
Abstract
The speed of sound in high-purity n-hexane and n-heptane was experimentally studied utilizing the double-path length pulse-echo technique. Measurements with each alkane were carried out at temperatures from (233 to 353) K with pressures up to 20 MPa. Considering the uncertainty contributions from temperature, pressure, path-length calibration, pulse timing and sample purity, the relative expanded combined uncertainty (k = 2) in the speed of sound in n-hexane ranges from (0.012 to 0.042) % over the investigated ranges of pressure and temperature; for n-heptane, the uncertainty varies from (0.014 to 0.018) %. The sound speed data measured in n-hexane were among the data used for the development of a new fundamental equation of state, which is, however, not described in this work. The experimental data of n-heptane can be considered appropriate for modeling purposes and validation of existing equations of state.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
The alkane n-hexane is currently being investigated as a possible working fluid for ORC-processes in geothermal power plants or in the field of waste heat recovery since the high critical temperature could lead to a better efficiency [1, 2]. Furthermore, it is relevant as a fuel component in the process chain of biodiesel [3] and in food processing technology for the extraction of vegetable oils [4, 5]. Additionally, n-hexane is relevant for a variety of industrial applications and is used as an extractant or reaction medium in polymerization [6]. As a constituent of natural gas, there is also a special interest in the thermodynamic properties of n-hexane for mixture models of such fuels.
The alkane n-heptane is mainly used as an extractant and solvent in the chemical industry and for vitamin [7] or vegetable oil extraction [8] in the food industry. It is also used in the extraction of bitumen from oil sands [9], the treatment of catalysts [10] and in polymerization processes [11]. Moreover, n-hexane is employed as a standard zero point for the octane number of engine fuels [12] and, thus, for the octane rating of petrol or rather the synthesis of knock resistant fuels [13].
A fundamental equation of state for n-hexane and n-heptane widely used in technical applications was developed by Span and Wagner in 2003 [14]. The aim of their work was to represent 15 non-polar and weakly polar substances with a generalized form of an equation of state so that it remains valid for an entire group of substances. This approach made it possible to describe similar substances for which only few measurement data were available. However, this model is not intended to serve as a reference equation for substances with a sufficient database. The equation for n-hexane by Span and Wagner [14] was developed without specific fitting of any speed of sound data, and only one data set by Muringer et al. [15] was used for the development of the equation for of n-heptane [14]. As a result, an uncertainty of 2 % for calculated sound speeds in the homogeneous liquid phase of n-heptane was reported. Against this background, new fundamental equations of state for n-hexane and n-heptane are under development at Ruhr University Bochum. For the development of the new equation for n-hexane by Thol et al. [16], as already implemented in REFPROP 10.0 [18] and TREND 4.0 [19], selected data points in the liquid phase of the speed of sound data sets by Daridon et al. [20] and Khasanshin et al. [21] were considered as primary data as well as the speed of sound data measured within the scope of the present work. The objective of our present study was to improve the data situation for the speed of sound in n-hexane at lower temperatures and pressures and to provide a consistent as well as reliable data set for modeling purposes.
For the development of the new fundamental equation of state for n-heptane, Thol et al. [17] used the data sets of Daridon et al. [22] and Muringer et al. [15] as well as selected data points of the speed of sound measurements by Baumhögger et al. [23] as primary data in the homogeneous liquid phase. The speed of sound data of n-heptane measured in the present work were not yet used in the development process of this new equation, which is also already implemented in REFPROP 10.0 [18] and TREND 4.0 [19]. However, our new data can still be used for validation of this equation as an independent and reliable data set or for further improvement in the future. Speed of sound data of other alkanes [n-pentane (Scholz et al. [24]) and n-hexane (this work)] measured with the same experimental setup and according to the same metrological protocols have already been used for the development of new fundamental equations of state by Thol et al. [16, 25]; therefore, we are confident that our results of speed of sound measurements in n-heptane are particularly suitable for the sake of validation or improvement of new models.
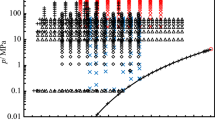
The (p, T) state points we investigated within the scope of the present work and further data from literature are presented in pressure versus temperature phase diagrams (Fig. 1) for n-hexane and n-heptane, respectively. Additionally, the available experimental (p, c, T) data from literature are summarized in Table 1 for n-hexane and in Table 2 for n-heptane, stating the number of points and ranges of temperature and pressure.

