
Abstract
A significant macrophage and T-cell infiltrate commonly occurs in inflammatory joint conditions such as rheumatoid arthritis that have significant bone destruction. Cytokines produced by activated macrophages and T cells are implicated in arthritis pathogenesis and are involved in osteoclast-mediated bone resorption. The scope of the present review is to analyze current knowledge and to provide a better understanding of how macrophage-derived factors promote the differentiation of a novel T-helper subset (Th17) that promotes osteoclast formation and activation.
Similar content being viewed by others


Introduction
Rheumatoid arthritis (RA) is a chronic inflammatory disease driven by immune dysregulation. The cause remains unknown, but environmental and genetic factors are believed to contribute to RA development. RA is characterized by joint inflammation, initially resulting in pain and swelling, and in a majority of the patients there is bone and cartilage erosion. The goal of current treatment regimens is to control inflammation and to retard the progression of structural damage of the joint bone structure as measured by X-ray analysis.
The RA joint synovial fluid and synovium contain a variety of hematopoeitic cells that are in direct proximity with the articular cartilage and underlying bone, and that contribute to the joint-destructive process. The present review will focus upon the bone-degrading cell, the osteoclast, and on how different T-cell subsets and their signature cytokines positively and negatively modulate osteoclast activity in autoimmune-driven inflammatory bone diseases. A specific T-cell subset, the Th17 cell, has a significant osteoclastogenic potential in the arthritic joint and may therefore provide a suitable target to combat arthritis and loss of joint function.
Osteoclast differentiation
Physiological and pathological bone resorption is carried out by a specialized cell called the osteoclast. Osteoclasts are large 20 to 100 μm multinucleated cells containing three to 100 nuclei with many mitochondria, lysosomes, dense granules, vesicles, and an extensive Golgi network required for the synthesis and secretion of factors required to degrade the bone matrix and subsequently phagocytose the resorbed products [1]. Tartrate-resistant acid phosphatase [2], cathepsin K [3], calcitonin receptor [4], and αvβ3 integrin [5] are characteristic gene products of the mature osteoclast [6].
The initial event in bone resorption is the attachment of the mature osteoclast to the bone matrix. Cell surface αvβ3 integrins bind to a variety of extracellular matrix proteins, including vitronectin, osteopontin, and bone sialoprotein. Arg-Gly-Asp-containing peptides, Arg-Gly-Asp mimetics, and blocking antibodies to αvβ3 integrins inhibit bone resorption in vitro and in vivo, suggesting that this integrin plays a key role in osteoclast function [7]. Once attached to bone, the osteoclast generates an isolated extracellular microenviron-ment between itself and the bone surface by creating a sealing zone structure unique to the osteoclast. A ring of filamentous actin associates with the intracellular proteins vinculin, talin, α-actinin, and cortactin, and the ring links to αvβ3 integrins that have bound various extracellular proteins. The ring appears when the osteoclast is immobilized on bone and during the resorption process. The ring disappears prior to osteoclast detachment from the eroded site and migration to another resorption site.
Resorption depends upon acidification of this extracellular compartment, leading to demineralization of the inorganic bone component and subsequent organic matrix degradation by cysteine proteases. Cathepsin K, an acid-activated cysteine proteinase, plays a critical and necessary role in the bone resorption process [8]. The expanded membrane within the filamentous actin ring creates additional surface area for massive H+ transport performed by the vacuolar (V-type) electrogenic H+-ATPase [9]. The proton source is carbonic acid produced by carbonic anhydrase type II [9]. The intra-cellular pH is balanced by a passive chloride–bicarbonate exchange in the basolateral membrane. Metalloproteinases secreted by vesicles into the sealing zone can also degrade the organic matrix, but their actions are only partly known. The resorbed material is transcytosed through the osteoclast [10, 11]. Although there are a few reports that macrophages and fibroblasts degrade or resorb mineralized bone in vitro [12, 13], it is widely accepted that osteoclasts are the only cells capable of lacunar resorption [6].
The osteoclast lineage is distinct from other mesenchymal cells of the bone (osteoblasts, chondrocytes, adipocytes, bone-marrow stromal cells, and fibroblasts). Osteoclasts are hematopoietic derived [14] and are from the colony-forming unit granulocyte–macrophage progenitor cells, which give rise to granulocytes and macrophages [15]. Monocytes are released from the bone marrow into the blood, where they home into different tissues and differentiate into tissue-resident macrophages. Multinucleated osteoclasts are formed under appropriate stimuli by the fusion of mononuclear precursors within the monocyte fraction of peripheral blood [16].
PU.1 is a monocyte/macrophage-specific transcription factor that acts as a master switch in programming hematopoietic cell commitment and differentiation [17]. PU.1 promotes the cell-type-specific expression of the myeloid lineage genes CD11b, CD11c, CD18, the granulocyte colony-stimulating factor (CSF) receptor, the granulocyte–macrophage CSF receptor, and the macrophage CSF receptor (c-fms) via binding to these genes' promoter regions [18]. Mice with homozygous PU.1 deficiency have osteopetrotic bones due to the lack of osteoclasts that would form from the myeloid lineage [19].
Osteoclast activation
Macrophage CSF and receptor activator of NFκB ligand (RANKL) are the most important factors known to date to drive osteoclast formation and activity [20, 21]. Macrophage CSF is a survival factor for osteoclast precursors due to upregulating Bcl-XL, inhibiting caspase-9 activation [22], and supporting mature osteoclast survival by preventing apoptosis [23]. Macrophage CSF stimulation also stimulates receptor activator of NFκB (RANK) expression in osteoclast precursor cells, thereby allowing RANKL to drive mature osteoclast formation [24]. RANKL is a transmembrane protein expressed by activated osteoblasts, synovial fibro-blasts, and T cells. It can also be proteolytically cleaved by TNF convertase (TACE) to generate a soluble molecule that has osteoclastic activity at distal sites [25, 26]. RANKL-induced osteoclastogenesis is inhibited by osteoprotegerin, a soluble decoy receptor for RANKL, which is also produced by a variety of cells including osteoblasts, synovial fibroblasts, B cells, and T cells [25–29]. Osteoprotegerin-deficient mice are severely osteoporotic [30], while osteoprotegerin-trans-genic mice are osteopetrotic [28].
The NFκB pathway is an integral component in the osteoclast differentiation pathway. RANKL activates NFκB both via the canonical pathway due to IκBα degradation to release p50/RelA and p50/c-Rel heterodimers and via the alternative or noncanonical pathway by promoting p100 processing into p52 [31]. TNF also stimulates NFκB activation via the canonical pathway [32], and a synergistic interaction between RANKL and TNF may account for the pathological osteoclastogenesis seen in RA [33, 34]. Elevated joint TNF activates TNF receptors expressed by osteoclast precursors [35] to drive NFκB and Jun N-terminal kinase activation using the TNF receptor adaptor proteins TRAF1, TRAF2, and TRAF6 [36–39]. Importantly, TNF induces osteoclastogenesis in RANK-deficient mice [40] and induces multinucleated cell formation from osteoclast precursors in the bone marrow macrophage population [41].
