
Abstract
The crystal structures of new N-phenyl-1,5-dimethyl-1H-imidazole-4-carboxamide 3-oxide derivatives are reported. The results of X-ray diffraction showed the existence of intramolecular hydrogen bonding between carboxamide nitrogen donors and N-oxide oxygen acceptors. The use of Quantum Theory of Atoms in Molecules allowed its classification as a strong interaction, with energy about 10 kcal/mol, and of intermediate character between closed shell and shared bonds. Comparison of experimental data and quantum theoretical calculations indicated that a substituent attached to the phenyl ring in the para position influences the strength and geometry of the title hydrogen bonding. Stronger π-electron-withdrawing properties of the higher energy substituent of the intramolecular hydrogen bond are observed. Among other intermolecular contacts in the studied crystals are C–H…O/C–H…N hydrogen bonds of imidazole carbon atoms and some π…π stacking interactions between aromatic molecular fragments. Their importance in stabilization of the crystal structure was confirmed by the results of Hirshfeld surface analysis.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
In recent years, interest in imidazole N-oxides has constantly increased. Because of regioselective reactivity, many N-oxide derivatives are used in the synthesis of a number of organic compounds [1–7]. Imidazole N-oxides act as precursors of biologically active molecules, transition metal chelating agents [8, 9], and inhibitors of the release of interleukin-1, cytokine release, and protein kinase p38 [10, 11]. Some imidazole N-oxide derivatives exhibit anti-inflammatory and antiprotozoal activity [10, 12, 13]. They may also be used as insecticides or as plant growth regulators [14–16].
Recently, the N-oxide group has been the subject of our studies in the context of its ability to act as an electron donor in various types of intermolecular interactions [17–19]. Various N-oxide interactions stabilizing crystal structures have been investigated [17]. On the basis of theoretical chemistry computations, it has been found that the N-oxide group may act as an effective Lewis base in both hydrogen and halogen bonds [18], even more effectively than the oxygen atom of a carbonyl or ether group. For example, the N-oxide group forms very strong hydrogen bonds with isocyanide with an energy close to 16 kcal/mol (DFT-B3LYP/aug-cc-pVTZ). The corresponding energy value for (H3C)2O and H2C=O molecules as oxygen proton acceptors is of about half of that, ∼7 and ∼9 kcal/mol, respectively. Interestingly, we recently found that imidazole N-oxide is able to interact with proton donors not only in strong intermolecular but also in intramolecular hydrogen bonds with energies of about 12 kcal/mol [19].
This paper is a continuation of our studies on hydrogen bonding of N-oxide groups. The previously obtained results caused our attention to turn to intramolecular interactions [19]. For the purpose of more detailed description of this kind of hydrogen bonding, a series of N-phenyl-1,5-dimethyl-1H-imidazole-4-carboxamide 3-oxide derivatives were synthesized according to a procedure described elsewhere [20–22] and analyzed by means of crystal X-ray diffraction methods. The compounds differ each other by a substituent in the para position of the phenyl ring (Fig. 1). According to our expectations, various substituents should influence the proton donating properties of the NH group via the resonance effect and in this way change the properties of N–H…ON (N-oxide) hydrogen bonding. The presented X-ray diffraction results are further compared with quantum chemical calculations in order to evaluate the influence of substituent effects on the energy of the investigated interaction.

