
Abstract
Water-sorption studies and certain organic chemistry reactions require water removal from cellulosic samples. This is hindered by the strong interaction of cellulosic materials with water, and it remains uncertain if a completely anhydrous state can be reached under common drying conditions. Here, different drying conditions were applied to wood and cellulose, and the residual moisture contents were quantified either gravimetrically or by coulometric Karl-Fischer titration. Vacuum-drying at 103 °C and ≤ 1 mbar for at least 360 min decreased the moisture content to ≤ 0.04%. However, in automated sorption balances, drying at atmospheric pressure under dry air or nitrogen flow left some samples with more than 1% moisture content. The residual moisture content obtained under dry gas flow was temperature dependent. Increasing the temperature up to 55 °C decreased the residual moisture content and cooling resulted in a moisture re-uptake, presumably due to small quantities of water vapor in the surrounding atmosphere. These effects must be considered in fundamental studies on water interactions of cellulosic materials.
Similar content being viewed by others


Explore related subjects
Find the latest articles, discoveries, and news in related topics.Avoid common mistakes on your manuscript.
Introduction
Cellulose-containing materials constantly interact with water from the surroundings because they contain polar groups (i.e. hydroxyl groups) that attract water (Berthold et al. 1996). Bound water significantly affects the physical and mechanical properties of cellulosic materials, which is particularly observed by the sorption-induced swelling and loss in stiffness (Pan and Zhong 2016; Chen et al. 2018; Nopens et al. 2019). Since our ambient atmosphere always contains water molecules, bound water is omnipresent in cellulosic materials and requires efforts to reach the anhydrous state.
Complete removal of bound water from cellulosic materials without causing deterioration is difficult, due to the strong interaction of cellulose with water (Rostom et al. 2021). Some organic chemistry reactions need to be performed in an anhydrous state, because residual water molecules make chemical reactions less favourable or act as competitors to hydroxyl groups (Steinmeier 2004). Incomplete water removal also results in an underestimation of the gravimetrically determined moisture content in water sorption studies, which denotes the ratio of the absorbed water mass to the anhydrous mass of the material. The error caused by residual water is much larger during the quantification of accessible hydroxyl groups following the deuterium-exchange approach (Pönni et al. 2014; Väisänen et al. 2018; Uimonen et al. 2020). In addition to the desired exchange of hydrogen to the heavier deuterium in accessible hydroxyl groups, incomplete water removal would also result in the replacement of residual bound water (H2O) by deuterated water (D2O) (Thybring et al. 2018). A correct determination of the anhydrous mass is thus essential in fundamental water sorption studies of cellulosic materials.
Various physical and chemical methods to determine the anhydrous mass of cellulosic materials exist. Residual moisture contents in cellulosic samples can be quantified by distillation using solvents such as xylene or toluene (Kollmann and Höckele 1962), or by titration using the Karl-Fischer reagent (Mitchell 1940). These methods provide accurate results and are insensitive to the evaporation of volatile compounds, but they are laborious, and the samples become unusable for further water sorption analyses (Kollmann and Höckele 1962). Some methods quantify the moisture content based on modern analytical techniques, such as nuclear magnetic resonance spectroscopy (Merela et al. 2009; Rostom et al. 2021), which are not routinely available in every laboratory. Hence, it is more common in water sorption studies to determine the anhydrous sample mass after removing water physically, especially by oven drying at 103 ± 2 °C, slightly above the atmospheric boiling point of water. Elevated temperatures can be avoided by vacuum drying or drying in anhydrous air or nitrogen at room temperature. The latter approaches are also applied in modern, automated sorption balances, which have been increasingly used for water sorption studies of cellulosic materials within the past decade (Thybring et al. 2018). However, it remains uncertain if drying under these conditions leads to complete water removal from cellulosic materials.
Previous studies provide evidence for drying-resistant water molecules in cellulosic materials. Kollmann and Höckele (1962) reported that small quantities of water (0.2–0.3% moisture content) remained in the wood after drying at a high vacuum as well as after oven-drying at 103 °C. Hatakeyama et al. (2000) found that a proportion of bound water in cellulose fibers has a vaporization temperature higher than 100 °C, which was dependent on the measurement conditions (nitrogen flow rate and heating rate). The anhydrous wood mass obtained by vacuum-drying may also vary depending on the previous sorption history due to strongly retained water (Hergt and Christensen 1965). Furthermore, the sample mass after conditioning at a constant climate is depending on the mass stability criterion applied (Glass et al. 2017, 2018). Insufficient holding times may thus result in residual moisture content, even if the applied climatic conditions were appropriate for complete drying.