Pressure vs. temperature phase diagram for n-hexane and n-heptane showing the (p, T) state points investigated in the present work and a selection of experimental (p, c, T) data from literature in the liquid phase. ◯ this work; △ Muringer et al. [15]; ▽ Khasanshin et al. [21]; □ Daridon et al. [20, 22]; + Baumhögger et al. [23]; ◊ further data from literature (see Table 1 and Table 2, respectively); ―, phase boundary and ☆, critical point, calculated with the equations of state by Thol et al. [16, 17]
2 Experimental Section
2.1 Apparatus Description
The measurements reported in this work were carried out with a speed of sound apparatus where the double-path pulse-echo technique is applied similar to the design of Meier and Kabelac [166]; thus far, our apparatus has been used for investigating the sound speed in several liquids [24, 167, 168]. Inside a cylindrical measuring cell, an x-cut quartz-crystal is used as a piezoelectric disc transducer, which is mounted perpendicular to the axis of the cell. The transducer is located axially off-center between two polished stainless steel reflectors that close the cell at each end. This results in two different path lengths of L1 = 20 mm and L2 = 30 mm. To conduct a measurement, a waveform generator (Agilent, model 3322A) generates a 30-cycle sinusoidal burst at a carrier frequency of 8 MHz, modulated by a half-cycle sin2 function that excites the transducer. The ultrasonic pulses originating from the excited transducer propagate into the fluid into both directions until they reach the two reflectors and are returned to the transducer. Due to the unequal path lengths, one of the two returning echoes arrives delayed by the time difference ∆techo. The electronic signal generated at the transducer is captured with a digital oscilloscope (Agilent, model MS6032A), which averages the waveforms of 16 consecutive pulses. The averaged waveform is treated with a bandpass filter, being based on a Fast–Fourier–Transform with a bandwidth of 1.6 MHz, to enhance the signal-to-noise ratio. Ultimately, the speed of sound c is determined by the following equation
where the time difference ∆techo between the two echoes is computed from the averaged waveform according to an algorithm by Dubberke et al. [169]. Equation 1 includes the time difference between the ideal and the real case of the wave propagation. The correction of these diffraction effects follows a method described by Harris [170] with values of τ/∆techo varying between (0.0016 and 0.0048) %. Since the nominal difference in the path lengths is influenced by the variation of temperature and pressure due to thermal expansion and pressure deformation, the actual path length difference can be determined by the following equation:
In the work we report here, ∆L0 is the path-length difference at the reference conditions of T0 = 293.15 K and p0 = 0.1 MPa. The mean value of the isobaric expansivity α at pressure p0 was determined by integration of values for the differential thermal expansion of stainless steel 1.4571 over the temperature interval [T0, T], according to Eq. 3:
Here, the coefficients ai as given in Table 3 are fixed values given by Meier [171], except a0 which is obtained from calibration (see Sect. 2.2). The mean compressibility β of the cell over the pressure interval [p0, p] at the temperature T is obtained from material-property data [172] as Young’s modulus E and Poisson’s ratio ν = 0.3 according to the following Eq. 4 with the coefficients bi given in Table 3:
The acoustic sensor of the speed of sound apparatus described above was housed in a stainless-steel pressure vessel that was completely immersed into a calibration bath thermostat (Fluke, model 7060). By using an ethylene glycol and water mixture, the temperature range from (233 to 353) K was realized. Temperature measurement was carried out with a long stem 25 Ω standard platinum resistance thermometer (SPRT, Rosemount Aerospace, model 162CE) in conjunction with a direct current thermometry bridge (Isotech, model TTI-2). With a calibration on ITS-90, a standard uncertainty in temperature measurement of u(T) = 0.004 K was achieved. The pressure was measured using two vibrating-quartz-crystal pressure transducers (Paroscientific, models 1000-500A and 1000-6 K), covering different ranges of pressure. A differential pressure indicator (Rosemount, model 3051) was employed to separate the pressure measuring circuit from the measuring cell, thus, preventing the pressure transducers from getting in contact with the liquid sample, but with nitrogen instead. With a second circulation bath thermostat, the pressure transducers and the differential pressure indicator were adjusted to a constant temperature in order to maintain a standard uncertainty of u(p) = 0.0024 MPa.
2.2 Calibration
While the time difference between the two echoes (Eq. 1) is measured at each state point, the actual difference in the path lengths is accurately calibrated only once prior to a complete series of measurements. The path length difference ΔL0 at the reference state and the coefficient a0 for the calculation of the mean isothermal expansion in Eq. 3 are determined from calibration measurements in high-purity water (see Table 4). In the present work, the calibration remains the same as presented in our previous work [24], where speed of sound measurements were carried out at nine state points with ambient pressure and temperatures of T = (274.15, 278.15, 283.15, 293.15, 303.15, 313.15, 333.15, 343.15 and 353.15) K. The parameters ΔL0 and a0 were regressed using Eqs. 1 to 3. Comparing the results of the calibration in water with values calculated from the IAPWS-95 equation of state by Wagner and Pruß [173], our experimental speed of sound data is reproduced within the uncertainty of the equation of state, which was reported to be 0.005 % in the temperature range of our calibration. Furthermore, reference data by Del Grosso and Mader [174] and Fujii and Masui [175] are in a good agreement with our speed of sound measurements in water.
2.3 Experimental Material
A detailed characterization of all substances used in this work can be found in Table 4. High-purity water with a stated purity of 99.9997 % by Sigma-Aldrich was used for the calibration. After the water had been decanted into a stainless-steel bottle, the sample was degassed by repeated cycles of freezing in liquid nitrogen and simultaneous evacuation. Due to their hygroscopic behavior, the two sample materials n-hexane and n-heptane were decanted within the dry inert nitrogen atmosphere of a glove box and then degassed in the same way. For both materials, a coulometric Karl Fischer titration was carried out by the supplier (Sigma-Aldrich or Honeywell, respectively), in which the water content was specified to be not more than 0.01 mass % for n-hexane or 0.005 mass % for the anhydrous n-heptane, respectively. The actual purity of the n-hexane sample was investigated with gas chromatography to be at least 99.73 mass %, using a flame ionization detector. No further analysis was carried out for n-heptane, since a purity of at least 99.84 mass % had already been certified by gas chromatography, conducted by the supplier.
2.4 Experimental Procedure
The experimental procedure applied for the speed of sound measurements presented in this work is the same for both substances. At first, the apparatus was thoroughly cleaned, and the measuring cell was evacuated to remove any residual contamination from previous studies. Before the cell was filled with the sample, the bath thermostat was set to the lowest temperature of 233 K, and the sample cylinder containing either n-hexane or n-heptane was heated to increase its vapor pressure. By taking advantage of the pressure and temperature gradient, the sample was then transferred into the measuring cell. Next, the pressure was increased to the highest pressure of 20 MPa with the aid of the hand pump being part of the sample manifold, and the system was left to equilibrate before measurements began. Speed of sound measurements for each sample were carried out along isotherms over a temperature range from (233 to 353) K, starting at T = 233 K and p = 20 MPa and were continued along isotherms with decreasing pressure.
2.5 Uncertainty Analysis
In order to obtain a complete working equation for the estimation of the uncertainty of the speed of sound in n-hexane and n-heptane (see Eq. 5), ΔL in Eq. 1 was substituted by the relation shown in Eq. 2. Additionally, Eq. 1 was formulated for the measurement in the calibration fluid, taking c0, Δt0 and τ0 into account and was further solved for ΔL0, allowing to eliminate this quantity by substitution in the previously developed equation.
The square of the standard uncertainty of the speed of sound is then given according to Eq. 6, with the small correction terms of the diffraction τ neglected for the case of the uncertainty analysis only.