IL-1 receptors are expressed on mature osteoclasts [42], and exogenous IL-1 induces NFκB activation via TRAF6 [43]. IL-1 signals feed into the tyrosine kinase pathways through a TRAF6–Src molecular complex, which regulates the cyto-skeletal reorganization essential for osteoclast activation and can enhance the ruffled border formation of the mature osteo-clast and hence its resorbing activity [44].
There is a significant macrophage infiltrate in the arthritic joint, and the extent of synovial macrophage infiltration correlates strongly with the degree of joint erosion in arthritis [45]. Synovial macrophages isolated from different types of arthritides differentiate in vitro to fully functional osteoclasts via RANKL stimulation as well as independently of the RANK/RANKL signaling pathway, via TNF and IL-1α signaling [46]. Stimulatory, costimulatory, and/or inhibitory signals may be provided by adjacent T cells present in the inflammatory infiltrate.
T-cell subsets and their action on osteoclasts
The seminal observation in 1972 that osteoclast activity was increased by leukocyte-derived factors [47] gave rise to the idea that T cells influenced osteoclast differentiation and activation. Similar to the increased macrophage infiltrate, activated T cells are also found in inflamed RA synovial tissue and CD4+ T-cell infiltration has become a hallmark of RA pathogenesis. This concept has become central to the understanding of RA, and a new field termed osteoimmunology has recently emerged. How T cells regulate osteoclastogenesis is only partly known. We provide a brief overview of four T-cell subsets and the factors that drive their differentiation into these subsets, and we then concentrate on how the signature cytokine(s) of these T cells impact inflammation-driven osteoclastogenesis in arthritis.
T-helper 17 cells
The Th17 lineage has only recently been recognized and the factors involved in its differentiation are still being identified and sorted out. If a naïve T cell is activated in the presence of transforming growth factor beta (TGFβ) plus IL-6 in the mouse or TGFβ plus an inflammatory stimuli in the human, then the resulting clonal memory T-cell population will be instructed to produce the Th17 signature cytokines IL-17A, IL-17F, IL-22, and IL-26 (there is no mouse IL-26). The inflammatory stimuli in the human setting can be IL-1β, IL-6, IL-21, and/or IL-23 [48–53].
IL-17A is the only Th17 signature cytokine currently known to influence the biology of osteoclasts. The cytokine's message is also present in RAG-deficient mice (that is, T-cell and B-cell deficient), consistent with IL-17A also being produced from nonlymphoid sources [54]. IL-17A receptors are single-pass transmembrane proteins expressed by many cell types, including synoviocytes [55], chondrocytes, [56] osteoblasts, [57], and osteoclasts (our data). Receptor activation results in NFκB activation and the phosphorylation and activation of the extracellular signal-regulated kinase, Jun N-terminal kinase, and p38 mitogen-activated protein kinase pathways [56].
Th17 cells express RANKL [58] and TNF [52, 59], which directly act on osteoclast precursors to induce osteoclasto-genesis. IL-17A induces RANKL expression by synovial fibro-blasts and osteoblasts to indirectly drive bone erosion [60] and to activate synovial macrophages to secrete the known osteoclastogenic factors TNF and IL-1β [61]. Synovial and synovial fluid macrophages can differentiate to fully functional bone-resorbing osteoclasts, and Th17-induced synovial macrophage-osteoclast differentiation may represent an important cellular mechanism in the bone destruction associated with RA [46, 62].
Several types of IL-17A antagonists have been used in a variety of animal arthritis models to address the efficacy of therapeutic IL-17A neutralization. Polyclonal anti-IL-17A antibody treatment after disease induction in the collagen-induced arthritis (CIA) model decreased clinical scores over 10 days of therapy compared with controls. Ankles and knees had reduced synovitis, cartilage damage, chondrocyte death, proteoglycan depletion, and bone erosion (histologically and radiographically) [63]. Polyclonal anti-IL-17A antibody also inhibited the antigen-induced arthritis model's knee swelling, proteoglycan depletion, and bone erosion in the smoldering knee following reintroduction of antigen [64]. Mice immunized with formalin-fixed Borrelia burgdorferi and later challenged with live B. burgdorferi display transient joint inflammation. Knee swelling was reduced with either anti-IL-17A or anti-IL-17RA therapy, and ankles and knees were free of histo-pathological changes including bone erosions following either therapy [65]. Lastly, rat adjuvant-induced arthritis models treated with an IL-17R–Fc fusion protein demonstrated decreased paw swelling, joint histopathology scores, and bone radiographic scores [66].
Mechanistically, the bone protection following therapeutic IL-17A neutralization has been attributed to normalizing excessive RANKL levels. Anti-IL-17A blockade in CIA and antigen-induced arthritis models that demonstrated decreased bone erosion correlated with decreased RANKL message and RANKL-positive cells in the joints [64, 67].
T-helper 1 cells
IL-12 is a master differentiation factor produced by activated antigen-presenting cells. If a naïve T cell is activated in the presence of IL-12, then the resulting clonal memory T-cell population is instructed to produce the Th1 signature cytokine IFNγ.
IFNγ is also produced by another hematopoeitic cell type, the natural killer cell, following its activation. Osteoclast precursors and mature osteoclasts express the IFNγ receptor and exogenous IFNγ in vitro inhibits murine osteoclasto-genesis by inducing the rapid degradation of the RANK adapter protein TRAF6, resulting in strong inhibition of RANKL-induced activation of NFκB and Jun N-terminal kinase [68].
Activated mouse Th1 cells inhibited osteoclastogenesis when mixed in cocultures with either RANKL-stimulated mouse bone marrow macrophages or osteoblasts stimulated with vitamin D3 and prostaglandin E2 [58]. Activated Th1 cells expressed significant IFNγ quantities, and the inhibitory effects of these cells on osteoclastogenesis were abrogated when using IFNγ receptor-deficient osteoclast precursors [58]. Although exogenous IFNγ inhibits RANKL-induced human osteoclasto-genesis, it was paradoxical that IFNγ-producing human T cells induced osteoclast formation in a RANKL-dependent mechanism (discussed below) [69]. The increased osteoclasto-genesis effect may well be due to a Th1 proinflammatory cytokine TNF, which is known to synergize with RANKL.
The Th1 master differentiation factor IL-12 has anti-osteo-clastogenic activities but the mechanism of inhibition remains unclear. Exogenous IL-12 inhibited RANKL-stimulated osteo-clast formation from splenocytes or from osteoblast and splenocyte cocultures. This IL-12-dependent inhibition, however, was not due to IFNγ [70]. IL-12 was also identified as the anti-osteoclast factor produced by Toll-like receptor 9 stimulation with CpG oligodeoxynucleotides that opposed RANKL-induced osteoclast differentiation [71]. CpG oligo-deoxynucleotides, known to mimic bacterial DNA, modulate osteoclastogenesis via interactions with osteoclast precursors and osteoblasts [72]. Mice specifically deficient in IL-12 (that is, IL-12p35-deficient mice) were not protected from CIA, but show exacerbated paw swelling responses following collagen challenge [73]. Similarly, mice lacking IFNγ signaling are not protected in the CIA model [74]. A Lyme's disease arthritis model using a two-step inactivated Borrelia vaccination followed by live Borrelia challenge shows a greater disease penetrance and severity when using IFNγ-deficient mice [65].