This study followed the hypothesis that common drying conditions applied in sorption studies are inadequate to reach an anhydrous state and leave significant amounts of water in different cellulosic samples. This was tested by exposing wood and cellulose samples to a range of temperatures (15–103 °C) under vacuum, dry air, or nitrogen flow. Residual moisture contents were quantified gravimetrically and by coulometric Karl-Fischer titration. The potential emission of volatile compounds from wood during drying was tested by thermogravimetric analysis coupled with mass spectrometry. The results show the limitations of reaching the anhydrous state of cellulosic materials in automated sorption balances, which should be considered when studying their water sorption behaviour.
Material and methods
Material
Wood samples were prepared from commercially kiln-dried Scots pine (Pinus sylvestris L.) sapwood and European beech (Fagus sylvestris L.) wood; hence one soft-and one hardwood species were tested. For measurements in the automated sorption balance, samples with dimensions of 20 × 20 × 1.5 mm3 (radial × tangential × longitudinal) were cut. For all other analyses, these samples were milled to wood flour using a rotary mill (SLf 90 S/4 T, Retsch GmbH, Germany). After fractioning, their main particle diameter was between 1 and 4 mm. All wood samples were conditioned at 20 °C and 65% RH before the experiments.
Microcrystalline cellulose (MCC) from pharmaceutical-grade wood pulp with a particle size range between 20 and 300 µm was purchased from ProUmid (Ulm, Germany). The MCC is typically used as a reference for humidity validation in automated sorption balances.
For the coulorimetric Karl-Fischer titration, the reagents Hydranal-Coulomat AG-Oven and Fluka Hydranal Coulomat AG (CAS 67-56-1, 111-42-2, 288-32-4, 7446-09-5, 68,007-08-9) were used. The setup for the colorimetric Karl-Fischer titration was verified using a lactose monohydrate standard (Hydranal-Water Standard KF-Oven; CAS 64044-51-5). All chemicals were purchased from Honeywell International Inc. (United States).
Thermogravimetric analysis coupled with mass spectrometry (TGA-MS)
Beech and pine wood flour were dried under nitrogen flow while mass changes and emitted volatiles were measured using a simultaneous thermal analyser (Netzsch STA 449 F3 Jupiter with a SiO furnace and TG sample holder) coupled to a mass spectrometer (QMS 403 Aëolos® II). Two temperature steps were applied, one step at 25 °C, close to typical room temperatures, and another step at 103 °C with reference to the oven-drying method (EN 13183–1 2002). A sample with an initial weight of ca. 90 (pine wood) or 140 mg (beech wood) was placed on an alumina slip-on plate (ø 17 mm; Netzsch) into the thermal analyser. The sample was first dried under nitrogen flow (purity ≥ 99.9999%) with 40 ml min−1 at 25 °C for 34 h (2040 min) before the temperature was increased to 103 °C at a rate of 5 °C min−1 and kept for another 24 h (1440 min). Blank measurements (without sample) were performed using the same conditions to correct for the density change of the inert gas (nitrogen) during the temperature rise and to determine the balance drift over the measurement period. The holding times were chosen to reach a nearly constant sample mass at the end of each temperature step. The sample mass change was calculated as moisture content using the mass at the end of the 103 °C step as the reference mass. Mass stability at the end of each temperature step was estimated by calculating the moisture content change over time (in μg g−1 min−1) within the final 1 h of each step by the slope of a linear regression line. During the measurements, molecular and fragment ions emitted from the wood samples were continuously measured by mass spectrometry. In addition to ions for water, we also measured ions for compounds that are typically emitted from wood during drying (McDonald et al. 2002; Shen et al. 2020), see Table 1.
Automated sorption balance
In preliminary tests, beech and pine samples (n = 5) were dried under dry airflow at 20 °C for 72 h in an automated sorption balance (SPSx-1µ-High-Load, ProUmid, Germany). The samples were then removed from the sorption balance, and the sample mass was determined again after drying in a conventional oven at 103 °C for ca. 48 h and cooling in a desiccator over phosphorus pentoxide (P2O5) at room temperature.