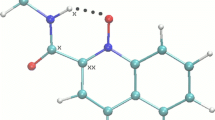
Molecular structures of the studied compounds: N-phenyl-1,5-dimethyl-1H-imidazole-4-carboxamide 3-oxide I (molecule IA) (a); N-(4-cyanophenyl)-1,5-dimethyl-1H-imidazole-4-carboxamide 3-oxide II (b); N-(4-trifluoromethylphenyl)-1,5-dimethyl-1H-imidazole-4-carboxamide 3-oxide III (c); N-(4-nitrophenyl)-1,5-dimethyl-1H-imidazole-4-carboxamide 3-oxide IV (d); N-(4-methoxyphenyl)-1,5-dimethyl-1H-imidazole-4-carboxamide 3-oxide V (e); N-(4-bromophenyl)-1,5-dimethyl-1H-imidazole-4-carboxamide 3-oxide VI (molecule VIA) (f). Anisotropic displacement parameters of non-hydrogen atoms are drawn as ellipsoids with 50% probability level. Blue dashed lines represent N-oxide intramolecular hydrogen bonds
Experimental details
X-ray crystal structure determination
The selected derivatives of 1H-imidazole N-oxide I-VI (see Scheme 1) were obtained by a method reported elsewhere [20–22]. Crystals suitable for X-ray measurements were obtained by slow evaporation of the solvent from ethanol solutions. X-ray diffraction measurements were made on a four-circle Oxford Diffraction Xcalibur diffractometer equipped with a two-dimensional area CCD detector using graphite monochromatized MoKα radiation and an Oxford Cryosystem low-temperature cooler device. Integration of the intensities and correction for Lorentz and polarization effects were performed using CrysAlis RED software [23]. Crystal structures of all the six compounds were solved by direct methods using SHELXS-97 [24] implemented in the WinGX [25] package of programs. Refinement of crystal structures was performed by the use of the full-matrix least-squares method and the F 2 method in the SHELXL-2014 program [24]. The positions of hydrogen atoms in aromatic rings and methyl groups were introduced in their calculated positions of idealized geometry. For the appropriate crystal structure determination of the two methyl groups of molecule IB, lying in the mirror plane m y , two sets of hydrogen atoms were introduced and constrained in staggered geometry (HFIX 123 instruction by SHELXL). The positions of hydrogen atoms in NH groups were found on a Fourier difference map and isotropically refined without any restraints. For p-trifluoromethyl derivative (III), two neighboring electron density maxima, which may correspond to the two positions of hydrogen atoms within the intramolecular hydrogen bonding bridge, were found on the difference Fourier map. In such cases, there are two procedures that may be chosen for hydrogen atom treatment. The first one includes retaining one hydrogen position and refining it. In the other, two different positions with partial occupancy factors are introduced and refined [26]. Both types of crystal structure refinements were tested. However, the latter strategy required using DIFX constraints, finally leading to artificial D–H distances and therefore to an artificial and constrained overall geometry of hydrogen bonding bridges. As refinements of the both models led to similar results (including R, wR 2, GooF, Δρ max /Δρ min [eÅ−3] descriptors of quality of crystal structure determination), a model with one H7 hydrogen atom position was chosen for III.

Structural formula of the studied N-phenyl-1,5-dimethyl-1H-imidazole-4-carboxamide 3-oxide derivatives
Details of X-ray measurements and crystal structure refinements are given in Table 1. The molecular geometries of all the derivatives were calculated with PLATON [27] and PARST [28]. Selected bond lengths, valence, and torsion angles are collected in Table 2. Atomic coordinates, displacement parameters, and structure factors of the analyzed crystal structures are deposited with the Cambridge Crystallographic Data Centre (CCDC) [29].
Hirshfeld surface analysis
Molecular Hirshfeld surfaces and fingerprint plots were generated using CrystalExplorer 3.0 [30, 31]. Using the automatic procedures implemented in the program, hydrogen atom bonds lengths were normalized to standard neutron values (C–H = 1.083 Å, O–H = 0.983 Å, N–H = 1.009 Å) [32]. The presented surfaces were mapped with normalized contact distance (d norm ), a parameter based on the distance from the Hirshfeld surface point to the nearest atom in the molecule (internal) (d i ), the nearest atom in another molecule (external) (d e ), and van der Waals radii of the corresponding pair of atoms (r i and r e ). The d norm parameter is given by the Eq. (1).
All Hirshfeld surfaces presented in this work are mapped with the standard red-white-blue color scheme, in which the white color represent contacts around the van der Waals separation, red is used for the shortest contacts with negative values of d norm , and blue corresponds to the longest intermolecular contacts with positive d norm . In all the figures, the d norm value ranges from −0.3 to 1.2.
Quantum theoretical calculations
Molecular geometries of the studied compounds were fully optimized with the use of DFT methods on the B3LYP/6–311++G** level of theory in the Gaussian 09 [33] set of codes. The frequency calculations allowed the conclusion that all of the resulting geometries are the ground state stationary points. For these obtained geometries, electron density analysis was performed according to Quantum Theory of Atoms in Molecules (QTAIM) [34]. Topological parameters of electron density in bond critical points were determined with the use of the AIMall program [35]. The energy of intramolecular hydrogen bonds was calculated using the Espinosa equation [36]:
where V BCP is the density of potential electron energy estimated at the considered hydrogen bond critical point.