Collectively, these data do not support that Th1 cells, nor their signature cytokine IFNγ or their master differentiation factor IL-12, are major drivers of inflammation-associated osteo-clastogenesis. These data instead support that this lineage may counteract other pro-osteoclast inflammation factors present in the RA joint, including Th17 products [75, 76].
T-helper 2 cells
The source of the Th2 master differentiation factor IL-4 during the naïve to memory transition is controversial, but mature Th2 cells, natural killer T cells, basophils, and mast cells produce IL-4 [77–79]. Th2 cells produce the signature cytokines IL-4, IL-5, and IL-13 when they encounter a nonself-antigen (or a cross-reacting self-antigen) being presented by an antigen-presenting cell. Little is know regarding the role, if any, that IL-5 plays in osteoclast biology, whereas IL-4 and IL-13 have been extensively studied. Mouse spleen cells and bone marrow macrophages express IL-4 and IL-13 receptors, and the addition of these factors decreased RANKL-stimulated tartrate-resistant acid phosphatase-positive multi-nucleated cell formation and cathepsin K message in spleno-cytes and bnone marrow macrophages [80]. IL-4 was more potent than IL-13 at inhibiting mouse osteoblast/osteoclast progenitor cocultures [81]. Mature osteoclasts express the IL-4 receptor, and exogenous IL-4 decreased tartrate-resistant acid phosphatase expression, actin ring formation, and bone resorption by osteoclasts [82]. Since IL-4 inhibits osteoclasto-genesis by inhibiting NFκB and mitogen-activated protein kinase signaling [83], the synergistic action of RANKL and TNF to stimulate osteoclast formation is also inhibited by IL-4 [84].
The Th2 signature cytokines IL-4 and IL-13 also use other mechanisms to inhibit osteoclastogenesis. IL-4 and IL-13 promote expression of the natural RANKL antagonist, osteo-protegerin, by endothelial cells and osteoblasts [81, 85]. IL-4 and IL-13 decreased RANKL and RANK protein in calvariae bone. Exogenous IL-4 addition to the CIA model had minimal impact on the outward signs of inflammation, but showed a bone-preserving biology including decreased bone erosion, tartrate-resistant acid phosphatase activity, and RANKL message [86, 87]. In total, these data do not support that Th2 cells or their signature cytokines IL-4 and IL-13 are major drivers of inflammation-associated osteoclastogenesis, but instead support that this lineage along with the Th1 lineage may counteract Th17 products and other pro-osteoclast inflammation factors present in the RA joint.
Regulatory T-cells
The regulatory T cell (Treg) lineage is composed of overlapping T-cell subsets whose role is to dampen the immune response to minimize tissue destruction. There are a number of mechanisms by which the Treg dampens the immune system, including some requiring cell-to-cell contact, but the present review will focus upon the Treg signature cytokines TGFβ and IL-10 [88].
There is support for Tregs inhibiting osteoclastogenesis, but there are discrepancies regarding the key mechanisms by which it occurs. Zaiss and colleagues concluded that CD4+CD25+Foxp3+ Tregs inhibited osteoclastogenesis in a cell-contact-dependent manner with a minor contribution by the signature cytokines IL-4, TGFβ, and IL-10 [89, 90]. In contrast, Kim and colleagues reported that human CD4+CD25+Foxp3+ Tregs inhibit osteoclast differentiation from peripheral blood mononuclear cells in a cell-contact-independent manner, but in an IL-4-dependent and TGFβ-dependent manner [91, 92].
Although IL-4 is not considered a Treg signature cytokine, it does inhibit osteoclastogenesis by inhibiting NFκB and mitogen-activated protein kinase signaling [83]. The role of TGFβ in bone turnover is quite complex, with conflicting in vivo and in vitro data on whether resorption is enhanced or inhibited [93]. IL-10, however, has a clear inhibitory effect on osteoclasts by downregulating NFATc1 [94].
Tregs are enriched in inflamed joints of patients with rheumatic disease [95], but the interaction of Tregs with activated monocytes in the joint might lead to diminished suppressive activity of Tregs in vivo – thus contributing to the chronic inflammation in RA [96]. Other workers have reported that Tregs have neither an inhibitory effect nor an enhancing effect on osteoclastogenesis. Collectively, these data do not support that Tregs nor their signature cytokines TGFβ and IL-10 are major drivers of inflammation-associated osteo-clastogenesis, but instead support that this lineage along with Th1 and Th2 signature cytokines may counteract Th17 and other pro-osteoclast inflammatory factors present in the RA joint.
Flavors of T-helper 17 cells
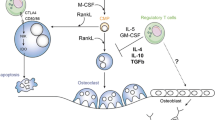
The schematic concept of separate nonoverlapping T-cell lineages identified by their unique cytokine repertoire, however, is hampered by reality (Figure 1). A subset of Th17 cells defined by their expression of the signature cytokine IL-17A also produce IFNγ, albeit less than that produced by Th1 cells [53]. The IL-17A/IFNγ double-producing human T cells arise from in vitro naïve to memory T-cell differentiation cultures and are found in peripheral blood memory T cells from healthy donors [50, 52, 53]. It could be envisioned that factors present in the microenvironment during the naïve to memory transition could alter either the number of double producers that arise or could alter the magnitude of IL-17A to IFNγ protein made by the double producers [49, 50, 53].
These different flavors of Th17 cells and, even more, the distinctly different T-cell subsets must be kept in mind when evaluating the literature precedent regarding T-cell effects on osteoclasts. The paradoxical observation that IFNγ-producing T cells promoted osteoclast formation, highlighted above, provides such an example. IFNγ-producing T cells and IFNγ-nonproducing T cells were cultured with human monocytes in the presence of macrophage CSF. To the authors' surprise, IFNγ-producing T cells induced osteoclastogenesis in a RANKL-dependent manner. The osteoclastogenesis was further enhanced when the anti-osteoclast factor IFNγ was neutralized, implicating other pro-osteoclast factor(s) being produced by IFNγ-producing T cells [69]. Th17 cells can be endowed with different cytokine repertoires and/or different secretion ratios between key cytokines based on master differentiation factors present in the microenvironment during initial antigen recognition [49]. All of the various osteoclast-related factors must be taken into account when hypothesizing the impact that an activated T cell may have in an inflamed joint.

Schematic representation of T cell differentiation and T-cell signature cytokines that inhibit or induce osteoclastogenesis. CFU-GM, granulocyte–macrophage colony-stimulating factor; RANKL, receptor activator of NFκB ligand; TGFβ, transforming growth factor beta; Treg, regulatory T cell.
Mouse models have been key to expanding our knowledge of various T-cell subsets, the biology of their signature cyto-kines, and how these cytokines affect osteoclasts. This is also true for the Th17 lineage; however, there are two keys areas where mouse studies must be analyzed with caution when translating to humans.
IL-6 (in combination of TGFβ) is required to differentiate activated naïve mouse CD4 T cells into memory IL-17A-producing Th17 cells [52]. Further exposure to IL-23 was required for full pathogenic activity in vivo (at least in animal central nervous system inflammation models). IL-6 is just one of a handful of proinflammatory factors (for example, IL-1β, IL-21, and IL-23) that can work singly and in combination to drive human Th17 development [49, 50, 52, 53]. This dichotomy may overemphasize the role of IL-6 (from mouse studies) in human Th17-mediated osteoclast formation.