The expanded combined uncertainty U(c) with a coverage factor k is then given by Eq. 7:
No corrections were made for the presence of impurities, therefore, Eq. 6 includes a term \(u\left( {c(w)} \right)\), which considers the contribution of impurities in the sample to the standard uncertainty of the speed of sound. The influence was quantified by a sensitivity analysis, carried out by using the respective mixture models as available in NIST’s REFPROP database 10.0 [18]. Impurities such as water or the respective isomers were investigated, and to be conservative, the largest influence was considered. This was approximately 64 % of the expanded uncertainty for n-hexane and about 45 % of the expanded uncertainty for n-heptane, at the state point presented in Table 5 or Table 6, respectively.
For both series of measurements, the respective median state point was chosen to exemplify the uncertainty budget for the speed of sound, which has been estimated for each state point individually. The expanded uncertainty of the speed of sound (k = 2) in n-hexane c = 1139 m s−1 at T = 293.20 K and p = 5 MPa is 0.219 m s−1, as presented in detail in Table 5, which corresponds to a relative expanded combined uncertainty of 0.019 %. Over the entire (p, T) region investigated, U(c) varies between (0.152 and 0.350) m s−1, i.e., the relative expanded combined uncertainty ranges from (0.012 to 0.042) %, with the contamination by 2-methylpentane being the dominant factor.
The speed of sound in n-heptane c = 1187 m s−1 at T = 293.20 K and p = 5 MPa has an expanded uncertainty (k = 2) of 0.181 m s−1 as shown in Table 6, which corresponds to a relative expanded combined uncertainty of 0.015 %. We find that U(c) varies between (0.157 and 0.217) m s−1 with the relative expanded combined uncertainty being roughly constant between (0.014 and 0.018) %. For the speed of sound data measured in n-heptane, the contribution from impurities and from calibration turned out to be the dominant factors.
3 Results
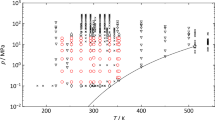
The (p, c, T) results of the speed of sound measurements are listed in Table 7 for n-hexane and in Table 8 for n-heptane, respectively, including the combined expanded uncertainty at each particular state point. Sound speeds were measured at eight (T, p) state points along each of the seven isotherms, leading to a total of 56 (p, c, T) data for each alkane. The resulting absolute speed of sound is plotted versus pressure in Fig. 2 for n-hexane and n-heptane, respectively. In both plots, measuring points at the same temperature are connected in order to visualize the course of each isotherm. We note that both measuring sets are internally consistent throughout the whole temperature and pressure range, and the speed of sound increases with decreasing temperature and increasing pressure. Speeds of sound in n-hexane, measured in the presented work, vary from 833 m s−1 (T = 353 K and p = 0.15 MPa) to 1489 m s−1 at (T = 233 K and p = 20 MPa) and from 899 m s−1 (T = 353 K and p = 0.15 MPa) to 1522 m s−1 at (T = 233 K and p = 20 MPa) for n-heptane.

Results of the speed of sound measurements in n-hexane and n-heptane. Absolute speeds of sound cexp are plotted versus pressure. ○ 233 K; □ 253 K; ▽ 273 K; ☆ 293 K; × 313 K; + 333 K; ◊ 353 K. Lines are shown only to guide the eye
4 Discussion
In Figs. 3 and 4, relative deviations of experimental speeds of sound in n-hexane and n-heptane from values calculated with the equations of state of Thol et al. [16, 17] (zero line) are plotted versus pressure along isotherms; the relative deviation of the equation of state by Span and Wagner [14] is furthermore illustrated as a dashed line. Moreover, relative deviations of other relevant speed of sound data from the literature are shown in Figs. 3 and 4.

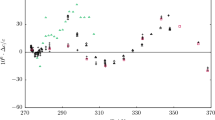
Results of the speed of sound measurements in n-hexane. Relative deviations of the experimental speeds of sound cexp from values cEOS calculated with the equation of state of Thol et al. [16] are plotted versus pressure: ○ this work; △ Khasanshin et al. [21]; ◊ Daridon et al. [20]; □ Ball and Trusler [30]; + Melikhov et al. [52]; ▽, Boelhouwer [32]; × Badalyan et al. [29]; ☆ Kagramayan et al. [45]; ✱ Kling et al. [50]; --- EOS of Span and Wagner [14]. Please note that different scales for the y-axes are used

Results of the speed of sound measurements in n-heptane. Relative deviations of the experimental speeds of sound cexp from values cEOS calculated with the equation of state of Thol et al. [17] are plotted versus pressure: ○ this work; △ Muringer et al. [15]; ◊ Daridon et al. [22]; □ Kiryakov et al. [99]; + Baumhögger [23]; ▽ Boelhouwer [32]; × Badalyan et al. [29]; ☆ Golik et al. [93]; ✱ Kling et al. [50]; --- EOS of Span and Wagner [14]. Please note that different scales for the y-axes are used
The data set of n-hexane (Fig. 3) obtained in the present work was used in the development of the equation of state of Thol et al. [16] and is represented by the equation within ± 0.015 % over the whole pressure range for temperatures between (273 and 353) K. These relative deviations are essentially within the estimated uncertainty of our experimental data. At lower temperatures, the experimental speeds of sound were weighted less in the fitting process, and the relative deviation increases up to 0.18 % at T = 233 K, thereby exceeding the experimental uncertainty, which is roughly about 0.012 % at these particular state points. Since the calibration with water was carried out starting at T = 274 K, the necessary extrapolation to T = 233 K might cause larger relative deviations. The data set by Daridon et al. [20] was also used in the development of the equation of state and is in very good agreement with our reported data within the entire overlapping range of pressure and temperature, starting at T = 293 K. The data set by Boelhouwer et al. [32] is also consistent with our speed of sound data in the overlapping temperature range from (253 to 333) K and confirms our data particularly at low temperatures; the data set is furthermore agreeing well with the data set by Daridon et al. [20] at higher temperatures and pressures. At T = (313, 333 and 353) K and p = (0.1 and 20) MPa or (10 and 20) MPa, respectively, a comparison with the data reported by Khasanshin et al. [21, 47], that was also partly used in the development of the equation of state, is feasible. At all six state points, this data set agrees within (0.04 to 0.08) % with our reported speed of sound data. The data sets by Ball and Trusler [30] and Melikhov et al. [52] show a different behavior compared to our data by intersecting the equation of state at pressures between (10 and 15) MPa and with relative deviations of approximately ± 0.25 % or rather ± 0.50 % at the investigated conditions.