The dynamic mass change of the samples during temperature exposure under dry air flow was determined in the same automated sorption balance at a resolution of 10 µg with ± 20 µg repeatability. For the generation of dry air, compressed air at a pressure of ca. 0.5 MPa was passed through a water separator and a compressed air filter (Ultra-Filter; Donaldson Europe B.V., Belgium). According to the manufacturer, the air filter reduced the residual water content to ≤ 0.11 g m−3, but the actual concentration of trace water in the dry air was not determined. The temperature was first held at 15 °C for 48 h before the temperature was increased by 10 °C every 24 h until reaching 55 °C, which was also held for 24 h before the temperature was decreased in the reverse order. The samples were placed on aluminium cups in a sample carousel, and the position of each sample was changed regularly by rotating the carousel. The initial sample mass was ca. 430, 340, and 172 mg for beech, pine, and MCC samples, respectively. During the experiment, the mass of each sample was recorded every 15 min and the dry airflow was stopped during the weighing procedure. An empty sample pan was measured to correct for a balance drift. The measurements were conducted with three replicates per wood species and one MCC replicate.
The moisture content (in %) was calculated by using the average sample mass at the end of the 55 °C step as the reference mass, which was determined from the final four measurements of this step. The change in moisture content over time (in μg g−1 min−1) was determined by linear regression with moisture content as the y-variable and time as the x-variable within a moving region of interest (ROI width = 180 min, ROI offset of 15 min). This rate of moisture content change was used as an indication of whether the samples approached an equilibrium state.
Additional sorption experiments to verify the temperature-dependent mass changes were conducted on beech and pine wood samples with two different automated sorption balances. The corresponding experimental details and results are shown in the supplementary material.
Vacuum-drying and coulometric Karl-Fischer titration
Approximately 50 mg of wood flour or MCC was placed into 6 ml glass vials that were sealed with aluminium septum caps. To allow for gas exchange, syringes (inner diameter 0.65 mm, outer diameter 0.90 mm, length 40 mm) were pierced into the septum of the vials just before the vials were placed into a 1 l autoclave glass reactor (Büchi Glas Uster AG, Switzerland) with a double jacketed glass shell for the flow of heat exchange liquids. Thermostats (EH heating circulators, JULABO GmbH, DE) were used for active heating, with polyethylene glycol 300 (CAS 25322-68-3, Carl Roth GmbH & Co. KG, Germany) as heat exchange liquid. The vacuum was generated with a rotary vane pump. A thermocouple and two separate pressure gauges measured temperature and pressure within the autoclave, respectively. After the drying time had elapsed, three purge-and-refill cycles were applied using nitrogen (purity 99.999%). The autoclave was opened at nitrogen atmospheric pressure, the vials were removed, and their syringes were pulled out immediately.
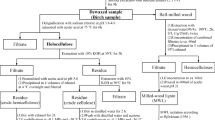
The moisture content of the samples was determined immediately after vacuum-drying by coulometric Karl-Fischer titration using the indirect oven method (see Fig. 1). First, the thermoprep oven (860 KF Thermoprep; Metrohm AG, Switzerland) was set to a temperature of 103 °C and a dry airflow of 100 ml min−1. After a sample vial was placed into the oven, a hollow two-way needle was lowered through the septum into the vial. Dry air entered the vial, took up water vapor released from the heated sample, and exited the vial into the coulometric titration unit (831 KF Coulometer; Metrohm AG, Switzerland) where the amount of water was determined electrochemically. For each vacuum drying run, three blank vials and three sample-containing vials were measured, and the data were analysed with the software Tiamo (Metrohm AG, Switzerland), which included automatic drift correction and blank subtraction. Average moisture contents were calculated based on the three samples from each vacuum drying run. The average coefficient of variation was 33%.

Basic design (not to scale) of the oven and the coulometric titration unit used for the coulometric Karl-Fischer titration by the indirect oven method
During the vacuum drying, the influence and 2-factor interactions of four different factors were investigated: (1) temperature, (2) time, (3) pressure, and (4) material (see Table 2). The pressure levels were chosen based on the capabilities of the vacuum pump. A maximum temperature of 103 °C was selected in reference to the oven-drying method (EN 13183–1 2002). The maximum duration of 1440 min (= 24 h) was chosen given typical drying schedules of cellulosic materials in automated sorption balances, which usually do not exceed a period of several hours. The design of experiments was used to reduce the number of combinations of a full factorial design. A randomized response surface methodology with a multiple regression analysis was applied using the software Design-Expert (Version 11, StatEase Inc., United States) to explore the influence and interactions of the four factors on the response variable (moisture content). This resulted in a total of 34 combinations. Details and results of the multiple regression analysis can be found in the supplementary material.
