
Abstract
Bacterial wilt (BW), caused by Ralstonia solanacearum species complex is one of the major biotic factors limiting tomato production in the humid tropics. Pyramiding of resistance genes through marker-assisted selection is an efficient way to develop durable BW resistant cultivars. Tomato line ‘Hawaii 7996’ (H7996) is a stable and robust resistance source against various strains of the species complex. Major BW resistance quantitative trait loci (QTLs) Bwr-12 and Bwr-6, and several minor or strain specific QTLs have been coarse-mapped in this line, but none has been fine-mapped and validated. The objective of the current study was to construct a high density genetic map using single-nucleotide polymorphism (SNP) markers derived from genotyping-by-sequencing, fine-map Bwr-12 and Bwr-6 and determine the effects of these QTLs using a near isogenic line (NIL) population. A high density genetic map using 1604 SNP markers with an average distance of 0.82 cM was developed for 188 F9 recombinant inbred lines derived from the cross H7996 × WVa700. A total of seven QTLs associated with BW resistance to race 1-phylotype I (R. pseudosolanacearum) or/and race 3-phylotype II (R. solanacearum) strains were located on chromosomes 6 (Bwr-6.1, 6.2, 6.3 and 6.4) and 12 (Bwr-12.1, Bwr-12.2 and Bwr-12.3) with logarithm of odds (LOD) scores of 6.2–15.6 and 6.2–31.1, explaining 14.2–33.4% and 15.9–53.9% of the total phenotypic variation contributed from H7996, respectively. To validate the genetic effects of the two QTL regions, a set of 80 BC3F3 NILs containing different sections of Bwr-6 with or without Bwr-12 was phenotyped for disease severity after challenge with either race 1-phylotype I Pss4 or race 3-phylotype II Pss1632 BW strains over two seasons. Bwr-6.1 specific to Pss4 and Bwr-6.3 specific to Pss1632 were mapped to an interval of 5.0 cM (P < 0.05) between 6_33,444,000_SLM6-47 and 6_33,868,000_SLM6-124 SNP marker, and to 2.7 cM (P < 0.01) between positions 6_35,949,000 _SLM6-107 to 6_36,750,000_SLM6-82 marker, respectively. In addition, the specific effect of Bwr-12 for resistance to Pss4 (LOD score of 5.8-16.1, P < 0.01) was confirmed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Bacterial wilt (BW) caused by the soil-borne bacterium Ralstonia solanacearum species complex is a major limiting factor for tomato production in the humid tropics, leading to wilting and plant death (Daunay et al. 2010; Hayward 1991; Huerta et al. 2015; Kelman 1953; Wang et al. 1998). The pathogen has a large host range encompassing more than 200 plant species, and is capable of reaching very high cell densities in the host xylem through rapid multiplication. The disease is reported to cause yield losses up to 91%, dependent of the tomato cultivar, climate, soil type, cropping pattern, and strain (Denny 2006; Elphinstone 2005; Krausz and Thurston 1975; Messiaen 1989). Based on host range, biochemical properties, geographical origin, multilocus sequence analysis and partial sequencing of the endoglucanase gene, R. solanacearum species complex has been delineated into five races, six biovars, four phylotypes, nine clades and 53 sequevars, respectively (Buddenhagen and Kelman1964; Fegan and Prior 2005; Hayward 1991; Poussier et al. 2000; Prior and Fegan 2005; Wicker et al. 2012). Safni et al. (2014) further divided the species complex into three species. In this study, race 1-phylotype I strains belonged to R. pseudosolanacearum and race 3-phylotype II strains belonged to R. solanacearum were used. Due to its broad genetic diversity, R. solanacearum species complex was ranked second in a list of the top ten most scientifically and economically impactful plant bacteria (Mansfield et al. 2012).
Because the pathogen can survive in the soil without host plants for long periods it is difficult to control by agronomical practices or chemical treatments. The use of genetic resistance is considered to be the cheapest, most efficient and environmentally friendliest approach to control bacterial wilt, but breeding for durable resistance against R. solanacearum species complex is challenging owing to the site-specific and strain-specific nature of resistance (Gaelle 2014; Genin 2010; Hanson et al. 1996; Lopes et al. 1994; Peter et al. 1993; Prior et al. 1990, 1994). Several screening tests for resistant host plants have been conducted using phylotype I or/and phylotype II strains (Carmeille et al. 2006a; Hai et al. 2008; Jaunet and Wang 1999; Kim et al. 2016; Scott et al. 2005; Wang et al. 1998) and various resistance sources were identified. Wang et al. (1998) found that Hawaii 7996 (H7996, Solanum lycopersicum) demonstrated high levels of resistance on the basis of trials conducted in 11 countries in Asia, America and Australia.
BW resistance QTLs in tomato have been mapped using various marker systems, such as simple sequence repeat (SSR), amplified fragment length polymorphic (AFLP), sequence characterized amplified region (SCAR), restriction fragment length polymorphism (RFLP), cleaved amplified polymorphic sequences (CAPS) markers (Ashrafi et al. 2009; Carmeille et al. 2006b; Danesh et al. 1994; Mangin et al. 1999; Miao et al. 2009; Thoquet et al. 1996a, b; Truong et al. 2015; Wang et al. 2000, 2013) and were located on different chromosomes, depending on the resistance sources and pathogen strains. Two resistance QTLs derived from L285 against UW365 (biovar 2, race 3) strain were situated on chromosomes 6 and 10 (Danesh et al. 1994). F3 and F2:3 H7996 × WVa700 populations inoculated by Pss4 (biovar 3, race 1-phylotype I), JT519 (biovar 3, race 1-phylotype I), GMI8217 (biovar 1) and JT516 (biovar 2, race 3-phylotype II) showed that resistance factors were located on five chromosomes (3, 4, 6, 8 and 12) (Carmeille et al. 2006b; Thoquet et al. 1996a, b; Wang et al. 2000). Wang et al. (2013) identified two major BW resistance QTLs in H7996, Bwr-12 and Bwr-6, based on ten screening trials with an F9 recombinant inbred line (RIL) population derived from a cross between H7996 and the BW susceptible line WVa700; screening trials were conducted in the field or in the greenhouse and included phylotype I and phylotype II strains. Bwr-12 and Bwr-6 located in a 2.8 and 15.5 cM interval on chromosome 12 and 6, respectively, controlled up to 56 and 22% of the phenotypic variation. These experiments also showed that Bwr-12 was effective for phylotype I, and Bwr-6 was associated with resistance to race 1-phylotype I and race 3-phylotype II strains (Wang et al. 2013). These results showed that resistance to BW in tomato is a complex strain-specific trait and is controlled by multiple genes.
Although several QTLs for resistance to R. solanacearum species complex have been mapped, none of them was fine-mapped, hindering efficient marker-assisted selection and gene cloning. Availability of a high quality reference genome and improved marker technologies such as single nucleotide polymorphism (SNP) markers capable of high-throughput genotyping made fine mapping of QTLs in segregating populations easier and allow for efficient introgression of favorable alleles from a donor to a recipient by both positive and background selection (Collard and Mackill 2008; Foolad 2007). Next-generation sequencing (NGS) technologies have facilitated the construction of reference genome sequences, which provide valuable information for understanding genomic variation and genome evolution (Feuillet et al. 2011). NGS technologies also are used for genotyping. Genotyping by sequencing (GBS) is the most efficient tool for simultaneous genome-wide SNP discovery and genotyping. GBS is simple, quick, and reproducible and SNP markers are obtained at relatively low cost in short time through multiplex sequencing of restriction site-associated DNA (Elshire et al. 2011). The draw backs of the method are often large amounts of missing data due to low coverage sequencing (Fu and Peterson 2011; Poland et al. 2012) and uneven genome coverage, due to the sequence specificity of the chosen restriction enzyme (Beissinger et al. 2013). GBS is used for genotyping for a wide range of purposes including for QTL mapping and genomic prediction in many crop and animal species, including for example rice (Spindel et al. 2013), tomato (Capel et al. 2015; Celik et al. 2017; Yu et al. 2016), grape (Marrano et al. 2017) and livestock species (Gurgul et al. 2018). GBS can be successfully adapted to model species like tomato with well-characterized reference genomes, as well as to crops without reference genome sequences (Berthouly-Salazar et al. 2016). The software Tassel for GBS analysis is a very powerful pipeline to efficiently handle studies where up to hundreds of thousands or even millions of SNPs are generated from up to 100,000 individuals (Glaubitz et al. 2014).