The speed of sound data from Kling et al. [50] and Kagramayan et al. [45] show a systematic offset from our data in the investigated ranges of pressure and temperature and differ by up to (0.64 or 0.74) %, respectively. The short multiparameter equation of state by Span and Wagner [14] deviates by up to − 2.5 % from our speed of sound data of n-hexane.
The speed of sound data of n-heptane reported in the scope of this work are presented in Fig. 4 and were not used for the development of the equation of state by Thol et al. [17]. Over the whole temperature and pressure range, an almost systematic offset can be observed with negative relative deviations between − 0.26 % at T = 253 K and p = 0.18 MPa and − 0.09 % at T = 353 K and p = 0.18 MPa. The trend of each isotherm is somehow comparable with the relative deviations of the model of Span and Wagner [14] from the equation of state by Thol et al. [17]: While the relative deviations are slightly decreasing with increasing pressure at T = (233 and 253) K and are almost constant at T = (273 and 293) K, the trend inverts at higher temperatures. However, the course of the relative deviations of the Span and Wagner model [14] is changing more significantly with relative deviations of up to 0.75 % at T = 353 K and p = 0.15 MPa. In contrast to the results of the speed of sound measurements in n-hexane, no inconsistency of the experimental data at T = 233 K is noticeable.
The speed of sound data reported by Baumhögger et al. [23], which were used in the development of the equation of state by Thol et al. [17], agree with our data within 0.08 % at T = 253 K and within 0.03 % at T = (293 and 353) K, over the entire pressure range up to 20 MPa. The data set by Daridon et al. [22] was also used for the development of the equation of state but shows an almost systematic offset of roughly 0.09 % at T = 293 K that increases with higher temperatures up to 0.22 % at T = 353 K compared to the data measured in the present work, even though both data sets show a similar trend. The speed of sound data by Muringer et al. [15] were used in the development of the new equation of state as well but are not in good agreement with our data presented here. Just two state points on each isotherm at p = (0.1 and 20) MPa are overlapping the pressure range of the present work, and the data show a positive offset from our data by (0.17 to 0.71) % at temperatures between (233 and 313) K.
5 Conclusion
We report speeds of sound measured over the temperature range from (233 to 353) K at pressures up to 20 MPa in samples of high-purity n-hexane and n-heptane. The speed of sound data in n-hexane were considered as primary data for the development of the new equation of state by Thol et al. [16]. This work’s data agree with the new equation of state within 0.015 % at T ≥ 273 K and within 0.18 % or better at lower temperatures. The overall relative expanded combined uncertainty (k = 2) of the n-hexane data ranges from (0.012 to 0.042) %. The new speed of sound data of n-heptane confirms the data set by Baumhögger et al. [23], which was already used in the development of the new equation of state by Thol et al. [17]. Both data sets agree within 0.08 % at T = 253 K and 0.03 % at T = (293 or 353) K, respectively. The overall relative expanded combined uncertainty (k = 2) of our n-heptane data ranges from (0.014 to 0.018) %.
References
I.H. Aljundi, Renew. Energy (2011). https://doi.org/10.1016/j.renene.2010.09.022
H.U. Helvaci, Z.A. Khan, Energy Convers. Manag. (2017). https://doi.org/10.1016/j.enconman.2017.02.011
G. Knothe, J. Van Gerpen, J. Krahl, The Biodiesel Handbook (AOCS Press, Illinois, 2005).
M. Saravanan, B.M. Bhosle, R. Subramanian, J. Food Eng. (2006). https://doi.org/10.1016/j.jfoodeng.2005.03.040
S.P.J. Kumar, S.R. Prasad, R. Banerjee, D.K. Agarwal, K.S. Kulkarni, K.V. Ramesh, Chem. Cent. J. (2017). https://doi.org/10.1186/s13065-017-0238-8
N. Metz, Dissertation, Johannes Gutenberg-Universität Mainz, 2009. https://d-nb.info/1004952295/34. Accessed 8 May 2020
O. Buddrick, O.A.H. Jones, P.D. Morrison, D.M. Small, RSC Adv. (2013). https://doi.org/10.1039/c3ra44442b
E.J. Conkerton, P.J. Wan, O.A. Richard, J. Am. Oil Chem. Soc. (1995). https://doi.org/10.1007/BF02542075
A. Hooshiar, P. Uhlik, Q. Liu, T.H. Etsell, D.G. Ivey, Fuel Process. Technol. (2012). https://doi.org/10.1016/j.fuproc.2011.10.008
H. Remmele, A. Köllhofer, H. Plenio, Organometallics (2003). https://doi.org/10.1021/om030450a
Y.-X. Chen, M.D. Rausch, J.C.W. Chien, J. Polym. Sci. A1 (1995) doi:https://doi.org/10.1002/pola.1995.080331301
G. Kalghatgi, H. Babiker, J. Badra, S.A.E. Int, J. Engines (2015). https://doi.org/10.4271/2015-01-0757
A. Chica, A. Corma, J. Catal. (1999). https://doi.org/10.1006/jcat.1999.2601
R. Span, W. Wagner, Int. J. Thermophys. 24, 41 (2003)
M.J.P. Muringer, N.J. Trappeniers, S.N. Biswas, Phys. Chem. Liq. (1985). https://doi.org/10.1080/00319108508080991
M. Thol, Y. Wang, E. W. Lemmon, R. Span, J. Phys. Chem. Ref. Data (to be submitted 2021)
M. Thol, D. Tenji, E.W. Lemmon, R. Span, Int. J. Thermophys. (to be submitted 2021)
E.W. Lemmon, I.H. Bell, M.L. Huber, M.O. McLinden, NIST Standard Reference Database 23: Reference Fluid Thermodynamic and Transport Properties-REFPROP (National Institute of Standards and Technology, Boulder, 2018).