Results and discussion
The results confirmed a significant effect of the drying conditions on residual water in dried wood and cellulose. Drying small wood samples in an automated sorption balance at 20 °C under dry air flow resulted in a larger sample mass than oven-drying at 103 °C for ca. 24 h followed by cooling over anhydrous phosphorus pentoxide (P2O5) in a desiccator. Using the oven-dry mass as a reference mass, the average residual moisture contents (n = 5) after drying in the sorption balance were determined at 1.2 and 1.1% for pine and beech, respectively. However, the data ranged from 0.3% (minimum) to 2% (maximum) residual moisture content.
One potential reason for the lower dry mass after oven-drying was the emission of wood constituents, in particular extractives, from the wood during drying at elevated temperatures in addition to the vaporization of water. Wood indeed emits some organic compounds during conventional kiln-drying (McDonald et al. 2002; Shen et al. 2020). Emissions also occur during water sorption measurements of wood at room temperature if a sample contains residual solvents from extraction procedures (Vahtikari et al. 2017) or if the extractive content is high (Wentzel et al. 2018). To test the potential emission of wood constituents during wood drying, beech and pine wood was dried under dry nitrogen gas flow at temperature steps of 25 and 103 °C in a TGA-MS instrument, which measured sample mass changes as well as emitted molecular and fragment ions for selected molecules (Fig. 2).

Analysis of pine (a and c) and beech (b and d) wood powders in a TGA-MS equipment: a and b Moisture content (in%) and temperature (in °C) over time (in min); c and d Change in ion current over time (min). The small ion current peaks starting at ca. 2040 min are highlighted
For better comparability with results from other measurements, sample mass changes during the TGA-MS measurements are shown as moisture content (Fig. 2a and b), which was calculated by using the sample mass at the end of the 103 °C step as reference mass. The moisture content decreased rapidly at the beginning of each temperature step but approached constant moisture contents towards the step ends. The rates of moisture content change within the final 1-h of each temperature step were lower than |1.5| µg g−1 min−1 for both wood species. The residual moisture contents at the end of the 25 °C step were 0.6 and 0.5% for pine and beech, respectively. These values should be regarded as lower levels, because the reference mass determined after exposure to 103 °C may have contained an undetermined quantity of residual moisture. Simultaneous analyses of the emitted gases by mass spectrometry showed that losses in moisture content correlated with the emission of water molecules (m/z = 16, 17, 18), without obvious contributions from other molecules within the heating period (Fig. 2c and d). In particular, the sharp decrease in moisture content upon ramping the temperature from 25 to 103 °C coincided with ion current peaks related to water. This showed that the temperature-dependent decrease in sample mass was caused by the vaporization of bound water without relevant emission of other compounds for hard-and softwood. Possibly, the kiln-drying of the boards before the wood sample preparation already removed some extractives that easily become volatile upon heating.
The temperature effect on the anhydrous mass was further analysed by measuring the dynamic mass changes of wood and MCC samples under dry airflow and stepwise changes of the temperature between 15 and 55 °C (Fig. 3). The temperature had a clear effect on the equilibrium moisture content. The moisture content decreased monotonically with increasing temperature from 15 °C up to the highest applied temperature of 55 °C (Fig. 3a). Decreasing the temperature had the opposite effect: the moisture content of wood and MCC increased monotonically. Figure 3b presents the average moisture content at the end of each step as a function of the temperature, and this shows a hysteresis between desorption (temperature increase) and absorption (temperature decrease) for the wood samples. The absolute hysteresis increased with decreasing temperature and reached 0.3–0.4%-points at 15 °C. This was similar to the hysteresis of wood between desorption of absorption measured under isothermal conditions (Fredriksson and Thybring 2018). The MCC sample showed no obvious difference between desorption and absorption. The reason for the absence of hysteresis is unclear because the sorption isotherm of MCC in the hygroscopic range at 25 °C shows a hysteresis effect (Espino-Pérez et al. 2016).