QTL validation is an essential step before using marker-assisted selection (MAS) to introgress QTLs into new genetic backgrounds (Hospital 2009). A large number of QTLs and molecular markers related with them have been identified and published, but only very few have been utilized in breeding programs. Failure to validate the QTLs and unexpected results in QTL introgression programs were mostly due to false positive QTLs, QTLs affected by environmental conditions, traits encoded by multiple genes, recombination between genes and the selectable markers, and epistatic effects with another QTL or with the genetic background (Collard and Mackill 2008; Hospital 2005). Near isogenic lines (NILs) have been used advantageously to identify and validate, as well as for fine mapping of QTLs. Moreover, NILs are highly useful to confirm that the QTL effect is indeed associated with the introgressed segments (Kinkade and Foolad 2013; Kongprakhon et al. 2009; Lavaud et al. 2015).
In this study, a high-density genetic map of a population derived from H7996 × WVa700 was constructed with more than 1000 SNP markers produced by GBS and markers flanking resistance regions to R. pseudosolanacearum (phylotype I) and R. solanacearum (phylotype II) were identified by QTL analysis. Furthermore, a NIL population containing different segments of Bwr-6 was phenotyped over two seasons using two strains (Pss4 in phylotype I and Pss1632 in phylotype II) and genotyped with CAPS and SSR markers located in BW resistance QTL regions, specifically in Bw-6, to validate the effect of resistance alleles at main resistance QTL.
Materials and methods
Confirmation of Bwr-12 and Bwr-6 in H7996
Bwr-12 and Bwr-6 was confirmed in the 188 F9 RILs developed from the cross between ‘Hawaii 7996’ (H7996, resistant, S. lycopersicum) and ‘West Virginia 700’ (WVa700, susceptible, S. pimpinellifolium) (HW), the same population previously used to map Bwr-12 and Bwr-6 (Wang et al. 2013). The RILs had been evaluated by Wang et al. (2013), in ten greenhouse or field trials, including five countries (Taiwan, Thailand, the Philippines, Indonesia and Reunion), six locations (World Vegetable Center (WorldVeg) and Taichung in Taiwan; Chiangmai in Thailand; UPLB in the Philippines; Purwakara in Indonesia; CIRAD in Reunion) and against eight pathogen strains (Pss4, Pss186, TC, Tm151, CM, PW, JT516 and JT519) and the phenotypic data were also used for current study (Table 1). The genetic parameters (estimates of phenotypic variation, coefficient of variation, genotypic and phenotypic coefficients of variation, broad sense heritability, and genetic advance) for bacterial wilt incidence in RIL population in various environments were estimated according to the methods suggested by Burton and de Vane (1953), Allard (1960), Johnson et al. (1955) after analysis of variance by means of R package ‘lme4’ (Table 2). All strains used in phenotyping were classified as race 1-phylotype I (R. pseudosolanacearum) except for JT516, which was race 3-phylotype II (R. solanacearum). DNA of 188 F9 RILs and parents was provided by the Bacteriology Unit of WorldVeg and used for GBS after quantification using a spectrophotometer (Beckman coulter, DU 800, Krefeld, Germany) and integrity check on 1% agarose gels. Genomic DNA from BC3F3 NIL plants including parents and controls was extracted from young leaves using DNeasy Plant Mini Kit (Qiagen Inc., Hilden, Germany) in 2016–2017.
Genotyping by sequencing (GBS)
Two 96-well plates containing 95 genomic DNAs from F9 RILs H7996 × WVa700 representing the resistance spectrum of the population and from parental plants was submitted to GBS analysis. Sequencing library preparation was done as described by Elshire et al. (2011) and single-end sequencing was performed on two lanes of an Illumina Hi-seq 2500 (Illumina Inc.) at the Genomics Core Facility, Biodiversity Research Center, Academia Sinica, Taiwan. FASTQ files for 100 bp single reads were analyzed using the Tassel5 GBS v2 Pipeline (Glaubitz et al. 2014). Quality trimming was conducted using the GBSSeqToTagDBPlugin with the following parameters: kmerLength 93, minKmerL 20, mnQS 20 and mxKmerNum 108. The alignment to the reference sequence of S. lycopersicum (https://www.maizegenetics.net/tassel) was done using the BWA aligner with n = 3 maxSeedDiff as the parameter for maximal sequence differences. The SAMToGBSdbPlugin was applied to determine the positions of tags on the reference genome by setting the parameters aProp to 0.05 and aLen to 70. SNPs were identified in the aligned tags by the DiscoverySNPCallerPlugin with 0.6 and 0.1 for minimum locus coverage and minimum minor allele frequency, respectively. The SNP data set was obtained by sequential application of the following three plugins: SNPQualityProfilterPlugin for quality control, UpdateSNPPositionQualityPlugin for verifying debugging SNPs/tags position with false for deleteOldData, and ProductionSNPCallerPluginV2 for the final SNP calling with kmerLength 93. The F9 lines were highly homozygous (93.97%) and heterozygous loci were changed to missing data. SNPs were filtered by setting 165 for minimum count out of 190 sequences with minimum and maximum frequency as 0.02 and 0.99, respectively.
Linkage map construction and QTL mapping
A linkage map for HW was constructed in two steps using JoinMap 4.0 (Van Ooijen 2011) with a minimum LOD-score of 3.0 in the maximum likelihood mode. First, a preliminary linkage map was constructed with 2951 polymorphic SNP markers obtained from GBS. Among the SNPs belonging to 13 genetic linkage groups (LG) obtained from first map construction, a total of 1347 redundant SNP markers co-localizing with other markers were removed. Finally, 1604 markers including SSR and CAPS/dCAPS obtained from genomic sequences of anchored BAC clones in tomato (Geethanjali et al. 2010, 2011) and SNP markers of this study, respectively were genetically mapped to 12 chromosomes. The 28 and 23 SSR markers located in Bwr-6 and Bwr-12, respectively (Wang et al. 2013), were added for linkage analysis and map construction. When several markers co-localized at a locus, only one of these markers was considered. Segregation distortion of individual markers was calculated by Chi square test in JoinMap. QTLs were detected using composite interval mapping (CIM) in QGene 4.0 (Joehanes and Nelson 2008) with stepwise cofactor selection and a scanning interval of 0.2 Mb for physical mapping and 2 cM for genetic mapping. Significance thresholds for P < 0.05 (RE-JT516) and 0.01 (nine trials except RE-JT516) of LOD (logarithm of odds) values were estimated by permutation tests. LOD thresholds ranged from 4.3 to 5.1 for CIM. The proportion of phenotypic variation and additive effects of the each QTL were obtained from CIM analysis. Data analysis was repeated using the R/qtl package and reference genome of S_lycopersicum_chromosomes.2.4 as well as 3.0 to confirm the presence of QTL and their interaction (Broman and Saunak 2009).
