suppress reactive oxygen species production and inflammasome activation in type 2 diabetic patients with pulmonary tuberculosis infection. Microbes Infect. 2015;17(3):195–204.
Watanabe K, Watanabe R, Konii H, et al. Counteractive effects of omentin-1 against atherogenesis. Cardiovasc Res. 2016;110(1):118–28.
Rao SS, Hu Y, Xie PL, et al. Omentin-1 prevents inflammation-induced osteoporosis by downregulating the pro-inflammatory cytokines. Bone Res. 2018;6:9.
Lin Y, Zhuang J, Li H, et al. Vaspin attenuates the progression of atherosclerosis by inhibiting ER stress-induced macrophage apoptosis in apoE-/- mice. Mol Med Rep. 2016;13(2):1509–16.
Saalbach A, Tremel J, Herbert D, et al. Anti-inflammatory action of keratinocyte-derived vaspin: relevance for the pathogenesis of psoriasis. Am J Pathol. 2016;186(3):639–51.
Moschen AR, Kaser A, Enrich B, et al. Visfatin, an adipocytokine with proinflammatory and immunomodulating properties. J Immunol. 2007;178(3):1748–58.
Li Y, Zhang Y, Dorweiler B, et al. Extracellular Nampt promotes macrophage survival via a nonenzymatic interleukin-6/STAT3 signaling mechanism. J Biol Chem. 2008;283(50):34833–43.
Jia SH, Li Y, Parodo J, et al. Pre-B cell colony-enhancing factor inhibits neutrophil apoptosis in experimental inflammation and clinical sepsis. J Clin Invest. 2004;113(9):1318–27.
Yun MR, Seo JM, Park HY. Visfatin contributes to the differentiation of monocytes into macrophages through the differential regulation of inflammatory cytokines in THP-1 cells. Cell Signal. 2014;26(4):705–15.
Vermi W, Riboldi E, Wittamer V, et al. Role of ChemR23 in directing the migration of myeloid and plasmacytoid dendritic cells to lymphoid organs and inflamed skin. J Exp Med. 2005;201(4):509–15.
Wittamer V, Franssen J-D, Vulcano M, et al. Specific recruitment of antigen-presenting cells by chemerin, a novel processed ligand from human inflammatory fluids. J Exp Med. 2003;198(7):977–85.
Laranjeira S, Regan-Komito D, Iqbal AJ, Greaves DR, Payne SJ, Orlowski P. A model for the optimization of anti-inflammatory treatment with chemerin. Interface Focus. 2018;8(1):20170007.
Izgüt-Uysal VN, Gemici B, Birsen I, Acar N, Üstünel I. The effect of apelin on the functions of peritoneal macrophages. Physiol Res. 2017;66(3):489–96.
Liu R, Zhao P, Zhang Q, et al. Adiponectin promotes fibroblast-like synoviocytes producing IL-6 to enhance T follicular helper cells response in rheumatoid arthritis. Clin Exp Rheumatol. 2020;38(1):11–8.
Heinlen L, Humphrey MB. Skeletal complications of rheumatoid arthritis. Osteoporos Int. 2017;28(10):2801–12.
Schett G, Gravallese E. Bone erosion in rheumatoid arthritis: mechanisms, diagnosis and treatment. Nat Rev Rheumatol. 2014;8(11):656–64.
Haugeberg G, Ørstavik RE, Kvien TK. Effects of rheumatoid arthritis on bone. Curr Opin Rheumatol. 2003;15(4):469–75.
Cici D, Corrado A, Rotondo C, Cantatore FP. Wnt signaling and biological therapy in rheumatoid arthritis and spondyloarthritis. Int J Mol Sci. 2019;20(22):5552.
Corrado A, Colia R, Rotondo C, Sanpaolo E, Cantatore FP. Changes in serum adipokines profile and insulin resistance in patients with rheumatoid arthritis treated with anti-TNF-α. Curr Med Res Opin. 2019;35(12):2197–205.
Franco-Trepat E, Alonso-Pérez A, Guillán-Fresco M, et al. Visfatin as a therapeutic target for rheumatoid arthritis. Expert Opin Ther Targets. 2019;23(7):607–18.