Secondly, Th17 cells from mouse autoimmune models rarely coexpress IFNγ – whereas this is more the norm when working with human Th17 cells from healthy donors. Whether this is due to an inherent mouse/human difference or due to different disease states remains to be seen. Human Th17 cells may therefore have a more prominent IFNγ anti-osteo-clastogenesis brake on the human cell's activity than a corresponding mouse Th17 cell. This would be consistent with some rodent arthritis models having an explosive bone eroding component, in comparison with the much slower bone erosion seen in RA patients. It is important to note that these above cautions relate to how a memory Th17 cell is formed. Once the Th17 cell is formed, however, the osteoclastogenic factors produced by the cell have similar biology between the mouse and the human (for example, RANKL, TNF, IL-17A).
T-helper 17 cells in RA
Multiple lines of evidence point to Th17's disease association with RA. IL-17A protein is present in both the synovium and the synovial fluid of rheumatoid patients [97–101], and a subset of T-cell lines expanded in vitro from RA synovium expressed IL-17A and IFNγ following activation [59, 102]. Classical IFNγ-only Th1 cells were also present and can be expanded from RA synovium [102, 103]. Exploratory medical studies, however, not only support IL-17A's disease association, but implicate IL-17A's correlation with poor disease prognosis. IL-17A message in synovial membrane biopsies was one factor (including TNF, IL-1β, and IL-10) that was predictive for subsequent bone erosion and joint damage as assessed by magnetic resonance imaging and radiography [104].
Additionally, Raza and colleagues prospectively collected synovial fluid samples within a few weeks after symptom onset from patients with early synovitis [105]. The patient's outcomes were subsequently determined 18 months after fluid acquisition to determine what early factors expression correlated with synovitis progression to RA. Patients were grouped into those who subsequently progressed to RA, those who subsequently progressed to non rheumatoid persistent synovitis, or those whose synovitis resolved. Importantly, IL-17A was one of a few factors (including IL-2, IL-4, IL-13, IL-15, basic fibroblast growth factor, and epidermal growth factor) whose expression in the earliest stages of disease was associated with subsequent progression to RA [105]. An IL-17A single nucleotide polymorphism was significantly associated with radiographic progression after 2 years [106].
Collectively, these data support that IL-17A is present in the inflamed synovium and that IL-17A expression levels correlate with poor prognosis and greater joint destruction.
In summary, Th17 cells are arrayed with factors that can directly and indirectly drive osteoclastogenesis. It is well appreciated within the T-cell development literature that master differentiation factors and signature cytokines of the Th1, Th2, and Treg lineage inhibit Th17 development. This same concept holds true from an osteoclastogenesis viewpoint since master differentiation factors and signature cyto-kines of the Th1, Th2, and Treg lineage inhibit osteoclasto-genesis and Th17 products stimulate osteoclastogenesis.
Conclusion
Th17 cells are a newly discovered T-cell lineage that plays a role in the adaptive immune response to extracellular pathogens. The repertoire of molecules at its disposal to combat these pathogens is formidable. When these products are unleashed within an inflamed joint, however, they can directly drive osteoclast precursors to differentiate into osteoclasts via RANKL-dependent and RANKL-independent pathways. Additionally, Th17 factors can act on other cell types to indirectly increase the osteoclastogenic potential in the inflamed joint microenvironment. Targeting this T-cell lineage and/or its osteoclastic factors may provide alternative strategies to combat inflammatory joint diseases compared with the gold-standard TNF antagonist therapies.
Abbreviations
- CIA:
-
collagen-induced arthritis
- CSF:
-
colony-stimulating factor
- IFN:
-
interferon
- IL:
-
interleukin
- NF:
-
nuclear factor
- RA:
-
rheumatoid arthritis
- RANK:
-
receptor activator of NFκB
- RANKL:
-
receptor activator of NFκB ligand
- Th:
-
T-helper cells
- TGFβ:
-
transforming growth factor beta
- TNF:
-
tumor necrosis factor
- Treg:
-
regulatory T cell.
References
Holtrop ME, King GJ: The ultrastructure of the osteoclast and its functional implications. Clin Orthop. 1977, 123: 177-196.
Minkin C: Bone acid phosphatase: tartrate-resistant acid phosphatase as a marker of osteoclast function. Calcif Tissue Int. 1982, 34: 285-290. 10.1007/BF02411252.
Drake FH, Dodds RA, James IE, Connor JR, Debouck C, Richardson S, Lee-Rykaczewski E, Coleman L, Rieman D, Barthlow R, Hastings G, Gowen M: Cathepsin K, but not cathepsins B, L, or S, is abundantly expressed in human osteoclasts. J Biol Chem. 1996, 271: 12511-12516. 10.1074/jbc.271.21.12511.
Hattersley G, Chambers TJ: Calcitonin receptors as markers for osteoclastic differentiation: correlation between generation of bone-resorptive cells and cells that express calcitonin receptors in mouse bone marrow cultures. Endocrinology. 1989, 125: 1606-1612.
Davies J, Warwick J, Totty N, Philp R, Helfrich M, Horton M: The osteoclast functional antigen, implicated in the regulation of bone resorption, is biochemically related to the vitronectin receptor. J Cell Biol. 1989, 109 (4 Pt 1): 1817-1826. 10.1083/jcb.109.4.1817.
Teitelbaum SL: Bone resorption by osteoclasts. Science. 2000, 289: 1504-1508. 10.1126/science.289.5484.1504.
Horton MA, Taylor ML, Arnett TR, Helfrich MH: Arg-Gly-Asp (RGD) peptides and the anti-vitronectin receptor antibody 23C6 inhibit dentine resorption and cell spreading by osteoclasts. Exp Cell Res. 1991, 195: 368-375. 10.1016/0014-4827(91)90386-9.
Zaidi M, Troen B, Moonga BS, Abe E: Cathepsin K, osteoclastic resorption, and osteoporosis therapy. J Bone Miner Res. 2001, 16: 1747-1749. 10.1359/jbmr.2001.16.10.1747.
Blair HC, Teitelbaum SL, Ghiselli R, Gluck S: Osteoclastic bone resorption by a polarized vacuolar proton pump. Science. 1989, 245: 855-857. 10.1126/science.2528207.
Salo J, Lehenkari P, Mulari M, Metsikko K, Vaananen HK: Removal of osteoclast bone resorption products by transcytosis. Science. 1997, 276: 270-273. 10.1126/science.276.5310.270.
Nesbitt SA, Horton MA: Trafficking of matrix collagens through bone-resorbing osteoclasts. Science. 1997, 276: 266-269. 10.1126/science.276.5310.266.
Eilon G, Mundy GR: Direct resorption of bone by human breast cancer cells in vitro. Nature. 1978, 276: 726-728. 10.1038/276726a0.