CAPS marker design
The SNP markers associated to BW resistance QTLs were converted to PCR-based CAPS or dCAPS markers using CAPS Designer (https://solgenomics.net/tools/caps_designer/caps_input.pl), SNP2CAPS (http://pgrc.ipk-gatersleben.de/snp2-caps/) (Thiel et al. 2004) and dCAPS Finder 2.0 (http://helix.wustl.edu/dcaps/dcaps.html) (Neff et al. 2002) (Table 3). Primer design and quality check was conducted by primer3 0.4.0 (http://bioinfo.ut.ee/primer3-0.4.0/primer3) and PCR Primer Stats (http://www.bioinformatics.org/sms2/pcr_primer_stats.html), respectively. The PCR mixture for CAPS amplification contained 40 ng plant genomic DNA, 0.25 μM of each forward and reverse primers, 200 μM dNTPs (FOcusBio, P-2.5 M), 10 × Supertherm GOLD buffer with 15 mM MgCl2 (Bersing, JMR-470), and 0.125 U of Supertherm GOLD Taq (hot start) polymerase (Bersing, JMR-851, Taiwan) in a total volume of 10 μL. Template DNA was initially denatured at 95 °C for 10 min, then 35 cycles for PCR amplification, using the following conditions: denaturation at 95 °C for 30 s, annealing at 50, 55, or 60 °C depending on the primer for 45 s, extension at 72 °C for 45 s, and then final extension at 72 °C for 5 min. The reaction mixture for the enzyme digestion contained: 2 μL PCR product, 7 μL ddH2O, and 0.3 μL restriction enzyme (10 U/μL), and incubation was done according to the instructions of the supplier for 14–16 h. Eleven CAPS/dCAPS markers were subsequently tested in the RIL parents, NIL parents, and BC3F3 NIL lines. The enzyme-digested products were visualized after electrophoresis on 1% agarose gels.
Fine-mapping and validation of Bwr-12 and Bwr-6 in the NIL population
To reduce the Bwr-6 QTL interval size and to validate the effects of resistance alleles at major QTLs identified in the RILs against phylotype I (R. pseudosolanacearum) and phylotype II (R. solanacearum) strains, backcrosses were carried out between the BW resistant F9 RIL NHG41 as donor line and the recurrent susceptible parent BL1413. Over all the backcross and selfing generations during NIL production, eight SSR markers (six for Bwr-6 and two for Bwr-12) from Wang et al. (2013) and four SNP markers designed from limited resequencing experiments of the QTL-6 interval in NHG41 and BL1413 were used for marker-assisted foreground selection. At each backcross generation, all progenies were genotyped to select plants carrying different fragments of Bwr-6 in absence or presence of Bwr-12. In addition, the backcrossed plants were screened with 24 background markers for maximal reconstitution of the recurrent BL1413 genotype. 76 BC3F1 progenies were then self-pollinated and a total of 156 BC3F2 progenies were screened for homozygous insertions of Bwr-6 fragments, followed by a second generation of self-pollination. BC3F3 lines carrying diverse fragments of Bwr-6 at homozygote state were selected. Out of the 145 BC3F3 NILs, 80 NILs with various marker Bwr-6 fragments at the QTL region were used for genotyping and phenotyping.
The NIL population was assessed for BW resistance at WorldVeg Taiwan using two pathogen strains, Pss4 (race 1-phylotype I; R. pseudosolanacearum) and Pss1632 (race 3-phylotype II; R. solanacearum). Entries were arranged in a random complete block design (RCBD) with three replications and six plants per replication in a plastic house at WorldVeg during (1) the cooler spring season from Mar. 1 through Apr. 11 (17.7–24.7 °C) and (2) the warmer autumn season from Sep. 19 through Oct. 31 (21.3–29.8 °C) in 2017, respectively. Both parental lines, WorldVeg lines CLN2026D and CLN2585D, susceptible checks WVa700, L390 and CLN286, and resistant checks H7996, CRA66 and L285 were included in each replication. Three to six seeds of each genotype were sown in plastic pots (8 cm diameter) which were placed in plastic containers (34 × 50 cm2) holding 24 pots arranged in four rows and six columns. Among germinated seedlings in a pot, the healthiest plant was left for artificial inoculation and the others were roughed around 2 weeks after sowing. Approximately 4-week-old plants were inoculated by drenching 30 ml bacterial suspension (108 CFU/ml) on the soil surface near the base of each plant. A wilting score was recorded once a week for 4 weeks on a scale of 0 to 5, where 0 = no visible wilt, 1 = one leaf wilted, 2 = two or three leaves wilted, 3 = all except top leaves wilted, 4 = all leaves wilted, and 5 = dead (Wang et al. 2000). Percentage of wilted plant (W) was calculated as described by Winstead and Kelman (1952) and Seleim et al. (2014) using the formula W = [(Nt − Nh)/Nt] × 100, where Nt is number of total plants and Nh is number of unwilted plants. W was arcsine square root-transformed for data normalization before QTL analysis using the PROC MIXED procedure of SAS (SAS 9.2, SAS Institute, Cary, USA).
SSR and CAPS/dCAPS genotyping
To verify the QTL effect against race 1-phylotype I Pss4 and race 3-phylotype II Pss1632 strains and to narrow down the Bwr-6 interval, totally 34 SSR (Wang et al. 2013) and CAPS/dCAPS markers located on chromosome 6 (17 markers) and 12 (17 markers) were used for genotyping. PCR reactions were carried out as described above and the PCR products were visualized on 6% acrylamide gels. Ten μL of the PCR product was mixed with 5 μL of the loading dye. Out of the 15 μL, 2 and 3–4 μL were loaded on the gel and run for 50 and 25 min for SSR and CAPS/dCAPS genotyping at 160 V, respectively.
Statistical analyses
R software was utilized to perform statistical analyses for analysis of variance (ANOVA), Duncan’s multiple range test (DMRT), single marker analysis (SMA) and correlation analysis. Broad-sense heritability for each season and each strain was calculated as described by Allard (1960).
Results
Confirmation of Bwr-12 and Bwr-6 in H7996
GBS-SNP identification
A total of ~ 214 million SE reads comprising 21.6 Gbp sequence information were generated for 188 F9 RILs and the parents using the Illumina sequencing platform (Table 4). After removing low-quality reads and barcode sequences, about 95% of the sequence data, in average 1.1 million reads per plant sample were analyzed. In total 98% of the bases had a sequencing quality over Q30 and 91.87% of the filtered reads with 3.0 of average depth were successfully mapped to the reference genome. All genotyped markers were attributed to 12 LGs representing the 12 chromosomes of tomato, spanning ~ 741 Mb with an average distance of 0.32 Mb between neighboring markers. The generated map covered 97.5% of the total physical distance of that the tomato reference genome (Table 5).
SNPs-based linkage map
Of the 2951 polymorphic SNPs, 1404 were used to develop a linkage map for the 188 RILs (Fig. 1). The genetic map spanned 1322 cM with 0.94 cM average genetic distance between two adjacent markers, suggesting that the marker density was sufficient to capture major genetic effects causing phenotypic variance for QTL analysis. However, gaps between markers of over 20 cM length were found on chromosomes 1, 4, 5 and 10 and the greatest gap was 33.5 cM from physical position 1.2 to 5.6 Mb at a distal region of chromosome 1. Significant segregation distortion (P < 0.05) in the RIL population concerned 1048 (45.2%) markers. The segregation distortion rates (SDRs) varied from chromosome to chromosome. The highest significant SDR was in chromosome 11, where –logP reached 24.7.