Mac Donald IJ, Liu SC, Huang CC, Kuo SJ, Tsai CH, Tang CH. Associations between adipokines in arthritic disease and implications for obesity. Int J Mol Sci. 2019;20(6):1505.
Cao H, Lin J, Chen W, Xu G, Sun C. Baseline adiponectin and leptin levels in predicting an increased risk of disease activity in rheumatoid arthritis: a meta-analysis and systematic review. Autoimmunity. 2016;49(8):547–53.
Kontny E, Janicka I, Skalska U, Mas̈liński W. The effect of multimeric adiponectin isoforms and leptin on the function of rheumatoid fibroblast-like synoviocytes. Scand J Rheumatol. 2015;44(5):363–8.
Skalska U, Kontny E. Adiponectin isoforms and leptin impact on rheumatoid adipose mesenchymal stem cells function. Stem Cells Int. 2016;2016:6532860.
Liu D, Luo S, Li Z. Multifaceted roles of adiponectin in rheumatoid arthritis. Int Immunopharmacol. 2015;28(2):1084–90.
Zhao T, Kecheng Y, Zhao X, et al. The higher serum endocan levels may be a risk factor for the onset of cardiovascular disease: a meta-analysis. Medicine (Baltimore). 2018;97(49):e13407.
Kim KS, Lee YA, Ji HI, et al. Increased expression of endocan in arthritic synovial tissues: effects of adiponectin on the expression of endocan in fibroblast-like synoviocytes. Mol Med Rep. 2015;11(4):2695–702.
Araki Y, Mimura T. Matrix metalloproteinase gene activation resulting from disordred epigenetic mechanisms in rheumatoid arthritis. Int J Mol Sci. 2017;18(5):905.
Krumbholz G, Junker S, Meier FMP, et al. Response of human rheumatoid arthritis osteoblasts and osteoclasts to adiponectin. Clin Exp Rheumatol. 2017;35(3):406–14.
Sun X, Feng X, Tan W, et al. Adiponectin exacerbates collagen-induced arthritis via enhancing Th17 response and prompting RANKL expression. Sci Rep. 2015;5(February):1–11.
Qian J, Xu L, Sun X, et al. Adiponectin aggravates bone erosion by promoting osteopontin production in synovial tissue of rheumatoid arthritis. Arthritis Res Ther. 2018;20(1):26.
Klein-Wieringa IR, Van Der Linden MPM, Knevel R, et al. Baseline serum adipokine levels predict radiographic progression in early rheumatoid arthritis. Arthritis Rheum. 2011;63(9):2567–74.
Giles JT, Van Der Heijde DM, Bathon JM. Association of circulating adiponectin levels with progression of radiographic joint destruction in rheumatoid arthritis. Ann Rheum Dis. 2011;70(9):1562–8.
van der Heijde D. How to read radiographs according to the Sharp/van der Heijde method. J Rheumatol. 2000;27(1):261–3.
Meyer M, Sellam J, Fellahi S, et al. Serum level of adiponectin is a surrogate independent biomarker of radiographic disease progression in early rheumatoid arthritis: results from the ESPOIR cohort. Arthritis Rheumatol. 2013;15:R210.
Olama SM, Senna MK, Elarman M. Synovial/Serum leptin ratio in rheumatoid arthritis: the association with activity and erosion. Rheumatol Int. 2012;32(3):683–90.
Lee YH, Bae SC. Circulating leptin level in rheumatoid arthritis and its correlation with disease activity: a meta-analysis. Z Rheumatol. 2016;75(10):1021–7.
Toussirot É, Michel F, Binda D, Dumoulin G. The role of leptin in the pathophysiology of rheumatoid arthritis. Life Sci. 2015;140:29–36.
Adamopoulos IE, Bowman EP. Immune regulation of bone loss by Th17 cells. Arthritis Res Ther. 2008;10(5):225.
El Azreq MA, Arseneault C, Boisvert M, et al. Cooperation between IL-7 receptor and integrin α2β1 (CD49b) drives Th17-mediated bone loss. J Immunol. 2015;195(9):4198–209.
Wang M, Tian T, Yu S, et al. Th17 and Treg cells in bone related diseases. Clin Dev Immunol. 2013;2013:203705.