Pap T, Claus A, Ohtsu S, Hummel KM, Schwartz P, Drynda S, Pap G, Machner A, Stein B, George M, Gay RE, Neumann W, Gay S, Aicher WK: Osteoclast-independent bone resorption by fibroblast-like cells. Arthritis Res Ther. 2003, 5: R163-R173. 10.1186/ar752.
Walker DG: Bone resorption restored in osteopetrotic mice by transplants of normal bone marrow and spleen cells. Science. 1975, 190: 784-785. 10.1126/science.1105786.
Kurihara N, Chenu C, Miller M, Civin C, Roodman GD: Identification of committed mononuclear precursors for osteoclast-like cells formed in long term human marrow cultures. Endocrinology. 1990, 126: 2733-2741.
Fujikawa Y, Quinn JM, Sabokbar A, McGee JO, Athanasou NA: The human osteoclast precursor circulates in the monocyte fraction. Endocrinology. 1996, 137: 4058-4060. 10.1210/en.137.9.4058.
Klemsz MJ, McKercher SR, Celada A, Van Beveren C, Maki RA: The macrophage and B cell-specific transcription factor PU.1 is related to the ets oncogene. Cell. 1990, 61: 113-124. 10.1016/0092-8674(90)90219-5.
Zhang DE, Hetherington CJ, Chen HM, Tenen DG: The macrophage transcription factor PU.1 directs tissue-specific expression of the macrophage colony-stimulating factor receptor. Mol Cell Biol. 1994, 14: 373-381.
Tondravi MM, McKercher SR, Anderson K, Erdmann JM, Quiroz M, Maki R, Teitelbaum SL: Osteopetrosis in mice lacking haematopoietic transcription factor PU.1. Nature. 1997, 386: 81-84. 10.1038/386081a0.
Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, Burgess T, Elliott R, Colombero A, Elliott G, Scully S, Hsu H, Sullivan J, Hawkins N, Davy E, Capparelli C, Eli A, Qian YX, Kaufman S, Sarosi I, Shalhoub V, Senaldi G, Guo J, Delaney J, Boyle WJ: Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998, 93: 165-176. 10.1016/S0092-8674(00)81569-X.
Quinn JM, Elliott J, Gillespie MT, Martin TJ: A combination of osteoclast differentiation factor and macrophage-colony stimulating factor is sufficient for both human and mouse osteoclast formation in vitro. Endocrinology. 1998, 139: 4424-4427. 10.1210/en.139.10.4424.
Woo KM, Kim HM, Ko JS: Macrophage colony-stimulating factor promotes the survival of osteoclast precursors by up-regulating Bcl-X(L). Exp Mol Med. 2002, 34: 340-346.
Fuller K, Owens JM, Jagger CJ, Wilson A, Moss R, Chambers TJ: Macrophage colony-stimulating factor stimulates survival and chemotactic behavior in isolated osteoclasts. J Exp Med. 1993, 178: 1733-1744. 10.1084/jem.178.5.1733.
Arai F, Miyamoto T, Ohneda O, Inada T, Sudo T, Brasel K, Miyata T, Anderson DM, Suda T: Commitment and differentiation of osteoclast precursor cells by the sequential expression of c-Fms and receptor activator of nuclear factor κB (RANK) receptors. J Exp Med. 1999, 190: 1741-1754. 10.1084/jem.190.12.1741.
Hofbauer LC, Khosla S, Dunstan CR, Lacey DL, Boyle WJ, Riggs BL: The roles of osteoprotegerin and osteoprotegerin ligand in the paracrine regulation of bone resorption. J Bone Miner Res. 2000, 15: 2-12. 10.1359/jbmr.2000.15.1.2.
Li Y, Toraldo G, Li A, Yang X, Zhang H, Qian W-P, Weitzmann MN: B cells and T cells are critical for the preservation of bone homeostasis and attainment of peak bone mass in vivo. Blood. 2007, 109: 3839-3848. 10.1182/blood-2006-07-037994.
Takayanagi H, Iizuka H, Juji T, Nakagawa T, Yamamoto A, Miyazaki T, Koshihara Y, Oda H, Nakamura K, Tanaka S: Involvement of receptor activator of nuclear factor kappaB ligand/osteoclast differentiation factor in osteoclastogenesis from synoviocytes in rheumatoid arthritis. Arthritis Rheum. 2000, 43: 259-269. 10.1002/1529-0131(200002)43:2<259::AID-ANR4>3.0.CO;2-W.
Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang MS, Luthy R, Nguyen HQ, Wooden S, Bennett L, Boone T, Shimamoto G, DeRose M, Elliott R, Colombero A, Tan HL, Trail G, Sullivan J, Davy E, Bucay N, Renshaw-Gegg L, Hughes TM, Hill D, Pattison W, Campbell P, Sander S, Van G, Tarpley J, Derby P, Lee R, Boyle WJ: Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell. 1997, 89: 309-319. 10.1016/S0092-8674(00)80209-3.
Gillespie MT: Impact of cytokines and T lymphocytes upon osteoclast differentiation and function. Arthritis Res Ther. 2007, 9: 103-10.1186/ar2141.
Bucay N, Sarosi I, Dunstan CR, Morony S, Tarpley J, Capparelli C, Scully S, Tan HL, Xu W, Lacey DL, Boyle WJ, Simonet WS: Osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev. 1998, 12: 1260-1268. 10.1101/gad.12.9.1260.
Beinke S, Ley SC: Functions of NF-κB1 and NF-κB2 in immune cell biology. Biochem J. 2004, 382 (Pt 2): 393-409.
Chaisson ML, Branstetter DG, Derry JM, Armstrong AP, Tometsko ME, Takeda K, Akira S, Dougall WC: Osteoclast differentiation is impaired in the absence of inhibitor of kappa B kinase alpha. J Biol Chem. 2004, 279: 54841-54848. 10.1074/jbc.M406392200.
Zhang G, Ghosh S: Toll-like receptor-mediated NF-kappaB activation: a phylogenetically conserved paradigm in innate immunity. J Clin Invest. 2001, 107: 13-19. 10.1172/JCI11837.
Lam J, Takeshita S, Barker JE, Kanagawa O, Ross FP, Teitelbaum SL: TNF-α induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J Clin Invest. 2000, 106: 1481-1488. 10.1172/JCI11176.
Abu-Amer Y, Erdmann J, Alexopoulou L, Kollias G, Ross FP, Teitelbaum SL: Tumor necrosis factor receptors types 1 and 2 differentially regulate osteoclastogenesis. J Biol Chem. 2000, 275: 27307-27310.
Liu ZG, Hsu H, Goeddel DV, Karin M: Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-κB activation prevents cell death. Cell. 1996, 87: 565-576. 10.1016/S0092-8674(00)81375-6.
Fuller K, Murphy C, Kirstein B, Fox SW, Chambers TJ: TNFα potently activates osteoclasts, through a direct action independent of and strongly synergistic with RANKL. Endocrinology. 2002, 143: 1108-1118. 10.1210/en.143.3.1108.
Rothe M, Wong SC, Henzel WJ, Goeddel DV: A novel family of putative signal transducers associated with the cytoplasmic domain of the 75 kDa tumor necrosis factor receptor. Cell. 1994, 78: 681-692. 10.1016/0092-8674(94)90532-0.