Genetic linkage map constructed using SNP markers including co-localized PCR-based markers (51 SSRs prefixed with SLM and 11 CAPS/dCAPS prefixed with HW). QTL regions identified in 10 individual trials named with ID-PW (Indonesia-Purwakarta), PH (the Philippines-UPLB)-Tm151, RE (Reunion-CIRAD)-JT516, RE (Reunion-CIRAD)-JT519, TH-CM (Thailand-Chiangmai), TW (Taiwan-WorldVeg)-Pss186, TW (Taiwan-WorldVeg)-Pss4a (in greenhouse), TW (Taiwan-WorldVeg)-Pss4b (in screenhouse), TW (Taiwan-WorldVeg)-Pss4c (colonization test) and TW-TC (Taiwan-Taichung) are shown as blue, yellow, orange, red, green, pink, sky blue, brown, grey and black, respectively. (Color figure online)
QTL mapping and detection of epistatic effects
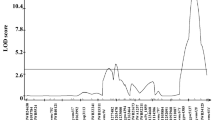
Seven significant QTL sites (four in chromosome 6 and three in chromosome 12) conferring resistance to bacterial wilt were contributed by ‘H7996’. Combining the phenotypic data obtained in 10 trials with the SNP genotyping data, the reported QTLs associated with BW resistance in tomato were corroborated (Wang et al. 2013), one on chromosome 6, hereafter referred to as Bwr-6, and the other on 12, hereafter referred to as Bwr-12 (Fig. 1). Four Bwr-6 with maximum LOD scores ranging from 6.2 to 15.6 (P < 0.01) that were detected with phenotypic data from seven independent trials were situated in 1.5 (Bwr-6.1), 2.5 (Bwr-6.2), 0.3 (Bwr-6.3) and 2.4 cM (Bwr-6.4) intervals located at 31.2–32.7, 36.9–39.4, 41.3–41.6 and 42.8–45.2 cM, respectively; the most significant Bwr-6.4 with 15.6 LOD score was located at 44.1 cM and ~ 35.3 Mb (Fig. 2). Of the four QTLs, Bwr-6.3 has been identified as a specific resistance site against phylotype II, the remaining were associated with defense to phylotype I strain. The phenotypic variation explained (PVE) of overall Bwr-6 ranged from 14.2 to 33.4% (Table 6). Compared to the four QTLs, Bwr-6a, 6b, 6c and 6d, based SSR markers, as reported by Wang et al. (2013) that were obtained from identical 10 phenotype datasets, each QTL region detected from GBS analysis apart from TW-Pss186 was not matched to them on the same genetic position. Especially, the Bwr-6.3 to phylotype II was fine-mapped on the narrower unique region, 0.03 Mb and 0.3 cM, between 34.85 Mb (41.3 cM) to 34.88 Mb (41.6 cM) than Bwr-6b located in a 0.47 Mb and 5.8 cM interval between SLM119 (34.41, 35.8) and SLM136 (34.88 Mb, 41.6 cM). Moreover, whole Bwr-6 segment excluded regions with an insignificant LOD score has downsized to 6.7 from 13.0 cM. The Bwr-12 was consistently associated with resistance to phylotype I strain in all trials, but not to the phylotype II strain of trial RE-JT516 conducted by CIRAD on Reunion. This QTL with a LOD score ranging from 6.2 to 31.1 and a PVE of 15.9% to 53.9%, was confirmed to be located at three sites of chromosome 12 at 28.5 to 31.9 cM (Bwr-12.1), 34.2 to 34.9 cM (Bwr-12.2) and 35.0 to 35.6 cM (Bwr-12.3). Among them, Bwr-12.1 detected from the ID-PW and TW-Pss4c trial was confirmed to be located a new region where Wang et al. (2013) didn’t report in the study. Considering the highest LOD score on chromosome 12, the SNP marker associated with resistance to phylotype I strain was located at ~ 34.5 cM in Bwr-12.2 (the vicinity of physical position ~ 2.9 Mb) (Fig. 2). By multiple QTL analysis using the R/qtl package, an additional QTL with a LOD score of 3.6 ~ 7.5 depending on interaction between QTL was found on chromosome 5 at 0.42 Mb near 05-1608068 SNP for only one trial (TW-Pss4b) (Table 7). Interactions between QTLs were tested to confirm epistatic effects of QTLs associated with BW resistance on chromosomes 5, 6 and 12. In seven trials more than one QTL regions were identified. In only one trial (TW-Pss4b) significant interaction (P < 0.05*) between Bwr-5 (50.44 Mb and 51.4 cM), Bwr-6 and Bwr-12 was detected from using 3.0 versions of the tomato reference genome (Table 7).

Mapping significant QTLs on chromosome 6 and 12 in an F9 RIL segregating tomato population. Marker names consist of chromosome number and physical position (bp) separated by an underscore and, if available, co-localized SSR or CAPS/dCAPS marker in parenthesis. A total of 16 QTL regions, seven for QTL-6 and nine for QTL-12 were identified in 10 trials. The trials are represented as different symbols in colors according to each trial. The left lateral axis shows genetic position (cM) and upper axis shows the LOD score
Fine-mapping and validation of BW resistance QTLs in the NIL population
To verify the phenotypic contribution of specific sections of Bwr-6 and of Bwr-12, a total of 80 BC3F3 NILs were developed from a cross between the BW resistant RIL ‘NHG41’ and the recurrent parent ‘BL1413’ through marker-assisted backcrossing. The NILs were homozygous for four different fragments of the original Bwr-6 interval reported by Wang et al. (2013) and either carried the R or S allele at the QTL Bwr-12. The NIL population and the parental lines were inoculated with Pss4 (phylotype I; R. pseudosolanacearum) and Pss1632 (phylotype II; R. solnacearum) strains in the spring (1st) and autumn (2nd) season, and the percentage of wilted plants (W) was assessed 4 weeks after inoculation. The temperatures during the two seasons were significantly (P < 10−8) cooler in spring (daily mean temperature of 17.7–24.7 °C) than in autumn 21.3–29.8 °C). Over two seasons, the susceptible checks ‘L390’ and ‘WVa700’ were highly susceptible with 52–74% W with Pss4 and 90% W with Pss1632 in the spring, and 82–90% W with Pss4 and 66–90% W with Pss1632 in the autumn (Table 8). High susceptibility of these lines indicated sufficient disease pressure in the experiments and increased wilting in autumn showed that elevated temperatures led to increased wilting. The W of resistant ‘NHG41’ inoculated with each strain did not show significant differences between trials with 16.2% W with Pss4 and 26.9% W with Pss1632 in the spring, and 0.3% W with Pss4 and 16.2% W with Pss1632 in the autumn, respectively. In contrast, susceptible ‘BL1413’ displayed more severe symptoms in the autumn than in spring, except with Pss1632. The NIL population inoculated with Pss1632 showed a continuous distribution of resistance. W through Pss1632 inoculation during the spring trial was nearly normal distributed among NILs, whereas during the autumn season, the disease symptoms were skewed towards susceptibility. In the Pss4 trial, W in spring was skewed towards the resistant parent, whereas disease incidence in the autumn season was skewed towards the susceptible parent. Distributions showed that disease incidence (W) was significantly (P < 0.01) higher in the hot season (autumn) than in the cooler season (spring, Fig. 3). Also the virulence of race 3-phylotype II Pss1632 strain was significantly (P < 0.01) greater in autumn (mean temperature: 21–30 °C) than in spring (mean temperature: 18–25 °C), similar to that reported using 45 race 3-phylotype II stains isolated in Taiwan by Lin et al. (2015). The variance components of the genotype × season and genotype × strain interaction were significant at P < 0.001 for W and, consequently, data from each of the two seasons and two strains were separated for QTL validation. Broad sense heritability estimates of the disease incidence for the Pss4 strain were higher, i.e., 0.61 in spring and 0.63 in autumn than 0.32 and 0.20 for Pss1632, respectively, which indicated that most of the phenotypic variation of resistance to Pss1632 strain observed resulted from environmental factors such as temperature. The Pearson correlation coefficients between trials were significant for season and pathogen strain combination analyzed in the BC3F3 NIL population (Table 9). Within Pss4 inoculation, spring and autumn trials were significantly correlated with each other (R2 = 0.58 (P < 0.0001)), while there was no significant correlation (P < 0.076) in Pss1632 inoculation across seasons, which confirmed that phenotypic data for validation of QTL effect should be treated separately for each season and strain, as the responses to these factors probably rely on different genetic loci.