Results and discussion
Comparison of molecular structures
Molecular drawings of all of the studied compounds are shown in Fig. 1. In two cases (I and VI), there are two distinct molecules A and B found in the asymmetric unit, but no significant differences in their conformations were found. In crystal structure I, the molecule B lies in a special position in the m y mirror plane, as presented in Fig. 2. Moreover, there is also one water molecule lying in the same mirror plane resulting in the final molar ratio 3:1 as shown in Table 1.

The arrangement of molecules in the unit cell of crystal structure I, indicating the special position of IB and water molecules on the m y mirror plane. The scheme of intermolecular hydrogen bonds in the final chain motif is represented by blue dotted lines
Selected geometrical parameters that allow comparison of the investigated derivatives are collected in Table 2 and in supplementary CIF files and indicate that the molecular structures of all of the analyzed compounds are very similar to each other. Detailed investigation was made of bonds including nitrogen and oxygen atoms that are able to participate in intermolecular hydrogen bonding type interactions. The N-oxide O1–N1 bond length varies from 1.332(2) Å for VIA to 1.351(3) Å for IB. A similar range of changes was observed for hetero bonds of the amide group: the C6–O6 bond length ranges from 1.221(2) Å in IV to 1.238(3) Å in VIA, while the C6–N7 bond ranges from 1.249(3) Å in VIA to 1.264(3) Å in II. However, the observed differences between these interatomic distances are usually not significant when taking into account the 3σ criterion, which is about 0.01 Å. In all of the structures these bonds are taking part is various interactions and playing the role of hydrogen bonding donors or acceptors, which can influence their lengths. This bond length alternation is accompanied by rather slight changes in valence angles. The ranges of geometrical parameters in the analyzed crystal structures are in agreement with that observed for the previously studied N-(4-fluorophenyl)-1,5-dimethyl-1H-imidazole-4-carboxamide 3-oxide [19].
All of the investigated molecules consist of three characteristic fragments: the phenyl ring (C8, C9, C10, C11, C12,C13—Ph plane) substituted at position 4 in compounds II–VI, a carboxamide bridge (C5, C6, O6, N7,C8—Carboxam plane), and an imidazole ring (N1, C2, N3, C4, C5—Im plane) substituted by two methyl groups. In all of the studied cases, these molecular fragments are very slightly twisted with respect to each other with various degrees of rotation. The only one exception is molecule IB which lies in a special position in the mirror plane m y and due to crystallographic symmetry, its structure is constrained to be planar. The angles between the calculated least-square planes of these fragments are also collected in Table 2.
Apart from IA, all angle values are below 10°, which defines the molecules as essentially planar. Relatively, the smallest values of the dihedral angles are observed for the carboxamide bridge-imidazole ring systems (from 7.4(1)° to 2.0(1)°). The greatest degree of twisting of terminal molecular fragments between the phenyl and imidazole rings is observed in IA (17.8(1)°) and the smallest in III (2.8(1)°). It is worth noting that among the investigated compounds, the smallest values of the discussed dihedral angles are observed for para-trifluoro and para-nitro derivatives (III and IV), raising the possibility of effective π-resonance involving the whole molecules and further influencing the proton donor/acceptor properties of O and N heteroatoms. A diagram presenting the relationships between O1–N1–C5–C6 torsion and the carboxamd/Im dihedral angles, describing molecular twisting, and the H…O distances of intramolecular hydrogen bonds to N-oxide groups are presented in Fig. 7a, b.
Intermolecular interactions
The presence of heteroatoms such as nitrogen, oxygen, and halogen in the molecules of the investigated compounds leads to a diversity of intermolecular interactions. Table 3 presents a summary of the geometrical parameters of these interactions. For intermolecular hydrogen bonds, the observed D…A distances are longer than those found for intramolecular interactions (see section Intramolecular hydrogen bonds, Table 4) suggesting that the latter interactions are much stronger.
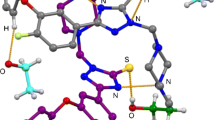
In crystal structure I, hydrogen bonds involving two independent molecules (A and B) and water molecules result in the formation of a chain of molecules extending along the [010] direction. The corresponding structural motif is shown in Fig. 2. Characteristic intermolecular interactions observed in the other crystal structures are presented in Fig. 3. It is worth noting that in all of the crystal structures except for II, there is a similar C2-H2…O6 intermolecular hydrogen bond with the imidazole carbon atom as a donor and the carbonyl of the amide group as an acceptor. In contrast, in II, the H2 atom is involved in a C2–H2…N16 interaction with the terminal cyano group.