R. Span, R. Beckmüller, T. Eckermann, S. Herrig, S. Hielscher, A. Jäger, T. Neumann, S. Pohl, B. Semrau, M. Thol, TREND. Thermodynamic Reference and Engineering Data (Lehrstuhl für Thermodynamik: Ruhr-Universität Bochum, Bochum, 2019)
J.L. Daridon, B. Lagourette, J.-P.E. Grolier, Int. J. Thermophys. (1998). https://doi.org/10.1023/A:1021403219658
T.S. Khasanshin, V.S. Samuilov, A.P. Shchemelev, J. Eng. Phys. Thermophy. (2008). https://doi.org/10.1007/s10891-008-0093-5
J.L. Daridon, B. Lagourette, A. Lagrabette, Phys. Chem. Liq. (1999). https://doi.org/10.1080/00319109908045122
E. Baumhögger, Personal Communication, Universität Paderborn (2017)
C.W. Scholz, Y. Sanchez-Vicente, T. Tananilgul, M. Thol, J.P.M. Trusler, M. Richter, J. Chem. Eng. Data (2020). https://doi.org/10.1021/acs.jced.0c00299
M. Thol, T. Uhde, M. Richter, Span, R. and Lemmon, E.W., J. Phys. Chem. Ref. Data (to be submitted 2020)
J. Allegra, S. Hawley, G. Holton, J. Acoust. Soc. Am. (1970). https://doi.org/10.1121/1.1911446
E. Alonso, H. Guerrero, D. Montaño, C. Lafuente, H. Artigas, Thermochim. Acta (2011). https://doi.org/10.1016/j.tca.2011.07.023
T.M. Aminabhavi, M.I. Aralaguppi, B. Gopalakrishna, R.S. Khinnavar, J. Chem. Eng. Data (1994). https://doi.org/10.1021/je00015a028
A.L. Badalyan, N.F. Otpushchennikov, Y.S. Shoitov, Uc. Zap.-Kursk. Gos. Ped. Inst. 91, 135 (1971)
S.J. Ball, J.P.M. Trusler, Int. J. Thermophys. (2001). https://doi.org/10.1023/A:1010770730612
M. Basu, T. Samanta, D. Das, J. Chem. Thermodyn. (2013). https://doi.org/10.1016/j.jct.2012.09.015
J.W.M. Boelhouwer, Physica (1967). https://doi.org/10.1016/0031-8914(67)90016-X
H.B. Bohidar, J. Appl. Phys. (1988). https://doi.org/10.1063/1.341780
M.F. Bolotnikov, Y.A. Neruchev, Y.F. Melikhov, V.N. Verveyko, M.V. Verveyko, J. Chem. Eng. Data (2005). https://doi.org/10.1021/je050060q
C.A. Cerdeiriña, C.A. Tovar, D. González-Salgado, E. Carballo, L. Romaní, Phys. Chem. Chem. Phys. (2001). https://doi.org/10.1039/b104891k
P.F. Cholpan, V.S. Sperkach, L.N. Garkusha, Fiz. Zhidk. Sostoyaniya 9, 79 (1981)
B.E. de Cominges, M.M. Piñeiro, L. Mosteiro, T.P. Iglesias, J.L. Legido, M.I. Paz Andrade, J. Chem. Eng. Data. (2001). https://doi.org/10.1021/je010039z
S.P. Dmitriev, in Ul'trazvuk i termodinamiceskie svojstva vescestva, ed. by Kursk State Pedagogical Inst. (Izd. KGPI) (Kursk, 1984), p. 213
H.F. Eden, E.G. Richardson, Acta. Acust. 10, 309 (1960)
G.V. Ermakov, R.G. Ismagilov, High Temp. 14, 975 (1976)
A. Gayol, M. Iglesias, J.M. Goenaga, R.G. Concha, J.M. Resa, J. Mol. Liq. (2007). https://doi.org/10.1016/j.molliq.2006.11.012
M. Gepert, S. Ernst, J. Solution Chem. (2003). https://doi.org/10.1023/B:JOSL.0000003152.24858.98
A.Z. Golik, Y.I. Kuzovkov, V.Y. Nagin, Ukr. Fiz. Zh. 32, 1524 (1987)
R. Ismagilov, G. Ermakov, High Temp. 20, 561 (1982)
L. Kagramanyan, A. Badalyan, Izv. Akad. Nauk. Arm. SSR Fiz. 13, 478 (1978)
V. Kannappan, R.J. Santhi, E.J.P. Malar, Phys. Chem. Liq. 40, 507 (2002)
T.S. Khasanshin, A.P. Shchemelev, High Temp. (2001). https://doi.org/10.1023/A:1004170530517
B.N. Kireev, N.F. Otpushchennikov, Russ. J. Phys. Chem. 48, 829 (1974)
B.S. Kiryakov, Y.S. Shoitov, Uc. Zap.-Kursk. Gos. Ped. Inst. 91, 209 (1971)
R. Kling, E. Nicolini, J. Tissot, Rech. Aérosp. 31, 31 (1953)
E. Mascato, A. Mariano, M.M. Piñeiro, J.L. Legido, M.I. Paz Andrade, J. Chem. Thermodyn. (2007). https://doi.org/10.1016/j.jct.2007.02.002
Y.F. Melikhov, G.A. Mel’nikov, V.M. Tutov, V.N. Verveiko, Izv. Vyssh. Uchebn. Zaved. Energy 34, 73 (1991)
G.A. Melnikov, V.N. Verveiko, N.F. Otpushchennikov, Russ. J. Phys. Chem. 62, 393 (1988)
Y.A. Neruchev, V.V. Zotov, N.F. Otpushchennikov, Russ. J. Phys. Chem. 43, 1597 (1969)
Y.A. Neruchev, M.F. Bolotnikov, V.V. Zotov, High Temp. 43, 274 (2005)
B. Orge, A. Rodríguez, J.M. Canosa, G. Marino, M. Iglesias, J. Tojo, J. Chem. Eng. Data (1999). https://doi.org/10.1021/je9900676
A. Rodrı́guez, J. Canosa, J. Tojo, J. Chem. Thermodyn. (2003). https://doi.org/10.1016/S0021-9614(03)00096-X
R.E. Rolling, C.J. Vogt, J. Basic Eng. 82, 635 (1960)