Frequency distribution of percentage of wilted plant of the 80 NILs for resistance to R. solanacearum Pss4 and Pss1632 strains according to season (spring and autumn). Mean values of the donor parent (NHG41), the recipient parent (BL1413) and NIL population are shown as ▽, ∨ and ◇ for the spring trial and ▼, ∀ and ◆ for the autumn trials, respectively
To explore whether the selected markers from Bwr-6 and Bwr-12 were associated with disease resistance, a single marker analysis was conducted with 34 PCR-based markers flanking the Bwr-6 (17: 12 SSR and 5 CAPS) and Bwr-12 (17: 11 SSR and 6 CAPS) loci and phenotyping data of W against race 1-phylotype I strain Pss4 and race 3-phylotype II strain Pss1632 over two seasons. A total of 16 markers located in the Bwr-12 region were confirmed over two seasons. They were significantly related with BW resistance against Pss4, but not Pss1632. Bwr-12.2 was located in a 0.6 cM interval between SNP12-3250000 (34.2 cM) and SNP12-2770472 (34.8 cM) in the F9 RIL population. However, the QTL region controlling BW caused by Pss4 (LOD score > 10; phenotypic variation explained > 47.5%) was extended to a 9.7 cM segment ranging from HW12-72 CAPS (32.3 cM) to SLM12-65 SSR (42.0 cM) (Fig. 4). In contrast to Bwr-12, Bwr-6 had low LOD scores of 3.9 and 2.3 and it was estimated that the resistance gene located in this region reduces BW incidence of Pss1632 and Pss4 in hotter condition, respectively. The effect of the QTL (LOD > 3.0) to phylotype II strain Pss1632 could be delimited to markers SLM6-110 (47.0 cM and 35.736 Mb) and SLM6-107 (49.7 cM and 35.949 Mb) spanning over 2.7 cM.

Single marker analysis for flanking markers situated on QTL-6 and QTL-12 region identified from F9 RIL population based on percentage of wilted plant (W) in plants challenged with race 1-phylotype I strain Pss4 and race 3-phylotype II strain Pss1632 over two seasons in the BC3F3 NIL population. Trial code consists of two parts, first the season (1st = spring or 2nd = autumn) followed by strain name (Pss4 or Pss1632). Y-axis shows the LOD score and X-axis shows the marker labels. Each marker is named as A_B_C_D, with ‘A’ being the chromosome number, ‘B’ the genetic position, ‘C’ the physical position, and ‘D’ co-localized SSR or CAPS/dCAPS marker with SNP
Discussion
Several QTL mapping studies have been performed to understand the genetic nature of BW resistance in tomato (Carmeille et al. 2006b; Danesh et al. 1994; Mangin et al. 1999; Thoquet et al. 1996a, b; Wang et al. 2000, 2013). QTLs around Bwr-6 and Bwr-12 have been detected in several studies and resulted in design of markers for selection in breeding. Marker-assisted selection for Bwr-12 and Bwr-6 is widely practiced in tomato breeding programs. However, QTL intervals were relatively large, especially for Bwr-6, and knowledge of the precise locations of these QTLs would lead to better marker, and reduction of linkage drag from the donor parent. Information of the specificity of resistance loci to different pathogen strains and their activity under different climatic condition would facilitate pyramiding the most promising QTLs in breeding. The current study used high density genetic and physical maps and phenotyping data from multilocation BW screening of the HW RIL population to fine-map Bwr-6 and Bwr-12, and then to confirm and validate the resistance loci in a NIL population.
A GBS approach resulted in over 10,000 polymorphic SNP markers that were ordered along the chromosomes of a reference sequence (Poland et al. 2012). In this study, over 87% of the produced reads could be aligned to the tomato reference genome (S_lycopersicum_chromosomes.2.40.fa). On the basis of stringent selection criteria including missing data percentage, minor allele frequency and percent heterozygosity for filtering SNPs, a panel of robust SNPs comprising about 19% of the 12,654 SNPs called by comparison of the S. lycopersicum genome sequences was used for constructing a genetic map. In spite of the dramatic reduction in marker number, a sufficient number of SNP markers (2317 SNPs) remained available for QTL mapping. The GBS approach of this work facilitated the construction of SNP-based physical and genetic maps of sufficient resolution for QTL mapping. A total of 1404 SNPs (11%) were finally used for constructing a dense genetic map for the F9 RIL population. polymorphic SNPs with below 3.0 LOD score or showing high SDR or low COR that especially situated in chromosome 6 and 11 was excluded to construct precise genetic map. The current dense map will be useful for the development of high-density consensus maps and fine-mapping of QTL.
Based on field and greenhouse evaluations conducted worldwide, ‘H7996’ was identified as a promising breeding resource with stable resistance to bacterial wilt (Wang et al. 1998, 2013). QTL mapping using Qgene 4.0 with reference genome of S. lycopersicum chromosome 2.4 version in a 188 F9 RIL population developed from a cross between ‘H7996’ and ‘WVa 700’ revealed two QTLs for BW resistance, one major QTL on chromosome 12 important for resistance to phylotype I isolates, and another QTL on chromosome 6 associated with resistance to both phylotype I and phylotype II isolates. These results corroborated findings of previous H7996 mapping studies (Carmeille et al. 2006b; Wang et al. 2000, 2013) except in this study the Bwr-12 region in trials PH-Tm151 and ID-PW were shifted toward the distal end of a 2.9 cM interval around SLM12-2 SSR marker located at the end of QTL region. Bwr-6.3 identified from JT-516 strain, the only race 3-phylotype II (R. solanacearum) strain among the 10 trials provided little resistance (LOD 6.4, P < 0.05) compared with the finding of Wang et al. (2013). Bwr-12 with a LOD score ranging from 7.1 to 31.1 and a PVE from 15.9 to 53.9%, respectively, provided strong and stable BW resistance to race 1-phylotype I strain, while a race 3-phylotype II isolate was affected by the only Bwr-6. Additional HW RIL phenotyping trials with phylotypes II, III, and IV would advance progress towards fine-mapping BW resistance. Several studies reported additional QTLs on chromosome 3, 4, 8, 10 and 11 connected with BW resistance (Carmeille et al. 2006b; Thoquet et al. 1996a, b; Wang et al. 2013) in the same HW mapping population but these QTLs were not detected in this study. Additional multi QTL analysis by R/qtl package with a new SNP set however revealed significant (P < 0.05) new QTL regions on chromosome 5 in trial TW-Pss4b (Table 7). In addition, this analysis confirmed a significant QTL epistasis (P < 0.05) between Bwr-5 and Bwr-6 at TW-Pss4b, and between Bwr-6 and Bwr-12 at ID-PW, TW-Pss186, TW-Pss4b, and TW-TC. QTLs identified in the ten trials were considered major when PVE was greater than 10%. The genetic effect of all QTLs was lower than the broad-sense heritability for disease resistance (0.64–0.94, Table 2), therefore we assume that additional minor QTLs contributed to resistance against race 3-phylotype II strains. Whole genome re-sequencing of RILs and read assembly could help detecting minor QTLs and elucidate their interaction with other QTLs. ‘H7996’ is not resistant to all isolates of R. solanacearum species complex and was susceptible to R. solanacearum UW551, a typical sequevar 1 (Race 3 biovar 2) strain (Gabriel et al. 2006) that causes losses in temperate zones and tropical highlands. This isolate killed about 80% of H7996 plants within 14 day post inoculation (Milling et al. 2011). Therefore, exploitation of new breeding sources especially against race 3-phylotype II is required to develop multi-phylotype/species resistance tomato cultivars.
BW susceptibility bio-assays on 80 BC3F3 NILs, parents and resistant/susceptible controls with race 1-phylotype I Pss4 (R. pseudosolanacearum) and race 3-phylotype II Pss1632 (R. solanacearum) strains over two seasons, confirmed the resistance phenotype of the NILs against two strains under two temperature regimes, the cooler spring season and the warmer autumn season. Mean values of NILs, the susceptible parent and controls showed different sensitivity patterns depending of the bacterial strain and season. Under higher temperatures (21–30 °C) the plant defense response against BW seems to be lower (Menna et al. 2015; Wang et al. 2009), while Pss4 and Pss1632 or other tropical strains such as of K60 and GMI1000 have reduced virulence in cooler conditions (18–25 °C) (Ciampi and Sequeira 1980; Ciampi et al. 1980; Huerta et al. 2015). Consequently, the percentage of wilted NILs were significantly higher in the relative warm conditions of the autumn trial than in the cool spring trial. Carmeille et al. (2006b) reported contrasting results that wilting symptoms in tomato were more severe during the cool season after inoculation with the race 3-phylotype II JT-1.1.1. 516 stain.