Kaji K, Katogi R, Azuma Y, Naito A, Inoue JI, Kudo A: Tumor necrosis factor alpha-induced osteoclastogenesis requires tumor necrosis factor receptor-associated factor 6. J Bone Miner Res. 2001, 16: 1593-1599. 10.1359/jbmr.2001.16.9.1593.
Li J, Sarosi I, Yan X-Q, Morony S, Capparelli C, Tan H-L, McCabe S, Elliott R, Scully S, Van G, Kaufman S, Juan SC, Sun Yu, Tarpley J, Martin L, Christensen K, McCabe J, Kostenuik P, Hsu H, Fletcher F, Dunstan CR, Lacey DL, Boyle WJ: RANK is the intrinsic hematopoietic cell surface receptor that controls osteoclastogenesis and regulation of bone mass and calcium metabolism. Proc Natl Acad Sci USA. 2000, 97: 1566-1571. 10.1073/pnas.97.4.1566.
Azuma Y, Kaji K, Katogi R, Takeshita S, Kudo A: Tumor necrosis factor-alpha induces differentiation of and bone resorption by osteoclasts. J Biol Chem. 2000, 275: 4858-4864. 10.1074/jbc.275.7.4858.
Xu LX, Kukita T, Nakano Y, Yu H, Hotokebuchi T, Kuratani T, Iijima T, Koga T: Osteoclasts in normal and adjuvant arthritis bone tissues express the mRNA for both type I and II interleukin-1 receptors. Lab Invest. 1996, 75: 677-687.
Ye H, Arron JR, Lamothe B, Cirilli M, Kobayashi T, Shevde NK, Segal D, Dzivenu OK, Vologodskaia M, Yim M, Du K, Singh S, Pike J, Wesley D, Bryant G, Choi Y, Wu H: Distinct molecular mechanism for initiating TRAF6 signalling. Nature. 2002, 418: 443-447. 10.1038/nature00888.
Nakamura I, Kadono Y, Takayanagi H, Jimi E, Miyazaki T, Oda H, Nakamura K, Tanaka S, Rodan GA, Duong LT: IL-1 regulates cytoskeletal organization in osteoclasts via TNF receptor-associated factor 6/c-Src complex. J Immunol. 2002, 168: 5103-5109.
Yanni G, Whelan A, Feighery C, Bresnihan B: Synovial tissue macrophages and joint erosion in rheumatoid arthritis. Ann Rheum Dis. 1994, 53: 39-44. 10.1136/ard.53.1.39.
Adamopoulos IE, Sabokbar A, Wordsworth BP, Carr A, Ferguson DJ, Athanasou NA: Synovial fluid macrophages are capable of osteoclast formation and resorption. J Pathol. 2006, 208: 35-43. 10.1002/path.1891.
Horton JE, Raisz LG, Simmons HA, Oppenheim JJ, Mergenhagen SE: Bone resorbing activity in supernatant fluid from cultured human peripheral blood leukocytes. Science. 1972, 177: 793-795. 10.1126/science.177.4051.793.
Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ: IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005, 201: 233-240. 10.1084/jem.20041257.
Volpe E, Servant N, Zollinger R, Bogiatzi SI, Hupe P, Barillot E, Soumelis V: A critical function for transforming growth factor-beta, interleukin 23 and proinflammatory cytokines in driving and modulating human T(H)-17 responses. Nat Immunol. 2008, 9: 650-657. 10.1038/ni.1613.
Manel N, Unutmaz D, Littman DR: differentiation of human T(H)-17 cells requires transforming growth factor-beta and induction of the nuclear receptor RORgammat. Nat Immunol. 2008, 9: 641-649. 10.1038/ni.1610.
Yang L, Anderson DE, Baecher-Allan C, Hastings WD, Bettelli E, Oukka M, Kuchroo VK, Hafler DA: IL-21 and TGF-beta are required for differentiation of human T(H)17 cells. Nature. 2008, 454: 350-352. 10.1038/nature07021.
Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F: Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol. 2007, 8: 942-949. 10.1038/ni1496.
Wilson NJ, Boniface K, Chan JR, McKenzie BS, Blumenschein WM, Mattson JD, Basham B, Smith K, Chen T, Morel F, Lecron JC, Kastelein RA, Cua DJ, McClanahan TK, Bowman EP, de Waal Malefyt R: Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat Immunol. 2007, 8: 950-957. 10.1038/ni1497.
Uhlig HH, McKenzie BS, Hue S, Thompson C, Joyce-Shaikh B, Stepankova R, Robinson N, Buonocore S, Tlaskalova-Hogenova H, Cua DJ, Powrie F: Differential activity of IL-12 and IL-23 in mucosal and systemic innate immune pathology. Immunity. 2006, 25: 309-318. 10.1016/j.immuni.2006.05.017.
Zrioual S, Toh ML, Tournadre A, Zhou Y, Cazalis MA, Pachot A, Miossec V, Miossec P: IL-17RA and IL-17RC receptors are essential for IL-17A-induced ELR+ CXC chemokine expression in synoviocytes and are overexpressed in rheumatoid blood. J Immunol. 2008, 180: 655-663.
Moseley TA, Haudenschild DR, Rose L, Reddi AH: Interleukin-17 family and IL-17 receptors. Cytokine Growth Factor Rev. 2003, 14: 155-174. 10.1016/S1359-6101(03)00002-9.
Van bezooijen RL, Farih-Sips HC, Papapoulos SE, Lowik CW: Interleukin-17: a new bone acting cytokine in vitro. J Bone Miner Res. 1999, 14: 1513-1521. 10.1359/jbmr.1999.14.9.1513.
Sato K, Suematsu A, Okamoto K, Yamaguchi A, Morishita Y, Kadono Y, Tanaka S, Kodama T, Akira S, Iwakura Y, Cua DJ, Takayanagi H: Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med. 2006, 203: 2673-2682. 10.1084/jem.20061775.
Pene J, Chevalier S, Preisser L, Venereau E, Guilleux MH, Ghannam S, Moles JP, Danger Y, Ravon E, Lesaux S, Yssel H, Gascan H: Chronically inflamed human tissues are infiltrated by highly differentiated Th17 lymphocytes. J Immunol. 2008, 180: 7423-7430.
Takayanagi H: Osteoimmunology: shared mechanisms and crosstalk between the immune and bone systems. Nat Rev Immunol. 2007, 7: 292-304. 10.1038/nri2062.
Jovanovic DV, Di Battista JA, Martel-Pelletier J, Jolicoeur FC, He Y, Zhang M, Mineau F, Pelletier JP: IL-17 stimulates the production and expression of proinflammatory cytokines, IL-beta and TNF-alpha, by human macrophages. J Immunol. 1998, 160: 3513-3521.
Fujikawa Y, Sabokbar A, Neale S, Athanasou NA: Human osteoclast formation and bone resorption by monocytes and synovial macrophages in rheumatoid arthritis. Ann Rheum Dis. 1996, 55: 816-822. 10.1136/ard.55.11.816.
Lubberts E, Koenders MI, Oppers-Walgreen B, Bersselaar van den L, Coenen-de Roo CJ, Joosten LA, Berg van den WB: Treatment with a neutralizing anti-murine interleukin-17 antibody after the onset of collagen-induced arthritis reduces joint inflammation, cartilage destruction, and bone erosion. Arthritis Rheum. 2004, 50: 650-659. 10.1002/art.20001.