V.N. Verveiko, G.A. Mel'nikov, N.F. Otpushchennikov, in Ul'trazvuk i termodinamiceskie svojstva vescestva, ed. by Kursk State Pedagogical Inst. (Izd. KGPI) (Kursk, 1986), p. 59
V.N. Verveiko, G.A. Mel'nikov, Y.F. Melikhov, Teplofiz. Svoistva Vesh. Mater. 30, 5 (1991)
X. Zheng, Y. Zhang, M. He, J. Chem. Eng. Data (2016). https://doi.org/10.1021/je501106d
V.V. Zotov, B.N. Kireev, Y.A. Neruchev, J. Appl. Mech. Technol. Phys. 15, 282 (1975)
E. Aicart, M.K. Kumaran, C.J. Halpin, G.C. Benson, J. Chem. Thermodyn. (1983). https://doi.org/10.1016/0021-9614(83)90125-8
Y. Akamatsu, H. Ogawa, S. Murakami, Thermochim. Acta (1987). https://doi.org/10.1016/0040-6031(87)88317-X
I. Alonso, I.G. de La Fuente, J.A. González, J.C. Cobos, J. Chem. Eng. Data (2013). https://doi.org/10.1021/je400134d
T.M. Aminabhavi, B. Gopalakrishna, J. Chem. Eng. Data (1995). https://doi.org/10.1021/je00019a022
M.I. Aralaguppi, T.M. Aminabhavi, R.H. Balundgi, S.S. Joshi, J. Phys. Chem. (1991). https://doi.org/10.1021/j100166a070
M.I. Aralaguppi, C.V. Jadar, T.M. Aminabhavi, J. Chem. Eng. Data (1999). https://doi.org/10.1021/je9802266
P. Bahadur, N.V. Sastry, Int. J. Thermophys. (2003). https://doi.org/10.1023/A:1022920005042
J.G. Baragi, M.I. Aralaguppi, M.Y. Kariduraganavar, S.S. Kulkarni, A.S. Kittur, T.M. Aminabhavi, J. Chem. Thermodyn. (2006). https://doi.org/10.1016/j.jct.2005.03.024
G.C. Benson, C.J. Halpin, A.J. Treszczanowicz, J. Chem. Thermodyn. (1981). https://doi.org/10.1016/0021-9614(81)90017-3
A. Blanco, A. Gayol, D. Gómez-Díaz, J.M. Navaza, Phys. Chem. Liq. (2013). https://doi.org/10.1080/00319104.2012.760086
A. Blanco, A. Gayol, D. Gómez, J.M. Navaza, Phys. Chem. Liq. (2013). https://doi.org/10.1080/00319104.2012.753601
N. Calvar, E. Gómez, B. González, Á. Domínguez, J. Chem. Thermodyn. (2009). https://doi.org/10.1016/j.jct.2009.03.009
J. Canosa, A. Rodriguez, J. Tojo, Fluid Phase Equilib. (1999). https://doi.org/10.1016/S0378-3812(99)00032-1
M. Chorążewski, M. Tkaczyk, J. Chem. Eng. Data (2006). https://doi.org/10.1021/je060200b
M. Chorążewski, P. Góralski, M. Hrynko, J.-P. Grolier, E. Wilhelm, J. Chem. Eng. Data (2010). https://doi.org/10.1021/je100610v
N.V. Choudary, A.P. Kudchadker, P.R. Naidu, Phys. Chem. Liq. (1986). https://doi.org/10.1080/00319108608078511
F.E. Danusso, Rendiconti Acad. Naz. Lincei Cl. Sci. Fis. Mat. Nat. 14, 81 (1953)
R. Dey, A. Chattopadhyay, A.K. Sharma, J.D. Pandey, J. Mol. Liq. (2009). https://doi.org/10.1016/j.molliq.2009.03.001
G. Dharmaraju, Narayana Swamy G., Raman G.K., Indian J. Chem. A 20, 1109 (1981)
M. Dzida, M. Cempa, J. Chem. Thermodyn. (2008). https://doi.org/10.1016/j.jct.2008.05.014
M. Dzida, S. Ernst, J. Chem. Eng. Data (2003). https://doi.org/10.1021/je030136n
M. Dzida, P. Góralski, J. Chem. Thermodyn. (2006). https://doi.org/10.1016/j.jct.2005.10.009
M. Dzida, L. Waleczek, J. Chem. Thermodyn. (2010). https://doi.org/10.1016/j.jct.2009.09.001
M. Dzida, A. Żak, S. Ernst, J. Chem. Thermodyn. (2005). https://doi.org/10.1016/j.jct.2004.10.001
G. Edgar, G. Calingaert, J. Am. Chem. Soc. (1929). https://doi.org/10.1021/ja01380a035
E.B. Freyer, J.C. Hubbard, D.H. Andrews, J. Am. Chem. Soc. (1929). https://doi.org/10.1021/ja01378a014
L.N. Garkusha, V.S. Sperkach, P.F. Cholpan, Fiz. Zhidk. Sostoyaniya 16, 80 (1988)
A. Gayol, Phys. Chem. Liq. (2012). https://doi.org/10.1080/00319104.2012.673612
A. Gayol, A. Touriño, M. Iglesias, Phys. Chem. Liq. (2010). https://doi.org/10.1080/00319104.2010.481764
O.Z. Golik, I.I. Ivanova, Zh. Fiz. Khim. 36, 1768 (1962)
A.Z. Golik, I.I. Adamenko, Makhno M.S., Fiz. Zhidk. Sostoyaniya 10, 3 (1982)
V.H. Hasanov, High Temp. (2012). https://doi.org/10.1134/S0018151X12010063
E. Junquera, G. Tardajos, E. Aicart, J. Chem. Thermodyn. (1988). https://doi.org/10.1016/0021-9614(88)90041-9
F. Kimura, A.J. Treszczanowicz, C.J. Halpin, G.C. Benson, J. Chem. Thermodyn. (1983). https://doi.org/10.1016/0021-9614(83)90048-4
F. Kimura, T. Sugiura, H. Ogawa, Thermochim. Acta (2013). https://doi.org/10.1016/j.tca.2013.09.017
B.S. Kiryakov, N.F. Otpushchennikov, P.P. Panin, Viniti 800–74, 1 (1974)
B.S. Kiryakov, N.F. Otpushchennikov, Uc. Zap.-Kursk. Gos. Ped. Inst. 91, 104 (1971)