Scheme of intermolecular interactions (represented by blue dotted lines) in the crystal structures: II (a); III (b); IV (c); V (d); and VI (e)
Some halogen bonding type Br…Br and Br…O interactions linking adjacent molecules are found in crystal structure VI. Moreover, there are many observed C/N/O…C/N/O intermolecular contacts that are shorter than the sum of the van der Waals radii (Table 3), which result from parallel arrangement of π systems (imidazole rings or amide moieties) and π…π stacking interactions.
The existence of the above-described intermolecular interactions can be confirmed by visual inspection of molecular Hirshfeld surfaces mapped with the d norm parameter. The corresponding molecular surfaces are shown in Fig. 4.

Hirshfeld surfaces calculated for molecules in the studied crystal structures: a IA, b IB, c II, d III, e IV, f V, g VIA, h VIB
In most cases, red areas, representing intermolecular contacts with distances shorter than the sum of van der Waals radii, can be seen close to imidazole H2 and carboxamide O6 atoms resulting from the mentioned C–H…O hydrogen bonds. Moreover, for the molecules of I, III, and IV, there are red areas near to the O1 N-oxide oxygen, which acts as a proton acceptor in C–H…ON intermolecular hydrogen bonds (compare with Table 4). Interestingly, as a result of F…F and Br…Br interactions in the crystal structures III and VI, there are some tiny light pink spots occurring around the halogen atoms in the corresponding molecular surfaces.
A percentage diagram summarizing the distribution of intermolecular contacts is presented in Fig. 5. N…H (red color in the diagram) and O…H (green color) contacts result from the formation of intermolecular hydrogen bonds with carbon donating atoms. This is especially recognized in molecule II. A very similar and relatively large distribution of both kinds of contacts results from C–H…O (13.0%) and C–H…N (15.5%) interactions as listed in Table 3. In the other structures, the distribution of C–H…O interatomic contacts is of similar range varying from 13.0 to 23.4% while C–H…N contacts are evidently rarer (from 1.5 to 6.1%), when compared with II, as there are no hydrogen bonds with a nitrogen atom as an acceptor. The dominant percentage of O…H contacts (37.7%) in IV is strictly connected with additional interactions of terminal oxygen atoms of para-nitro substituents.

Distribution of intermolecular contacts on the basis of Hirshfeld surface analysis
In turn, for molecule III, a large proportion of all intermolecular contacts (about 33%) result from close neighboring of fluorine and hydrogen atoms, and there are similarly many bromine-hydrogen contacts in VI (almost 20%), which are represented in orange and dark blue in the diagram. In both derivatives, halogen atoms are in the para position in the phenyl ring; so, there are no steric hindrances to hide them from intermolecular interactions. The rather large contribution of C/N/O…C/N/O contacts found for structures II, IV, and VI is connected with the above-mentioned π stacking interactions.
Intramolecular hydrogen bonds
Our recent studies have shown that the N-oxide group may act as an effective proton acceptor in hydrogen bonding [17–19]. Also, in case of the crystal structures investigated here, the N-oxide group contributes to these kinds of interaction. As a result, an intramolecular hydrogen bond between an N7–H7 donor of the carboxamide group and O1 of the N-oxide group, as a hydrogen bonding acceptor, is observed for all of the studied compounds (Fig. 1). In all cases, the positions of the H7 atom were found in the Fourier difference map.
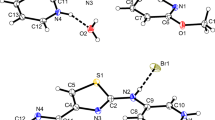
There are revealed electron density maxima corresponding to the positions of the hydrogen atom in intramolecular hydrogen bonding bridges for all of the investigated crystal structures as seen in Fig. 6. In the case of p-nitro- and p-bromine-derivatives (IV and VI), the observed peaks of electron density maxima are fuzzy and shifted towards the N-oxide O1 oxygen atom. Similar pictures of Fourier difference maps in the area of intramolecular hydrogen bonding bridges are known for benzopyrane derivatives [37–41], and they have been treated as evidence of hydrogen bonding strengthening and possible dynamic proton transfer reaction.