Koenders MI, Lubberts E, Oppers-Walgreen B, Bersselaar van den L, Helsen MM, Di Padova FE, Boots AM, Gram H, Joosten LA, Berg van den WB: Blocking of interleukin-17 during reactivation of experimental arthritis prevents joint inflammation and bone erosion by decreasing RANKL and interleukin-1. Am J Pathol. 2005, 167: 141-149.
Burchill MA, Nardelli DT, England DM, DeCoster DJ, Christopherson JA, Callister SM, Schell RF: Inhibition of interleukin-17 prevents the development of arthritis in vaccinated mice challenged with Borrelia burgdorferi. Infect Immun. 2003, 71: 3437-3442. 10.1128/IAI.71.6.3437-3442.2003.
Bush KA, Farmer KM, Walker JS, Kirkham BW: Reduction of joint inflammation and bone erosion in rat adjuvant arthritis by treatment with interleukin-17 receptor IgG1 Fc fusion protein. Arthritis Rheum. 2002, 46: 802-805. 10.1002/art.10173.
Nakae S, Saijo S, Horai R, Sudo K, Mori S, Iwakura Y: IL-17 production from activated T cells is required for the spontaneous development of destructive arthritis in mice deficient in IL-1 receptor antagonist. Proc Natl Acad Sci USA. 2003, 100: 5986-5990. 10.1073/pnas.1035999100.
Takayanagi H, Ogasawara K, Hida S, Chiba T, Murata S, Sato K, Takaoka A, Yokochi T, Oda H, Tanaka K, Nakamura K, Taniguchi T: T-cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN-γ. Nature. 2000, 408: 600-605. 10.1038/35046102.
Kotake S, Nanke Y, Mogi M, Kawamoto M, Furuya T, Yago T, Kobashigawa T, Togari A, Kamatani N: IFN-gamma-producing human T cells directly induce osteoclastogenesis from human monocytes via the expression of RANKL. Eur J Immunol. 2005, 35: 3353-3363. 10.1002/eji.200526141.
Horwood NJ, Elliott J, Martin TJ, Gillespie MT: Alone and in synergy with IL-18 inhibits osteoclast formation in vitro. J Immunol. 2001, 166: 4915-4921.
Amcheslavsky A, Bar-Shavit Z: Interleukin (IL)-12 mediates the anti-osteoclastogenic activity of CpG-oligodeoxynucleotides. J Cell Physiol. 2006, 207: 244-250. 10.1002/jcp.20563.
Zou W, Amcheslavsky A, Bar-Shavit Z: CpG oligodeoxynucleotides modulate the osteoclastogenic activity of osteoblasts via Toll-like receptor 9. J Biol Chem. 2003, 278: 16732-16740. 10.1074/jbc.M212473200.
Murphy CA, Langrish CL, Chen Y, Blumenschein W, McClanahan T, Kastelein RA, Sedgwick JD, Cua DJ: Divergent pro- and anti-inflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J Exp Med. 2003, 198: 1951-1957. 10.1084/jem.20030896.
Chu CQ, Song Z, Mayton L, Wu B, Wooley PH: IFNγ deficient C57BL/6 (H-2b) mice develop collagen induced arthritis with predominant usage of T cell receptor Vβ6 and Vβ8 in arthritic joints. Ann Rheum Dis. 2003, 62: 983-990. 10.1136/ard.62.10.983.
Kotake S, Schumacher HR, Yarboro CH, Arayssi TK, Pando JA, Kanik KS, Gourley MF, Klippel JH, Wilder RL: In vivo gene expression of type 1 and type 2 cytokines in synovial tissues from patients in early stages of rheumatoid, reactive, and undifferentiated arthritis. Proc Assoc Am Phys. 1997, 109: 286-301.
Manoury-Schwartz B, Chiocchia G, Bessis N, Abehsira-Amar O, Batteux F, Muller S, Huang S, Boissier MC, Fournier C: High susceptibility to collagen-induced arthritis in mice lacking IFN-gamma receptors. J Immunol. 1997, 158: 5501-5506.
Boulay JL, Paul WE: The interleukin-4 family of lymphokines. Curr Opin Immunol. 1992, 4: 294-298. 10.1016/0952-7915(92)90079-T.
Romagnani S: Regulation of the T cell response. Clin Exp Allergy. 2006, 36: 1357-1366. 10.1111/j.1365-2222.2006.02606.x.
Chen H, Paul WE: Cultured NK1.1+ CD4+ T cells produce large amounts of IL-4 and IFN-γ upon activation by anti-CD3 or CD1. J Immunol. 1997, 159: 2240-2249.
Palmqvist P, Lundberg P, Persson E, Johansson A, Lundgren I, Lie A, Conaway HH, Lerner UH: Inhibition of hormone and cytokine-stimulated osteoclastogenesis and bone resorption by interleukin-4 and interleukin-13 is associated with increased osteoprotegerin and decreased RANKL and RANK in a STAT6-dependent pathway. J Biol Chem. 2006, 281: 2414-2429. 10.1074/jbc.M510160200.
Yamada A, Takami M, Kawawa T, Yasuhara R, Zhao B, Mochizuki A, Miyamoto Y, Eto T, Yasuda H, Nakamichi Y, et al: Interleukin-4 inhibition of osteoclast differentiation is stronger than that of interleukin-13 and they are equivalent for induction of osteoprotegerin production from osteoblasts. Immunology. 2007, 120: 573-579. 10.1111/j.1365-2567.2006.02538.x.
Mangashetti LS, Khapli SM, Wani MR: IL-4 inhibits bone-resorbing activity of mature osteoclasts by affecting NF-κB and Ca2+ signaling. J Immunol. 2005, 175: 917-925.
Wei S, Wang MWH, Teitelbaum SL, Ross FP: Interleukin-4 reversibly inhibits osteoclastogenesis via inhibition of NF-κB and mitogen-activated protein kinase signaling. J Biol Chem. 2002, 277: 6622-6630. 10.1074/jbc.M104957200.
Kitaura H, Nagata N, Fujimura Y, Hotokezaka H, Tatamiya M, Nakao N, Yoshida N, Nakayama K: Interleukin-4 directly inhibits tumor necrosis factor-α-mediated osteoclast formation in mouse bone marrow macrophages. Immunol Lett. 2003, 88: 193-198. 10.1016/S0165-2478(03)00082-8.
Stein NC, Kreutzmann C, Zimmermann SP, Niebergall U, Hellmeyer L, Goettsch C, Schoppet M, Hofbauer LC: Interleukin-4 and interleukin-13 stimulate the osteoclast inhibitor osteo-protegerin by human endothelial cells via the STAT6 pathway. J Bone Miner Res. 2008, 23: 750-758. 10.1359/jbmr.080203.
Joosten LA, Lubberts E, Durez P, Helsen MM, Jacobs MJ, Goldman M, Berg van den WB: Role of interleukin-4 and interleukin-10 in murine collagen-induced arthritis. Protective effect of interleukin-4 and interleukin-10 treatment on cartilage destruction. Arthritis Rheum. 1997, 40: 249-260. 10.1002/art.1780400209.