O. Kiyohara, G.C. Benson, J. Chem. Thermodyn. (1979). https://doi.org/10.1016/0021-9614(79)90067-3
M.K. Kumaran, F. Kimura, C.J. Halpin, G.C. Benson, J. Chem. Thermodyn. (1984). https://doi.org/10.1016/0021-9614(84)90050-8
D.J. Luning Prak, J.S. Cowart, P.C. Trulove, J. Chem. Eng. Data (2014). https://doi.org/10.1021/je5007532
K. Malakondaiah, V. Hyder Khan, S.V. Subrahmanyam, J. Acoust. Soc. India 6, 51 (1978)
G. Marino, M.M. Piñeiro, M. Iglesias, B. Orge, J. Tojo, J. Chem. Eng. Data (2001). https://doi.org/10.1021/je000200q
G.W. Marks, J. Acoust. Soc. Am. (1955). https://doi.org/10.1121/1.1907993
L. Martínez-Baños, C. Rivas, J.M. Embid, S. Otín, J. Chem. Eng. Data (2013). https://doi.org/10.1021/je300775u
G.R. Naidu, P.R. Naidu, Acta. Chim. Hung. 108, 85 (1981)
H. Nakai, H. Soejima, K. Tamura, H. Ogawa, S. Murakami, Y. Toshiyasu, Thermochim. Acta (1991). https://doi.org/10.1016/0040-6031(91)80441-K
J. Nath, J. Chem. Thermodyn. (1996). https://doi.org/10.1006/jcht.1996.0046
J. Nath, J. Chem. Thermodyn. (1997). https://doi.org/10.1006/jcht.1997.0200
J. Nath, J. Chem. Thermodyn. (1998). https://doi.org/10.1006/jcht.1998.0358
J. Nath, J. Chem. Thermodyn. (2002). https://doi.org/10.1016/S0021-9614(02)00255-0
G. Natta, M. Baccaredda, Rendi. Aeead. Nazionaledei Lincei 4, 360 (1948)
J.N. Nayak, M.I. Aralaguppi, T.M. Aminabhavi, J. Chem. Eng. Data (2001). https://doi.org/10.1021/je010020w
S.M. Nayeem, M. Kondaiah, K. Sreekanth, M. Srinivasa Reddy, D. Krishna Rao, J. Therm. Anal. Calorim. (2016). https://doi.org/10.1007/s10973-015-4994-1
K. Nishikawa, K. Tamura, S. Murakami, J. Chem. Thermodyn. (1998). https://doi.org/10.1006/jcht.1997.0296
K. Ohomuro, K. Tamura, S. Murakami, J. Chem. Thermodyn. (1987). https://doi.org/10.1016/0021-9614(87)90107-8
B. Orge, M. Iglesias, A. Rodriguez, J.M. Canosa, J. Tojo, Fluid Phase Equilib. (1997). https://doi.org/10.1016/S0378-3812(97)00031-9
S.L. Oswal, M.M. Maisuria, J. Mol. Liq. (2002). https://doi.org/10.1016/S0167-7322(02)00021-1
J.D. Pandey, P. Jain, V. Vyas, Can. J. Chem. (1994). https://doi.org/10.1139/v94-315
D. Papaioannou, D. Ziakas, C. Panayiotou, J. Chem. Eng. Data (1991). https://doi.org/10.1021/je00001a011
J.M. Pardo, D. González-Salgado, C.A. Tovar, C.A. Cerdeiriña, E. Carballo, L. Romani, Can. J. Chem. (2002). https://doi.org/10.1139/v02-030
S. Parthasarathy, Proc. Indian Acad. Sci. A 2, 497 (1935)
A.B. Pereiro, A. Rodriguez, J. Chem. Thermodyn. (2007). https://doi.org/10.1016/j.jct.2007.02.008
A.B. Pereiro, A. Rodriguez, Phys. Chem. Liq. (2008). https://doi.org/10.1080/00319100701459327
M.J.W. Povey, S.A. Hindle, J.D. Kennedy, Z. Stec, R.G. Taylor, Phys. Chem. Chem. Phys. (2003). https://doi.org/10.1039/b206425a
S. Prakash, S.K. Singh, N. Prasad, O. Prakash, Acoust. Lett. 4, 132y4 (1981)
N. Prasad, S.K. Singh, S. Singh, S. Prakash, Indian J. Phys. B 52, 69 (1978)
A. Przybyła, M. Chorążewski, E. Zorębski, W. Marczak, Int. J. Thermophys. (2011). https://doi.org/10.1007/s10765-010-0773-1
R.D. Rai, R.K. Shukla, A.K. Shukla, J.D. Pandey, J. Chem. Thermodyn. (1989). https://doi.org/10.1016/0021-9614(89)90122-5
S.K. Raikar, T.M. Aminabhavi, S.B. Harogoppad, R.H. Balundgi, Indian J. Technol. 31, 581 (1993)
E. Rajagopal, S.V. Subrahmanyam, J. Acoust. Soc. India 6, 45 (1978)
V. Rajendran, J. Indian, Pure Appl. Phys. 34, 52 (1996)
R.M. Rama, Indian J. Phys. 14, 109 (1940)
K.P.C. Rao, K.S. Reddy, Phys. Chem. Liq. (1988). https://doi.org/10.1080/00319108808078565
M.V. Rathnam, S. Mankumare, M.S.S. Kumar, J. Chem. Eng. Data (2010). https://doi.org/10.1021/je9006597
M.V. Rathnam, S. Mankumare, K. Jain, M.S.S. Kumar, J. Solution Chem. (2012). https://doi.org/10.1007/s10953-012-9802-y
P. Reddy, J.D. Raal, D. Ramjugernath, Fluid Phase Equilib. (2013). https://doi.org/10.1016/j.fluid.2013.07.044
A. Rodriguez, J. Canosa, J. Tojo, J. Chem. Eng. Data (1999). https://doi.org/10.1021/je990009k
X. Rosario Rajkumar, K.V. Raman, S.J. Arulraj, J. Indian, Pure Appl. Phys. 22, 241 (1984)
V.K. Sachdeva, V.S. Nanda, J. Chem. Phys. (1981). https://doi.org/10.1063/1.442654
N.V. Sastry, P.N. Dave, Int. J. Thermophys. (1996). https://doi.org/10.1007/BF01438671