Ruscitti P, Di Benedetto P, Berardicurti O, et al. Adipocytokines in rheumatoid arthritis: the hidden link between inflammation and cardiometabolic comorbidities. J Immunol Res. 2018;2018:8410182.
Kassem E, Mahmoud L, Salah W. Study of resistin and YKL-40 in rheumatoid arthritis. J Am Sci. 2010;6(10):1004–12.
Bokarewa M, Nagaev I, Dahlberg L, Smith U, Tarkowski A. Resistin, an adipokine with potent proinflammatory properties. J Immunol. 2005;174(9):5789–95.
Su CM, Huang CY, Tang CH. Characteristics of resistin in rheumatoid arthritis angiogenesis. Biomark Med. 2016;10(6):651–60.
Li X, Islam S, Xiong M, et al. Epigenetic regulation of NfatC1 transcription and osteoclastogenesis by nicotinamide phosphoribosyl transferase in the pathogenesis of arthritis. Cell Death Discov. 2019;5:62.
Busso N, Karababa M, Nobile M, et al. Pharmacological inhibition of nicotinamide phosphoribosyltransferase/visfatin enzymatic activity identifies a new inflammatory pathway linked to NAD. PLoS One. 2008;3(5):e2267.
Mirfeizi Z, Noubakht Z, Rezaie AE, Jokar MH, Sarabi ZS. Plasma levels of leptin and visfatin in rheumatoid arthritis patients; is there any relationship with joint damage? Iran J Basic Med Sci. 2014;17(9):662–6.
Tolusso B, Gigante MR, Alivernini S, et al. Chemerin and PEDF are metaflammation-related biomarkers of disease activity and obesity in rheumatoid arthritis. Front Med. 2018;5:207.
Senolt L, Polanská M, Filková M, et al. Vaspin and omentin: new adipokines differentially regulated at the site of inflammation in rheumatoid arthritis. Ann Rheum Dis. 2010;69(7):1410–1.
Maijer KI, Neumann E, Müller-Ladner U, et al. Serum vaspin levels are associated with the development of clinically manifest arthritis in autoantibody-positive individuals. PLoS One. 2015;10(12):e0144932.
Magrey MN, Khan MA. The paradox of bone formation and bone loss in ankylosing spondylitis: evolving new concepts of bone formation and future trends in management. Curr Rheumatol Rep. 2017;19(4):17.
Park JH, Lee SG, Jeon YK, Park EK, Suh YS, Kim HO. Relationship between serum adipokine levels and radiographic progression in patients with ankylosing spondylitis: A preliminary 2-year longitudinal study. Medicine (Baltimore). 2017;96(33):e7854.
Syrbe U, Callhoff J, Conrad K, et al. Serum adipokine levels in patients with ankylosing spondylitis and their relationship to clinical parameters and radiographic spinal progression. Arthritis Rheumatol. 2015;67(3):678–85.
Neve A, Maruotti N, Corrado A, Cantatore FP. Pathogenesis of ligaments ossification in spondyloarthritis: insights and doubts. Ann Med. 2017;49(3):196–205.
Yang J, Zhang X, Ma Y, et al. Serum levels of leptin, adiponectin and resistin in patients with ankylosing spondylitis: a systematic review and meta-analysis. Int Immunopharmacol. 2017;52:310–7.
Gonzalez-Lopez L, Fajardo-Robledo NS, Miriam Saldaña-Cruz A, et al. Association of adipokines, interleukin-6, and tumor necrosis factor-α concentrations with clinical characteristics and presence of spinal syndesmophytes in patients with ankylosing spondylitis: a cross-sectional study. J Int Med Res. 2017;45(3):1024–35.
Creemers MCW, Franssen MJAM, van’t Hof MA, Gribnau FWJ, van de Putte LBA, van Riel PLCM. Assessment of outcome in ankylosing spondylitis: an extended radiographic scoring system. Ann Rheum Dis. 2005;64(1):127–9.
Derdemezis CS, Filippatos TD, Voulgari P V, Tselepis AD, Drosos AA, Kiortsis DN. Leptin and adiponectin levels in patients with ankylosing spondylitis. The effect of infliximab treatment. Clin Exp Rheumatol. 2010;28(6):880–3.