Lubberts E, Joosten LA, Chabaud M, Bersselaar van Den L, Oppers B, Coenen-De Roo CJ, Richards CD, Miossec P, Berg van Den WB: IL-4 gene therapy for collagen arthritis suppresses synovial IL-17 and osteoprotegerin ligand and prevents bone erosion. J Clin Invest. 2000, 105: 1697-1710. 10.1172/JCI7739.
Lohr J, Knoechel B, Abbas AK: Regulatory T cells in the periphery. Immunol Rev. 2006, 212: 149-162. 10.1111/j.0105-2896.2006.00414.x.
Zaiss MM, Axmann R, Zwerina J, Polzer K, Guckel E, Skapenko A, Schulze-Koops H, Horwood N, Cope A, Schett G: Treg cells suppress osteoclast formation: a new link between the immune system and bone. Arthritis Rheum. 2007, 56: 4104-4112. 10.1002/art.23138.
Zaiss MM, Zwerina J, Polzer K, Gückel E, Skapenko A, Schulze-Koops H, Horwood N, Cope A, Schett G: Treg cells suppress osteoclast formation: a new link between the immune system and bone. Arthritis Rheum. 2007, 56: 4104-4112. 10.1002/art.23138.
Kim YG, Lee CK, Nah SS, Mun SH, Yoo B, Moon HB: Human CD4+CD25+ regulatory T cells inhibit the differentiation of osteoclasts from peripheral blood mononuclear cells. Biochem Biophys Res Commun. 2007, 357: 1046-1052. 10.1016/j.bbrc.2007.04.042.
Morgan ME, Sutmuller RP, Witteveen HJ, van Duivenvoorde LM, Zanelli E, Melief CJ, Snijders A, Offringa R, de Vries RR, Toes RE: CD25+ cell depletion hastens the onset of severe disease in collagen-induced arthritis. Arthritis Rheum. 2003, 48: 1452-1460. 10.1002/art.11063.
Lari R, Fleetwood AJ, Kitchener PD, Cook AD, Pavasovic D, Hertzog PJ, Hamilton JA: Macrophage lineage phenotypes and osteoclastogenesis – complexity in the control by GM-CSF and TGF-β. Bone. 2007, 40: 323-336. 10.1016/j.bone.2006.09.003.
Evans KE, Fox SW: Interleukin-10 inhibits osteoclastogenesis by reducing NFATc1 expression and preventing its translocation to the nucleus. BMC Cell Biol. 2007, 8: 4-10.1186/1471-2121-8-4.
van Amelsfort JM, Jacobs KM, Bijlsma JW, Lafeber FP, Taams LS: CD4(+)CD25(+) regulatory T cells in rheumatoid arthritis: differences in the presence, phenotype, and function between peripheral blood and synovial fluid. Arthritis Rheum. 2004, 50: 2775-2785. 10.1002/art.20499.
van Amelsfort JM, van Roon JA, Noordegraaf M, Jacobs KM, Bijlsma JW, Lafeber FP, Taams LS: Proinflammatory mediator-induced reversal of CD4+,CD25+ regulatory T cell-mediated suppression in rheumatoid arthritis. Arthritis Rheum. 2007, 56: 732-742. 10.1002/art.22414.
Chabaud M, Durand JM, Buchs N, Fossiez F, Page G, Frappart L, Miossec P: A T cell-derived proinflammatory cytokine produced by the rheumatoid synovium. Arthritis Rheum. 1999, 42: 963-970. 10.1002/1529-0131(199905)42:5<963::AID-ANR15>3.0.CO;2-E.
Kotake S, Udagawa N, Takahashi N, Matsuzaki K, Itoh K, Ishiyama S, Saito S, Inoue K, Kamatani N, Gillespie MT, Martin TJ, Suda T: IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J Clin Invest. 1999, 103: 1345-1352. 10.1172/JCI5703.
Nistala K, Moncrieffe H, Newton KR, Varsani H, Hunter P, Wedderburn LR: Interleukin-17-producing T cells are enriched in the joints of children with arthritis, but have a reciprocal relationship to regulatory T cell numbers. Arthritis Rheum. 2008, 58: 875-887. 10.1002/art.23291.
Parsonage G, Filer A, Bik M, Hardie D, Lax S, Howlett K, Church LD, Raza K, Wong SH, Trebilcock E, Scheel-Toellner D, Salmon M, Lord JM, Buckley CD: Prolonged, granulocyte-macrophage colony-stimulating factor-dependent, neutrophil survival following rheumatoid synovial fibroblast activation by IL-17 and TNFα. Arthritis Res Ther. 2008, 10: R47-10.1186/ar2406.
Hirota K, Hashimoto M, Yoshitomi H, Tanaka S, Nomura T, Yamaguchi T, Iwakura Y, Sakaguchi N, Sakaguchi S: T cell self-reactivity forms a cytokine milieu for spontaneous development of IL-17+ Th cells that cause autoimmune arthritis. J Exp Med. 2007, 204: 41-47. 10.1084/jem.20062259.
Aarvak T, Chabaud M, Miossec P, Natvig JB: IL-17 is produced by some proinflammatory Th1/Th0 cells but not by Th2 cells. J Immunol. 1999, 162: 1246-1251.
Yamada H, Nakashima Y, Okazaki K, Mawatari T, Fukushi JI, Kaibara N, Hori A, Iwamoto Y, Yoshikai Y: Th1 but not Th17 cells predominate in the joints of patients with rheumatoid arthritis. Ann Rheum Dis. 2007, 67: 1299-1304. 10.1136/ard.2007.080341.
Kirkham BW, Lassere MN, Edmonds JP, Juhasz KM, Bird PA, Lee CS, Shnier R, Portek IJ: Synovial membrane cytokine expression is predictive of joint damage progression in rheumatoid arthritis: a two-year prospective study (the DAMAGE study cohort). Arthritis Rheum. 2006, 54: 1122-1131. 10.1002/art.21749.
Raza K, Falciani F, Curnow SJ, Ross EJ, Lee CY, Akbar AN, Lord JM, Gordon C, Buckley CD, Salmon M: Early rheumatoid arthritis is characterized by a distinct and transient synovial fluid cytokine profile of T cell and stromal cell origin. Arthritis Res Ther. 2005, 7: R784-R795. 10.1186/ar1733.
Furuya T, Hakoda M, Ichikawa N, Higami K, Nanke Y, Yago T, Kamatani N, Kotake S: Associations between HLA-DRB1, RANK, RANKL, OPG, and IL-17 genotypes and disease severity phenotypes in Japanese patients with early rheumatoid arthritis. Clin Rheumatol. 2007, 26: 2137-2141. 10.1007/s10067-007-0745-4.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
IEA and EPB are employees of Schering-Plough Corporation and therefore, receive salary from and/or hold stock in Schering-Plough.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Adamopoulos, I.E., Bowman, E.P. Immune regulation of bone loss by Th17 cells. Arthritis Res Ther 10, 225 (2008). https://doi.org/10.1186/ar2502
Published:
DOI: https://doi.org/10.1186/ar2502