N.V. Sastry, M.M. Raj, J. Chem. Eng. Data (1996). https://doi.org/10.1021/je950172p
N.V. Sastry, M.K. Valand, J. Chem. Eng. Data (1996). https://doi.org/10.1021/je960135d
N.V. Sastry, Fluid Phase Equilib. (1997). https://doi.org/10.1016/S0378-3812(96)03162-7
N.V. Sastry, N.J. Jain, A. George, P. Bahadur, Fluid Phase Equilib. (1999). https://doi.org/10.1016/S0378-3812(99)00232-0
W. Schaaffs, Z. Phys. Chem. 194, 28 (1944)
D. Sharma, J.S. Yadav, K.C. Singh, V.K. Sharma, J. Solution Chem. (2008). https://doi.org/10.1007/s10953-008-9294-y
D. Sharma, J.S. Yadav, S. Kumar, K.C. Singh, V.K. Sharma, Thermochim. Acta (2008). https://doi.org/10.1016/j.tca.2008.06.004
R.K. Siwach, D. Sharma, V.K. Sharma, Thermochim. Acta (2010). https://doi.org/10.1016/j.tca.2010.03.012
V.S. Sperkach, P.F. Cholpan, Sinilo V.N., Zolotar A.V., Fiz. Zhidk. Sostoyaniya 7, 110 (1979)
M. Sreenivasulu, P.R. Naidu, Aust. J. Chem. (1979). https://doi.org/10.1071/CH9790471
T. Sugiura, H. Ogawa, J. Chem. Thermodyn. (2009). https://doi.org/10.1016/j.jct.2009.06.001
K. Tamura, K. Ohomuro, S. Murakami, J. Chem. Thermodyn. (1983). https://doi.org/10.1016/0021-9614(83)90092-7
K. Tamura, K. Ohomuro, S. Murakami, J. Chem. Thermodyn. (1984). https://doi.org/10.1016/0021-9614(84)90143-5
K. Tamura, S. Murakami, M. Fukumori, Y. Akagi, Y. Kawasaki, J. Solution Chem. (1994). https://doi.org/10.1007/BF00974099
G. Tardajos, M. Diaz Pena, E. Aicart, J. Chem. Thermodyn. (1986). https://doi.org/10.1016/0021-9614(86)90071-6
A. Touriño, A. Gayol, G. Marino, M. Iglesias, Phys. Chem. Liq. (2004). https://doi.org/10.1080/003191000410001658876
A.J. Treszczanowicz, C.J. Halpin, G.C. Benson, J. Chem. Eng. Data (1982). https://doi.org/10.1021/je00029a026
V.M. Tutov, N.F. Otpushennikov, Ukr. Fiz. Zh. 11, 1253 (1966)
N. Utter, M.R. Kling, C.R. Hebd, Acad. Sci. 227, 41 (1948)
E.B. Wilson, W.T. Richards, J. Phys. Chem. (1931). https://doi.org/10.1021/j150334a017
P.D.G. Wilson, N.R. Gladwell, D.J. Hibberd, M.M. Robins, Ultrasonics (1991). https://doi.org/10.1016/0041-624X(91)90060-L
V.V. Zotov, Y.A. Neruchev, N.F. Otpushennikov, Inzh.-Fiz. Zh. 15, 890 (1968)
V.V. Zotov, Y.F. Melikhov, G.A. Melnikov, Y.A. Neruchev, Sound Velocity in Liquid Hydrocarbons (Kursk, 1995)
K. Meier, S. Kabelac, Rev. Sci. Instrum. (2006). https://doi.org/10.1063/1.2400019
R. Wegge, M. Richter, R. Span, J. Chem. Eng. Data (2015). https://doi.org/10.1021/je501065g
R. Wegge, M. Richter, R. Span, Fluid Phase Equilib. (2016). https://doi.org/10.1016/j.fluid.2015.10.034
F.H. Dubberke, E. Baumhögger, J. Vrabec, Rev. Sci. Instrum. (2015). https://doi.org/10.1063/1.4921478
G.R. Harris, J. Acoust. Soc. Am. (1981). https://doi.org/10.1121/1.386671
K. Meier, Dissertation, Helmut-Schmidt-Universität - Universität der Bundeswehr Hamburg, 2006. https://www.hsu-hh.de/thermodynamik/wp-content/uploads/sites/741/2017/09/Habilitationsschrift-Meier.pdf. Accessed 8 May 2020
Verein Deutscher Eisenhüttenleute, Stahl-Eisen-Werkstoffblätter (1997)
W. Wagner, A. Pruß, J. Phys. Chem. Ref. Data (2002). https://doi.org/10.1063/1.1461829
V.A. Del Grosso, C.W. Mader, J. Acoust. Soc. Am. (1972). https://doi.org/10.1121/1.1913258
K.-I. Fujii, R. Masui, J. Acoust. Soc. Am. (1993). https://doi.org/10.1121/1.405661
Acknowledgements
The experimental part of this work was carried out in the labs of Prof. Roland Span’s thermodynamics group at Ruhr University Bochum (RUB). We thank Dr. Monika Thol of RUB for helpful discussions regarding the present measurements. Moreover, we are thankful to the group of Professor Ulf-Peter Apfel (faculty of chemistry and biochemistry at RUB) for providing the glove box for the sample transfer and for supporting us with the GC-analysis of the n-hexane sample.
Funding
Open Access funding enabled and organized by Projekt DEAL..
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Scholz, C.W., Richter, M. Speeds of Sound in n-Hexane and n-Heptane at Temperatures from (233.33 to 353.21) K and Pressures up to 20 MPa. Int J Thermophys 42, 18 (2021). https://doi.org/10.1007/s10765-020-02778-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10765-020-02778-6