Difference Fourier maps in the region of intramolecular N7–H7…O1–N1 hydrogen bonding: I (molecule IA) (a); II (b); III (c); IV (d); V (e); VI (molecule VIA) (f). The color code is shown by the color bar in eÅ−3, contour level: 0.1 eÅ−3. Maps are calculated for models without H7 hydrogen atoms
The situation of p-trifluoromethyl derivative (III) is very interesting. Two almost distinct peaks of maximum electron density are shown corresponding to the positions of the hydrogen atoms. However, for the reasons described in the experimental section and to be consistent with the other crystal structures, the model with one H7 atom position in III has been presented. It is worth mentioning that the picture in Fig. 6 c could be also treated as evidence of proton movement within the intramolecular hydrogen bridge.
The changes in the H…O distance are directly connected with hydrogen position and possible proton transfer in the discussed intramolecular hydrogen bonding bridge. Therefore, a set of correlations between this distance and other parameters are presented in Fig. 7. Firstly, clear trends between O1–N1–C5–C6 torsion (a) and carboxamd/Im dihedral (b) angles, describing molecular twisting, and hydrogen bonding distance can be seen, although no statistically important correlations can be found here. Secondly, proton transfer should influence properties of acceptor N-oxide group. Indeed, relationships between the H…O distance and the N1–O1 bond length (c) and the O1 axis of the anisotropic displacement parameter in the direction of the H7 atom (d) again indicate some kind of interrelation.

Relationships between H7…O1 hydrogen bonding distances and O1–N1–C5–C6 torsion angles (a); carboxamd/Im dihedral angle (b); N1–O1 bond length (c); and O1 axis of the anisotropic displacement parameter in the direction of the H7 atom (d)
As a result of N7–H7…O1–N1 intramolecular hydrogen bond formation, a characteristic pattern of chelate cyclic rings, denoted as S(6) according to Etter graph-set notation [42], is closed in all the crystal structures (Fig. 2). Geometrical parameters characterizing these interactions are collected in Table 3. The positions of the N7 atoms were refined, leading to a set of NH distances varying from 0.88(3) Å to 1.03(4) Å. Relatively long NH distances, but equal to each other within experimental error, were found for the p-bromo derivative (VI). The observed elongation of the NH distance is accompanied by shortening of the H…O distance within the hydrogen bridge. Comparing the data collected in Table 4, it can be clearly seen that the shortest distances are found for compounds with strong electron-withdrawing substituents in the para position of the phenyl ring, such as trifluoromethyl (III) and nitro (IV) groups.
The most reliable geometrical parameter associated with the hydrogen bond strength is the distance between the donor atom (N7 nitrogen) and the acceptor (O1 oxygen atom). In all cases, this interatomic N…O distance is evidently shorter than the sum of their van der Waals radii, reported as 3.16 Å [43]. The shortest hydrogen bonds, with N…O distances around 2.60 Å, are observed in structures III, IV, and V. In turn, the longest, with a corresponding distance of 2.655(2) Å, is observed for the unsubstituted derivative I. D–H…A angles of hydrogen bridges vary from 145(2)° for IA to 158(2)° for III. The latter is very close to the value known from the literature for the p-fluoro derivative [19]. Therefore, larger values of these angles are observed for the shortest hydrogen bonds, and smaller angles are observed for longer hydrogen bonds.
Summarizing, it may be stated that the differences in the intramolecular hydrogen bridges, observed for each crystal structure, are noticeable. Taking into account that the molecular fragment directly involved in the S(6) hydrogen bonding pattern is rather rigid, due to possible π-conjugation, and that the compounds differ from each other only at the position of para substituent in the phenyl ring, we have performed quantum theoretical calculations to analyze whether such differences will be also reproduced for isolated molecules in the gas phase. We also investigated the possible influence of the mentioned substituents on the geometry and energy of the interaction. The energies of the investigated intramolecular hydrogen bonds were approximated by the use of Espinosa equation [39].
Table 4, alongside the experimental data, shows selected parameters of hydrogen bridges in the investigated model systems. In general, the geometrical characteristics are qualitatively comparable with the experimental data. As can be seen, the differences in the numerical values, although slightly smaller than these found for the experimental data, are still noticeable. It can therefore be concluded that not only are the packing effects are responsible for the observed differences in the geometrical parameters of hydrogen bonding, but also some intramolecular effects resulting from the various substituents attached to the phenyl ring can influence the geometry of the hydrogen bonding bridges.