The broad heritability estimate of resistance in the NIL population was 0.61 in the spring and 0.63 in the autumn infected with Pss4, and 0.32 in the spring and 0.20 in the autumn inoculated with Pss1632, suggesting that resistance to the phylotype II strain was mainly affected by non-genetic effects, especially temperature, in contrast to phylotype I. It was also found that the phenotypic coefficient of variation for Pss1632 had twofold higher values than the genetic coefficient of variation in both seasons. Both QTLs Bwr-6 and Bwr-12 contributed 54% for Pss4, but only 19% for Pss1632 of the phenotypic effect on the resistance. Thus it is suggested that there are additional genetic factors playing a role on the resistance, which might have been lost during backcrossing. The minor QTLs on chromosome 5 detected by R/qtl is a candidate for explaining this effect. Consistent with previous studies, major genes have been identified on chromosome 6 and 12. The new information of the current work is the fine mapping of specific Bwr-6 sites conferring resistance to Pss1632 and Pss4. Resistance to Pss1632 (R. solanacearum) was mapped to an interval of 2.7 cM between 6_35, 736,000 _SLM6-110 to 6_35,949,000_SLM6-107 SNP marker, while resistance to Pss4 (R. pseudosolanacearum) with a low LOD value (< 3.0) was associated with a 5.0 cM interval between 6_33,444,000_SLM6-47 and 6_33,868,000_SLM6-124. Bwr-12 had a significant effect, LOD scores of 5.8–16.1 (P < 0.01) to control BW disease caused by the Pss4 strain was confirmed to be located in ~ 30 cM wide interval ranged from 12_1,981,000_SLM12-54 to 12_4,209,000_SLM12-69 marker in the NIL population. The NIL population was designed to fine-map Bwr-6 and not Bwr-12, therefore the Bwr-12 interval remained very large in this population. Previous reports showed that Bwr-12 is confined to phylotype I defense response (Carmeille et al. 2006a, b; Wang et al. 2000, 2013). Recently it was reported by Kim et al. (2018) that genes encoding leucine-rich repeat (LRR) receptor-like proteins are located in the Bwr-12 region and may be associated with resistance. A dense genetic map with markers flanking and within QTL regions will help to improve the efficiency to breed tomato resistant to BW.
References
Allard RW (1960) Principles of plant breeding. Wiley, New York
Ashrafi H, Kinkade M, Foolad MR (2009) A new genetic linkage map of tomato based on a Solanum lycopersicum × S. pimpinellifolium RIL population displaying locations of candidate pathogen response genes. Genome 52:935–956. https://doi.org/10.1139/G09-065
Beissinger TM, Hirsch CN, Sekhon RS, Foerster JM, Johnson JM, Muttoni G, Vaillancourt B, Buell CR, Kaeppler SM, de Leon N (2013) Marker density and read depth for genotyping populations using genotyping-by-sequencing. Genetics 193:1073–1081. https://doi.org/10.1534/genetics.112.147710
Berthouly-Salazar C, Mariac C, Couderc M, Pouzadoux J, Floc’h JB, Vigouroux Y (2016) Genotyping-by-sequencing SNP identification for crops without a reference genome: using transcriptome based mapping as an alternative strategy. Front Plant Sci 7:777. https://doi.org/10.3389/fpls.2016.00777
Broman KW, Saunak S (2009) A guide to QTL mapping with R/qtl. Springer, New York. https://doi.org/10.1007/978-0-387-92125-9
Buddenhagen I, Kelman A (1964) Biological and physiological aspects of bacterial wilt caused by Pseudomonas solanacearum. Annu Rev Phytopathol 2:203–230
Burton GW, de Vane EH (1953) Estimating heritability in tall fescue (Festuca arundinacea) from replicated clonal material. Agron J 45:478–481
Capel C, Fernández del Carmen A, Alba JM, Lima-Silva V, Hernández-Gras F, Salinas M, Boronat A, Angosto T, Botella MA, Fernández-Muñoz R, Granell A, Capel J, Lozano R (2015) Wide-genome QTL mapping of fruit quality traits in a tomato RIL population derived from the wild-relative species Solanum pimpinellifolium L. Theor Appl Genet 128:2019–2035. https://doi.org/10.1007/s00122-015-2563-4
Carmeille A, Prior P, Kodja H, Chiroleu F, Luisetti J, Besse P (2006a) Evaluation of resistance to race 3, biovar 2 of Ralstonia solanacearum in tomato germplasm. J Phytopathol 154:398–402. https://doi.org/10.1111/j.1439-0434.2006.01112.x
Carmeille A, Caranta C, Dintinger J, Prior P, Luisetti J, Besse P (2006b) Identification of QTLs for Ralstonia solanacearum race 3-phylotype II resistance in tomato. Theor Appl Genet 113:110–121. https://doi.org/10.1007/s00122-006-0277-3
Celik I, Gurbuz N, Uncu AT, Frary A, Doganlar S (2017) Genome-wide SNP discovery and QTL mapping for fruit quality traits in inbred backcross lines (IBLs) of solanum pimpinellifolium using genotyping by sequencing. BMC Genom 18:1. https://doi.org/10.1186/s12864-016-3406-7
Ciampi L, Sequeira L (1980) Influence of temperature on virulence of Race 3 strains of Pseudomonas solanacearum. Am Potato J 57:307–317
Ciampi L, Sequeira L, French ER (1980) Latent infection of potato tubers by Pseudomonas solanacearum. Am Potato J 57:377–386
Collard BC, Mackill DJ (2008) Marker-assisted selection: an approach for precision plant breeding in the twenty-first century. Philos Trans R Soc Lond B Biol Sci 363:557–572. https://doi.org/10.1098/rstb.2007.2170
Danesh D, Aarons S, McGill GE, Young ND (1994) Genetic dissection of oligogenic resistance to bacterial wilt in tomato. Mol Plant Microbe Interact 7:464–471. https://doi.org/10.1094/mpmi-7-0464
Daunay MC, Laterrot H, Scott JW, Hanson P, Wang JF (2010) Tomato resistance to bacterial wilt caused by Ralstonia solanaearum E.F. Smith: ancestry and peculiarities. TGC Rep 60:6–40
Denny TP (2006) Plant pathogenic Ralstonia species. In: Gnanamanickam SS (ed) Plant-associated bacteria. Springer, Dordrecht, pp 573–644
Elphinstone JG (2005) The current bacterial wilt situation: a global overview. In: Allen C, Prior P, Hayward AC (eds) Bacterial wilt disease and the Ralstonia solanacearum species complex. American Phytopathological Society Press, St. Paul, pp 9–28
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, Mitchell SE (2011) A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 6(5):e19379. https://doi.org/10.1371/journal.pone.0019379
Fegan M, Prior P (2005) How complex is the Ralstonia solanacearum species complex’’. In: Allen C, Prior P, Hayward AC (eds) Bacterial wilt: the disease and Ralstonia solanacearum species complex. APS Press, St. Paul, pp 449–461
Feuillet C, Leach JE, Rogers J, Schnable PS, Eversole K (2011) Crop genome sequencing: lessons and rationales. Trends Plant Sci 16:77–88. https://doi.org/10.1016/j.tplants.2010.10.005
Foolad MR (2007) Genome mapping and molecular breeding of tomato. Int J Plant Genom 2007:64358. https://doi.org/10.1155/2007/64358
Fu YB, Peterson GW (2011) Genetic diversity analysis with 454 pyrosequencing and genomic reduction confirmed the eastern and western division in the cultivated barley gene pool. Plant Genome 4:226–237. https://doi.org/10.3835/plantgenome2011.08.0022