Hartl A, Sieper J, Syrbe U, et al. Serum levels of leptin and high molecular weight adiponectin are inversely associated with radiographic spinal progression in patients with ankylosing spondylitis: Results from the ENRADAS trial. Arthritis Res Ther. 2017;19(1):140.
Ramiro S, Stolwijk C, van Tubergen A, et al. Evolution of radiographic damage in ankylosing spondylitis: a 12 year prospective follow-up of the OASIS study. Ann Rheum Dis. 2015;74(1):52–9.
Feldtkeller E, Bruckel J, Khan MA. Scientific contributions of ankylosing spondylitis patient advocacy groups. Curr Opin Rheumatol. 2000;12(4):239–47.
Xue Y, Jiang L, Cheng Q, et al. Adipokines in psoriatic arthritis patients: the correlations with osteoclast precursors and bone erosions. PLoS One. 2012;7(10):e46740.
Chen XX, Yang T. Roles of leptin in bone metabolism and bone diseases. J Bone Miner Metab. 2015;33(5):474–85.
Mei YJ, Wang P, Chen LJ, Li ZJ. Plasma/serum leptin levels in patients with ankylosing spondylitis: a systematic review and meta-analysis. Arch Med Res. 2016;47(2):111–7.
Kim KJ, Kim JY, Park SJ, et al. Serum leptin levels are associated with the presence of syndesmophytes in male patients with ankylosing spondylitis. Clin Rheumatol. 2012;31(8):1231–8.
Colak S, Omma A, Sandikci SC, Yucel C, Omma T, Turhan T. Vaspin, neutrophil gelatinase-associated lipocalin and apolipoprotein levels in patients with psoriatic arthritis. Bratisl Lek Listy. 2019;120(1):65–9.
Corrado A, Maruotti N, Cantatore FP. Osteoblast role in rheumatic diseases. Int J Mol Sci. 2017;18(6):1272.
Tang Q, Hu ZC, Shen LY, Shang P, Xu HZ, Liu HX. Association of osteoarthritis and circulating adiponectin levels: a systematic review and meta-analysis. Lipids Health Dis. 2018;17(1):189.
Ku JH, Lee CK, Joo BS, et al. Correlation of synovial fluid leptin concentrations with the severity of osteoarthritis. Clin Rheumatol. 2009;28(12):1431–5.
Scotece M, Pérez T, Conde J, et al. Adipokines induce pro-inflammatory factors in activated Cd4+ T cells from osteoarthritis patient. J Orthop Res. 2017;35(6):1299–303.
Griffin TM, Huebner JL, Kraus VB, Guilak F. Extreme obesity due to impaired leptin signaling in mice does not cause knee osteoarthritis. Arthritis Rheum. 2009;60(10):2935–44.
Mutabaruka MS, Aoulad Aissa M, Delalandre A, Lavigne M, Lajeunesse D. Local leptin production in osteoarthritis subchondral osteoblasts may be responsible for their abnormal phenotypic expression. Arthritis Res Ther. 2010;12(1):R20.
Corrado A, Sanpaolo ER, Rotondo C, Cantatore FP. Pattern of adipokine expression in osteoblasts from osteoporotic and osteoarthritic bone. J Gerontol Geriatr. 2019;67(4):212–9.
Li XC, Tian F, Wang F. Clinical significance of resistin expression in osteoarthritis: a meta-analysis. Biomed Res Int. 2014;2014:208016.
Song Y, Guan J, Wang H, et al. Possible involvement of serum and synovial fluid resistin in knee osteoarthritis: cartilage damage, clinical, and radiological links. J Clin Lab Anal. 2016;30(5):437–43.
Koskinen A, Vuolteenaho K, Moilanen T, Moilanen E. Resistin as a factor in osteoarthritis: synovial fluid resistin concentrations correlate positively with interleukin 6 and matrix metalloproteinases MMP-1 and MMP-3. Scand J Rheumatol. 2014;43(3):249–53.
Zhang Z, Xing X, Hensley G, et al. Resistin induces expression of proinflammatory cytokines and chemokines in human articular chondrocytes via transcription and messenger RNA stabilization. Arthritis Rheum. 2010;62(7):1993–2003.