On the basis of theoretical calculations, the shortest distances observed for model systems III and IV (similar to the corresponding crystal structures) may indicate that strong π-electron-withdrawing substituents stabilize the hydrogen bond. Other electron-withdrawing substituents still stabilize it but less effectively, due to less efficient π-electron interactions with the rest of the molecule. Finally, the weakest hydrogen bond is observed in I, with an unsubstituted phenyl ring. The above conclusions are confirmed by the hydrogen bonding interaction energy values, which are collected in Table 5. It can be stated that due to π-electron conjugation, the substituents in the para position of the phenyl ring may effectively influence the proton-donating properties of the N–H group, by their electron-withdrawing substituent effects, as illustrated in Scheme 2.

Two possible resonance structures of I–VI. The structure with formal charge separation (b) increases the proton-donating properties of NH group, stabilizing the considered intramolecular NH…ON hydrogen bond
The other QTAIM parameters (Table 5) are also in line with the N–H…O hydrogen bond strength analysis. The highest values for electron density in hydrogen bonding bond critical points (BCP) of III and IV follow the interaction energy estimations. Positive values of the electron density Laplacian (∇2 ρBCP) and negative values of total electron energy density (HBCP) allow the considered interactions to be classified as strong hydrogen bonds with intermediate character between the typical closed-shell and shared bonds [44, 45]. The ability to form strong hydrogen bonds with an N-oxide group as a proton acceptor has been already reported by us on the basis of experimental research on pyridine N-oxides [17].
aEnergy of hydrogen bonds estimated by the Espinosa equation [39]
Conclusions
The use of X-ray diffraction methods allowed us to confirm the existence of very strong intramolecular hydrogen bonds in the crystal structures of a series of N-phenyl-1,5-dimethyl-1H-imidazole-4-carboxamide 3-oxide derivatives. For all the structures, a similar intramolecular S(6) hydrogen bond between a carboxamide N7–H7 proton donor and an N-oxide O1 acceptor is observed. Comparison of the geometrical parameters of this hydrogen bridge obtained using X-ray diffraction and quantum theoretical calculations leads to the conclusion that π-electron-withdrawing groups substituted in the para position of the phenyl ring stabilize the intramolecular hydrogen bond, due to π-electron conjugation and its influence on NH proton donating properties. On the basis of QTAIM analysis, the studied intramolecular hydrogen bonds, with energies about 10 kcal/mol, are classified as strong hydrogen bonds with intermediate character between shared and closed shell interactions. In the case of the strongest hydrogen bonds, analysis of difference Fourier maps indicates possible proton transfer within the hydrogen bridge.
Analysis of intermolecular interactions in the crystal state shows that the N-oxide group may act as a proton acceptor not only in intramolecular hydrogen bonds but also in some intermolecular interactions. Moreover, the C2 atom of the imidazole ring has proved to be a good proton donor in C–H…O and C–H…N hydrogen bonding to amide or cyano groups. Some intermolecular contacts of halogen atoms were also observed in all of the analyzed crystal structures. Among them are F…F, Br…Br, and Br…O. The geometrical parameters of the latter indicate its halogen bonding character.
Finally, it can be stated that by using various methods (i.e., structural analysis, Hirshfeld analysis, DFT, and QTAIM calculations), we obtained consistent results, which appropriately support the above conclusions.
References
Balaban AT, Halls PJ, Katritzky AR (1968) Chem Ind 5:651
Laufer S, Wagner G, Kotschenreuther D (2002) Angew Chem Int Ed Engl 41:2290
Lettau H (1970) Z Chem 10:211
Mloston G, Gendek T, Heimgartner H (2000) Tetrahedron 56:5405
Volkamer K, Baumgärtel H, Zimmermann H (1967) Angew Chem 79:947