Gabriel DW, Allen C, Schell M, Denny TP, Greenberg JT et al (2006) Identification of open reading frames unique to a select agent: Ralstonia solanacearum Race 3 Biovar 2. Mol Plant-Microbe Interact 19:69–79. https://doi.org/10.1094/MPMI-19-0069
Gaelle H (2014) Breeding for resistances to Ralstonia solanacearum. Front Plant Sci 5:715. https://doi.org/10.3389/fpls.2014.00715
Geethanjali S, Chen KY, Pastrana DV, Wang JF (2010) Development and characterization of tomato SSR markers from genomic sequences of anchored BAC clones on chromosome 6. Euphytica 173:85–97. https://doi.org/10.1007/s10681-010-0125-z
Geethanjali S, Kadirvel P, Pena R, Rao ES, Wang JF (2011) Development of tomato SSR markers from anchored BAC clones of chromosome 12 and their application for genetic diversity analysis and linkage mapping. Euphytica 178:283–295. https://doi.org/10.1007/s10681-010-0331-8
Genin S (2010) Molecular traits controlling host range and adaptation to plants in Ralstonia solanacearum. New Phytol 187(4):920–928
Glaubitz J, Casstevens T, Lu F et al (2014) TASSEL-GBS: a high capacity genotyping by sequencing analysis pipeline. PLoS ONE 9(2):e90346. https://doi.org/10.1371/journal.pone.0090346
Gurgul A, Miksza-Cybulska A, Szmatola T, Jasielczuk I, Piestrzynska-Kajtoch A, Fornal A, Semik-Gurgul E, Bugno-Poniewierska M (2018) Genotyping-by-sequencing performance in selected livestock species. Genomics 111:186–195. https://doi.org/10.1016/j.ygeno.2018.02.002
Hai TTH, Esch E, Wang JF (2008) Resistance to Taiwanese race 1 strains of Ralstonia solanacearum in wild tomato germplasm. Eur J Plant Pathol 122:471–479. https://doi.org/10.1007/s10658-008-9314-1
Hanson PM, Wang J, Licardo O, Haudin MS, Hartman GL, Lin Y, Chen J (1996) Variable reaction of tomato lines to bacterial wilt evaluated at several locations in southeast Asia. HortSci 31:143–146. https://doi.org/10.21273/HORTSCI.31.1.143
Hayward AC (1991) Biology and epidemiology of bacterial wilt caused by Pseudomonas Solanacearum. Annu Rev Phytopathol 29:65–87. https://doi.org/10.1146/annurev.py.29.090191.000433
Hospital F (2005) Selection in backcross programmes. Philos Trans R Soc B-Biol Sci 360:1503–1511. https://doi.org/10.1098/rstb.2005.1670
Hospital F (2009) Challenges for effective marker-assisted selection in plants. Genetica 136:303–310. https://doi.org/10.1007/s10709-008-9307-1
Huerta AI, Milling A, Allen C (2015) Tropical strains of Ralstonia solanacearum outcompete race 3 biovar 2 strains at lowland tropical temperatures. Appl Environ Microbiol 81:3542–3551. https://doi.org/10.1128/AEM.04123-14
Jaunet TX, Wang JF (1999) Variation in genotype and aggressiveness of Ralstonia solanacearum race 1 isolated from tomato in Taiwan. Phytopathology 89(4):320–327. https://doi.org/10.1094/PHYTO.1999.89.4.320
Joehanes R, Nelson JC (2008) QGene 4.0 an extensible Java QTL-analysis platform. Bioinformatics 24:2788–2789. https://doi.org/10.1093/bioinformatics/btn523
Johnson HW, Suleiman TM, Lusas EW (1955) Estimates of genetic and environmental variability in soybeans. Agron J 47:314–318
Kelman A (1953) The bacterial wilt caused by Pseudomonas solanacearum: a literature review and bibliography. N C Agric Exp Stn Tech Bul N 99:194
Kim SG, Hur OS, Ro NY, Ko HC, Rhee JH, Sung JS, Ryu KY, Lee SY, Baek HJ (2016) Evaluation of resistance to Ralstonia solanacearum in tomato genetic resources at seedling stage. Plant Pathol J 32:58–64. https://doi.org/10.5423/PPJ.NT.06.2015.0121
Kim B, Hwang IS, Lee HJ, Lee JM, Seo E, Choi D, Oh CS (2018) Identification of a molecular marker tightly linked to bacterial wilt resistance in tomato by genome-wide SNP analysis. Theor Appl Genet. https://doi.org/10.1007/s00122-018-3054-1
Kinkade MP, Foolad MR (2013) Validation and fine mapping of lyc12.1, a QTL for increased tomato fruit lycopene content. Theor Appl Genet 126:2163–2175. https://doi.org/10.1007/s00122-013-2126-5
Kongprakhon P, Cuesta-Marcos A, Hayes PM, Richardson KL, Sirithunya P, Sato K, Steffenson B, Toojinda T (2009) Validation of rice blast resistance genes in barley using a QTL mapping population and near-isolines. Breed Sci 59:341–349. https://doi.org/10.1270/jsbbs.59.341
Krausz JP, Thurston HD (1975) Breakdown of resistance to Pseudomonas solanacearum in tomato. Phytopathology 65:1272–1274. https://doi.org/10.1094/Phyto-65-1272
Lavaud C, Boutet G, Moussart A, Poncet C (2015) Validation of QTL for resistance to Aphanomyces euteiches in different pea genetic backgrounds using near-isogenic lines. Theor Appl Genet 128:2273–2288. https://doi.org/10.1007/s00122-015-2583-0
Lin CH, Wang JF, Wu YF, Cheng AH (2015) Phylotype II race 3 biovar 2 strain of Ralstonia solanacearum in Taiwan: survival potential and virulence on tomato, eggplant, and pepper. Acta Hort 1069:293–300. https://doi.org/10.17660/actahortic.2015.1069.42
Lopes CA, Quezado-Soares AM, Melo PED (1994) Differential resistance of tomato cultigens to biovar I and III of Pseudomonas solanacearum. Plant Dis 78:320–327. https://doi.org/10.1094/PD-78-1091
Mangin B, Thoquet P, Olivier J, Grimsley NH (1999) Temporal and multiple quantitative trait loci analyses of resistance to bacterial wilt in tomato permit the resolution of linked loci. Genetics 151:1165–1172
Mansfield J, Genin S, Magori S, Citovsky V, Sriariyanum M, Ronald P, Dow M, Verdier V, Beer SV, Machado MA, Toth I, Salmond G, Foster GD (2012) Top 10 plant pathogenic bacteria in molecular plant pathology. Mol Plant Pathol 13:614–629. https://doi.org/10.1111/j.1364-3703.2012.00804.x
Marrano A, Birolo G, Prazzoli ML, Lorenzi S, Valle G, Grando MS (2017) SNP-Discovery by RAD-Sequencing in a germplasm collection of wild and cultivated grapevines (V. vinifera L.). PLoS ONE 12(1):e0170655. https://doi.org/10.1371/journal.pone.0170655
Menna A, Nguyen D, Guttman DS, Desveaux D (2015) Elevated temperature differentially influences effector-triggered immunity outputs in Arabidopsis. Front Plant Sci 6:995. https://doi.org/10.3389/fpls.2015.00995
Messiaen CM (1989) Environmental influences on the severity of tomato bacterial wilt in the French West Indies: Interactions with varietal resistance. Pp 235-238 In: Green SK, Griggs TD, Mc Lean BT (Eds) Tomato and pepper production in the tropics. Proceeding on international symposium on integrated management practices. AVRDC, Shanhua
Miao L, Shou S, Cai J, Jiang F, Zhu Z, Li H (2009) Identification of two AFLP markers linked to bacterial wilt resistance in tomato and conversion to SCAR markers. Mol Biol Rep 36:479–486. https://doi.org/10.1007/s11033-007-9204-1
Milling A, Babujee L, Allen C (2011) Ralstonia solanacearum extracellular polysaccharide is a specific elicitor of defense responses in wilt-resistant tomato plants. PLoS ONE 6(1):e15853. https://doi.org/10.1371/journal.pone.0015853
Neff MM, Turk E, Kalishman M (2002) Web-based primer design for single nucleotide polymorphism analysis. Trends Genetics 18:613–615. https://doi.org/10.1016/S0168-9525(02)02820-2