Lee JH, Ort T, Ma K, et al. Resistin is elevated following traumatic joint injury and causes matrix degradation and release of inflammatory cytokines from articular cartilage in vitro. Osteoarthr Cartil. 2009;17(5):613–20.
Ma J, Niu DS, Wan NJ, Qin Y, Guo CJ. Elevated chemerin levels in synovial fluid and synovial membrane from patients with knee osteoarthritis. Int J Clin Exp Pathol. 2015;8(10):13393–8.
Huang K, Du G, Li L, Liang H, Zhang B. Association of chemerin levels in synovial fluid with the severity of knee osteoarthritis. Biomarkers. 2012;17(1):16–20.
Şimşek Kaya G, Yapıcı Yavuz G, Kızıltunç A. Expression of chemerin in the synovial fluid of patients with temporomandibular joint disorders. J Oral Rehabil. 2018;45(4):289–94.
Ma J, Ren L, Guo CJ, Wan NJ, Niu DS. Chemerin affects the metabolic and proliferative capabilities of chondrocytes by increasing the phosphorylation of AKT/ERK. Eur Rev Med Pharmacol Sci. 2018;22(12):3656–62.
Gosset M, Berenbaum F, Salvat C, et al. Crucial role of visfatin/pre–B cell colony-enhancing factor in matrix degradation and prostaglandin E2 synthesis in chondrocytes: possible influence on osteoarthritis. Arthritis Rheum. 2008;58(5):1399–409.
Liao L, Chen Y, Wang W. The current progress in understanding the molecular functions and mechanisms of visfatin in osteoarthritis. J Bone Miner Metab. 2016;34(5):485–90.
Bao JP, Jiang LF, Chen WP, Hu PF, Wu LD. Expression of vaspin in the joint and the levels in the serum and synovial fluid of patients with osteoarthritis. Int J Clin Exp Med. 2014;7(10):3447–53.
Li ZG, Zhao DW, Xia CJ, et al. Decreased synovial fluid omentin-1 concentrations reflect symptomatic severity in patients with knee osteoarthritis. Scand J Clin Lab Invest. 2012;72(8):623–8.
Xu L, Zhu G, Wang L, Wang D, Jiang X. Synovial fluid omentin-1 levels are inversely correlated with radiographic severity of knee osteoarthritis. J Investig Med. 2012;60(3):583–6.
Li J, Chen X, Lu L, Yu X. The relationship between bone marrow adipose tissue and bone metabolism in postmenopausal osteoporosis. Cytokine Growth Factor Rev. 2020;52:88–98.
Franco-Trepat E, Guillán-Fresco M, Alonso-Pérez A, et al. Visfatin connection: present and future in osteoarthritis and osteoporosis. J Clin Med. 2019;8(8):1178.
Beekman KM, Zwaagstra M, Veldhuis-Vlug AG, et al. Ovariectomy increases RANKL protein expression in bone marrow adipocytes of C3H/HeJ mice. Am J Physiol Metab. 2019;317(6):E1050–4.
Beekman KM, Veldhuis-Vlug AG, den Heijer M, et al. The effect of raloxifene on bone marrow adipose tissue and bone turnover in postmenopausal women with osteoporosis. Bone. 2019;118:62–8.
Limonard EJ, Veldhuis-Vlug AG, van Dussen L, et al. Short-term effect of estrogen on human bone marrow fat. J Bone Miner Res. 2015;30(11):2058–66.
Li GW, Xu Z, Chen QW, et al. Quantitative evaluation of vertebral marrow adipose tissue in postmenopausal female using MRI chemical shift-based water–fat separation. Clin Radiol. 2014;69(3):254–62.
Pino AM, Ríos S, Astudillo P, et al. Concentration of adipogenic and proinflammatory cytokines in the bone marrow supernatant fluid of osteoporotic women. J Bone Miner Res. 2010;25(3):492–8.
Funding
Open access funding provided by Università di Foggia within the CRUI-CARE Agreement.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cici, D., Corrado, A., Rotondo, C. et al. Adipokines and Chronic Rheumatic Diseases: from Inflammation to Bone Involvement. Clinic Rev Bone Miner Metab 18, 58–71 (2020). https://doi.org/10.1007/s12018-021-09275-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12018-021-09275-w