Volkamer K, Zimmerman H (1970) Chem Ber 103:296
Mlostoń G, Jasiński M, Wróblewska A, Heimgartner H (2016) Curr Org Chem 20:1359–1369
Witschel M (2009) Bioorg Med Chem 17:4221
Lin YS, Liu CW, Tsai TYR (2005) Tetrahedron Lett 46:1859
Laufer SA, Zimmermann W, Ruff KJ (2004) J Med Chem 47:6311
Begtrup M (2012) Adv in Heterocyc Chem 106:1–109
Cerecetto H, González M (2002) Current Topic Med Chem 2:1185
Cosar C, Ganter P, Jolou L (1961) Presse Med 69:1069
Marmalis P, La Croix ES, Mhasalker SE (1973) Ger Often Pat2 254:474
Creutzburg D (1980) Ger Pat (East) 140:966
Pozharskiĭ AF, T-Soldatenkov A, Katritzky AR (1997) Heterocycles in life and society: an introduction to heterocyclic chemistry and biochemistry and the role of heterocycles in science, technology, medicine, and agriculture. Wiley, New York
Rybarczyk-Pirek AJ, Łukomska-Rogala M, Wojtulewski S, Palusiak M (2015) CrystGrowth Des 15:5802–5815
Łukomska M, Rybarczyk-Pirek AJ, Jabłoński M, Palusiak M (2015) Phys Chem Chem Phys 17:16375–16387
Rybarczyk-Pirek AJ, Łukomska M, Ejsmont K, Jasiński M, Palusiak M (2014) Struct Chem 25:979–989
Mlostoń G, Romański J, Jasiński M, Heimgartner H (2009) Tetrahedron Asymmetry 20:1073
G. Mlostoń, M. Jasiński, Heimgartner H, (2011) Eur J Org. Chem 2542
Mlostoń G, Jasiński M (2010) Collect Czechoslov Chem Commun 75:871
CrysAlis PRO; Agilent, Agilent Technologies: Yarnton, England, 2011
Sheldrick GM (2008) Acta Crystallogr Sect A 64:112
Farrugia LJ (1999) J Appl Crystallogr 32:837–838
Rybarczyk-Pirek Struct AJ (2012) Chem 23:1739–1749
Spek AL (2009) Acta Cryst D65:148–155
Nardelli M (1995) J Appl Crystallogr 28:659
the Cambridge Crystallographic Data Centre, 12, Union Road, Cambridge CB2 1EZ, UK; http://www.ccdc.cam.ac.uk/conts/retrieving.html
Spackman MA, Jayatilaka D (2009) CrystEngComm 11:19–32
McKinnon JJ, Fabbiani FPA, Spackman MA (2007) Cryst Growth Des 7:755–769
Allen FH, Kennard O, Watson DG, Brammer L, Orpen AG, Taylor R (1987) J Chem Soc Perkin Trans 2:S1
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery Jr JA, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar S, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2009) Gaussian 09, Revision A.1. Gaussian, Inc., Wallingford
Bader RFW (1994) Atoms in molecules - a quantum theory. Oxford University Press, Oxford
Keith TA (2010) AIMAll program Version 10.12.11; http://aim.tkgristmill.com
Espinosa E, Molins E, Lecomte C (1998) Chem Phys Lett 285:170–173
Rybarczyk-Pirek AJ, Dubis AT, Grabowski SJ, Nawrot-Modranka Chemical J (2006) Physics 320:247–258
Rybarczyk-Pirek AJ, Grabowski SJ, Nawrot-Modranka J, Phys J (2003) Chem. A 107:9232–9239
Rybarczyk-Pirek AJ, Grabowski SJ, Małecka M, Nawrot-Modranka J, Phys J (2002) Chem A 106:11956–11962
Rybarczyk-Pirek AJ, Małecka M, Grabowski SJ, Nawrot-Modranka Acta J (2002) Crystallogr. C58:o405–o406
Rybarczyk AJ, Olszak TA, Małecka M, Nawrot-Modranka Acta J (1999) Crystallogr C55:1313–1315
Etter MC, MacDonald JC, Bernstein J (1990) Acta Crystallogr Sect B 46:25
Alvarez S (2013) Dalton Trans 42:8617
Palusiak M, Krygowski TM (2007) Chem Eur J 13:7996–8006
Rybarczyk-Pirek AJ, Małecka M, Palusiak Cryst M (2016) Grow Des 16:6841–6848
Acknowledgments
The theoretical computations using the Gaussian 09 set of codes were carried out in the Wrocław Center for Networking and Supercomputing (http://www.wcss.wroc.pl). Access to the HPC machines and licensed software is gratefully acknowledged. The authors acknowledge financial support from the National Science Centre of Poland (OPUS grant: 2015/19/B/ST4/01773). This work was partially supported by a grant for young scientists from the University of Łódź (Project id: B1411100000471.02 Project No. 1152).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Łukomska-Rogala, M., Rybarczyk-Pirek, A.J., Ejsmont, K. et al. Non-covalent interactions of N-phenyl-1,5-dimethyl-1H-imidazole-4-carboxamide 3-oxide derivatives—a case of intramolecular N-oxide hydrogen bonds. Struct Chem 28, 1229–1241 (2017). https://doi.org/10.1007/s11224-017-0935-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-017-0935-x