Peter KV, Gopalakrishnan TR, Rajan S, Sadhan KPG (1993) Breeding for resistance to bacterial wilt in tomato, eggplant and pepper. In: Hartman GL, Hayward AC (eds) Bacterial wilt. ACIAR proceedings No. 45. ACIAR, Canberra, pp 183–190
Poland J, Endelman J, Dawson J, Rutkoski J, Wu S, Manes Y, Dreisigacker S, Crossa J, Sánchez-Villeda H, Sorrells M et al (2012) Genomic selection in wheat breeding using genotyping-by-sequencing. Plant Genome 5:103–113. https://doi.org/10.3835/plantgenome2012.06.0006
Poussier S, Prior P, Luisetti J, Hayward AC, Fegan M (2000) Partial sequencing of the hrpB and endoglucanase genes confirms and expands the known diversity within the Ralstonia solanacearum species complex. Syst Appl Microbiol 23:479–486. https://doi.org/10.1016/S0723-2020(00)80021-1
Prior P, Fegan M (2005) Recent development in the phylogeny and classification of Ralstonia solanacearum. Acta Hort 695:127–136. https://doi.org/10.17660/ActaHortic.2005.695.14
Prior P, Steva H, Cadet P (1990) Aggressiveness of strains of Pseudomonas solanacearum from the French West Indies on tomato. Plant Dis 74:962–965. https://doi.org/10.1094/PD-74-0962
Prior P, Grimault V, Schmit J (1994) Resistance to bacterial wilt (Pseudomonas solanacearum) in tomato: present status and prospects. In: Hayward AC, Hartman GL (eds) Bacterial Wilt, the disease and its causative agent Pseudomonas solanacearum. CAB International, Wallingford, pp 209–223
Safni I, Cleenwerck I, De Vos P, Fegan M, Sly L, Kappler U (2014) Polyphasic taxonomic revision of the Ralstonia solanacearum species complex: proposal to emend the descriptions of Ralstonia solanacearum and Ralstonia syzygii and reclassify current R. syzygii strains as Ralstonia syzygii subsp. syzygii subsp. nov., R. solanacearum phylotype IV strains as Ralstonia syzygii subsp. indonesiensis subsp. nov., banana blood disease bacterium strains as Ralstonia syzygii subsp. celebesensis subsp. nov. and R. solanacearum phylotype I and III strains as Ralstonia pseudosolanacearum sp. nov. Int J Syst Evol Microbiol 64:3087–3103. https://doi.org/10.1099/ijs.0.066712-0
Scott JW, Wang JW, Hanson PM (2005) Breeding tomatoes for resistance to bacterial wilt, a global view. Acta Hort 695:161–172. https://doi.org/10.17660/ActaHortic.2005.695.18
Seleim MA, Abo-Elyousr KA, Abd-El-Moneem KM, Saead FA (2014) First report of bacterial wilt caused by Ralstonia solanacearum biovar 2 race 1 on tomato in Egypt. Plant Pathol J 30:299–303. https://doi.org/10.5423/PPJ.NT.10.2013.0101
Spindel J, Wright M, Chen C, Cobb J, Gage J, Harrington S, Lorieux M, Ahmadi N, McCouch S (2013) Bridging the genotyping gap: using genotyping by sequencing (GBS) to add high-density SNP markers and new value to traditional bi-parental mapping and breeding populations. Theor Appl Genet 126:2699–2716. https://doi.org/10.1007/s00122-013-2166-x
Thiel T, Kota R, Grosse I, Stein N, Graner A (2004) SNP2CAPS: a SNP and INDEL analysis tool for CAPS marker development. Nucleic Acids Res 32:e5. https://doi.org/10.1093/nar/gnh006
Thoquet P, Olivier J, Sperisen C, Rogowsky P, Laterrot H, Grimsley N (1996a) Quantitative trait loci determining resistance to bacterial wilt in tomato cultivar Hawaii7996. Mol Plant-Microbe Interact 9:826–836. https://doi.org/10.1094/MPMI-9-0826
Thoquet P, Olivier J, Sperisen C, Rogowsky P, Prior P, Anais G, Mangin B, Bazin B, Nazer R, Grimsley N (1996b) Polygenic resistance of tomato plants to bacterial wilt in the French West Indies. Mol Plant-Microbe Interact 9:837–842. https://doi.org/10.1094/MPMI-9-0837
Truong HTH, Kim SY, Tran HN, Nguyen TTT, Nguyen LT, Hoang TK (2015) Development of a SCAR marker linked to bacterial wilt (Ralstonia solanacearum) resistance in tomato line Hawaii 7996 using bulked-segregant analysis. Hortic Environ Biotechnol 56:506–515. https://doi.org/10.1007/s13580-015-1050-9
Van Ooijen JW (2011) Multipoint maximum likelihood mapping in a full-sib family of an outbreeding species. Genetic Res 93:343–349. https://doi.org/10.1017/S0016672311000279
Wang JF, Hanson PM, Barens JA (1998) Worldwide evaluation of international set of resistance sources to bacterial wilt in tomato. In: Prior P, Allen C, Elphinstone J (eds) Bacterial Wilt disease: molecular and ecological aspects. Springer, Berlin, pp 269–275. https://doi.org/10.1007/978-3-662-03592-4_39
Wang JF, Olivier J, Thoquet P, Mangin B, Sauviac L, Grimsley NH (2000) Resistance of tomato line Hawaii7996 to Ralstonia solanacearum Pss4 in Taiwan is controlled mainly by a major strain-specific locus. Mol Plant-Microbe Interact 13:6–13. https://doi.org/10.1094/MPMI.2000.13.1.6
Wang Y, Bao Z, Zhu Y, Hua J (2009) Analysis of temperature modulation of plant defense against biotrophic microbes. Mol Plant Microbe Interact 22:498–506. https://doi.org/10.1094/MPMI-22-5-0498
Wang JF, Ho FI, Truong HTH, Hugan SM, Balatero CH, Dittapongpitch V, Hidayati N (2013) Identification of major QTLs associated with stable resistance of tomato cultivar ‘Hawaii 7996’ to Ralstonia solanacearum. Euphytica 190:241–252. https://doi.org/10.1007/s10681-012-0830-x
Wicker E, Lefeuvre P, Cambiaire JC, Lemaire C, Poussier S, Prior P (2012) Contrasting recombination patterns and demographic histories of the plant pathogen Ralstonia solanacearum inferred from MLSA. ISME J 6:961–974. https://doi.org/10.1038/ismej.2011.160
Winstead NN, Kelman A (1952) Inoculation techniques for evaluating resistance to Pseudomonas solanacearum. Phytopathology 42:628–634
Yu LX, Zheng P, Zhang T, Rodringuez J, Main D (2016) Genotyping-by-sequencing-based genome-wide association studies on Verticillium wilt resistance in autotetraploid alfalfa (Medicago sativa L.). Mol Plant Pathol 18:187–194. https://doi.org/10.1111/mpp.12389
Acknowledgements
This study was supported by the Rural Administration Development of Korea Rep. (10000279-01) and the Council of Agriculture of Taiwan (10000299-01). The works were performed at the World Vegetable Center. The core donors to the World Vegetable Center are the Republic of China (Taiwan), UK Department for International Development (UK/DFID), United States Agency for International Development (USAID), Australian Centre for International Agricultural Research (ACIAR) Germany, Thailand, Philippines, Korea, and Japan. The authors gratefully thank Burlakoti Pragyan for technical assistance and Chiou Fen Hsu for resistance assay on NIL population.
Author information
Authors and Affiliations
Contributions
Conceptualization, ISS and RS; genotyping and QTL mapping, J-CH and S-MH; resistance assay for RIL, J-FW and NIL, J-RC; data analysis, ISS and J-FW; writing—original draft preparation, ISS; writing—review and editing, J-FW, PH and RS.
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shin, I.S., Hsu, JC., Huang, SM. et al. Construction of a single nucleotide polymorphism marker based QTL map and validation of resistance loci to bacterial wilt caused by Ralstonia solanacearum species complex in tomato. Euphytica 216, 54 (2020). https://doi.org/10.1007/s10681-020-2576-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10681-020-2576-1