Temperature-induced sample mass changes measured in an automated sorption balance under dry air flow: a Moisture content and temperature over time; b average moisture content at the end of each temperature step in dependence on the temperature; and c rate of moisture content change and temperature over time (beech/pine n = 3; cellulose n = 1). The illustration shows the basic design of the automated sorption balance in the top and side views
At each temperature step, the moisture content changed rapidly at the initial stage but approached a constant value towards the end of each step (Fig. 3a). Short holding times under constant climatic conditions can produce errors in equilibrium moisture content. Glass et al. (2018) recommended holding each conditioning step until the rate of moisture content change over time (measured in a 2-h window) decreases to |3| µg g−1 min−1 to limit the average error in equilibrium moisture content to less than 0.3%. In the present study, the rate of moisture content change of the wood samples decreased to less than |1| µg g−1 min−1 towards the end of each step during desorption (temperature increase) and to less than |0.5| µg g−1 min−1 during absorption (temperature decrease), as shown in Fig. 3c. The MCC sample approached a rate of 0 µg g−1 min−1, but higher data scattering was recorded due to the smaller sample mass compared to wood samples. It is thus concluded that different thermodynamic equilibria were approached at each temperature step, presumably due to some quantities of water vapor within the system. Although a direct effect of the temperature on the residual moisture content at 15–55 °C cannot be ruled out, the main effect was likely trace amounts of water in the dry airflow that led to a variation in relative humidity when the temperature was changed.
Additional sorption experiments confirmed the temperature-dependent sample mass change for other automated sorption balances with dry nitrogen flow, see Supplementary Fig. S.1 and Fig. S.2. These experiments also resulted in a moisture content decrease with increasing temperature and a re-increase in moisture content upon cooling. However, the magnitude of the residual moisture content differed, and one reason could be variations in the amount of trace water within the gas flow of the different devices. Changes in the gas flow rate also influenced the results because a nitrogen flow rate of 100 cm3 min−1 at 15 °C resulted in a lower sample mass compared to a flow rate of 200 cm3 min−1 at 25 °C (Supplementary Fig. S.1). However, it should be noted that the additional sorption experiments were conducted without correction of a balance drift and that the sorption balances used in the additional experiments are mainly designed for isothermal measurements. An aluminium reference indeed showed mass changes from one temperature step to the next, which were unrelated to the ab-or desorption of water (Supplementary Fig. S.2). Although the mass changes of the pine wood sample exceeded those of the aluminium reference, the measured moisture contents at different temperature levels must thus be interpreted with caution. Furthermore, the reference mass used for the moisture content calculation may still contain unknown quantities of water; hence, residual moisture contents determined gravimetrically should be seen as lower limits.
In the vacuum-drying experiments, the residual moisture content was quantified by Karl-Fischer titration (Fischer 1935). The impact of four different factors (temperature, duration, pressure, and material) during drying on the moisture content was analysed and a multiple regression analysis identified the relative impact of these factors based on their coefficients in a coded factor equation (Supplementary Table S.1). This showed a significant impact of temperature and time and an even higher impact of the factor “pressure” on the residual moisture content. The large impact of the pressure also became obvious by the measured residual moisture content when comparing drying at 150 mbar (Fig. 4 a) and at ≤ 1 mbar (Fig. 4 b) within similar drying times and temperatures. This was not surprising when considering that drying at 150 mbar required temperatures above ca. 54 °C to cause rapid vaporization by exceeding the water boiling point. Presumably, this had a large impact on the drying rate. However, the time-dependent moisture content data shows that even at temperatures exceeding the boiling point, larger moisture contents were measured at 150 mbar than at ≤ 1 mbar after drying for 1440 min. According to Chen (1997), the partial pressure of water vapor is nearly the same as the total pressure during the vacuum-drying of wood, because the volume of water vapor from evaporation is much greater than the volume of air inside the wood. It is thus likely that the pressure dependence of the residual wood moisture content, even at temperatures above the boiling point, was related to the amount of water vapor within the system.

Moisture contents (in %) as a function of vacuum-drying pressure, time, temperature, and sample type (pine, beech, and MCC). The pressure was set either to 150 mbar (a) or ≤ 1 mbar (b). The moisture content range below 0.4% is highlighted
Although no full factorial design was applied, some interesting trends could be observed from the measured moisture contents after vacuum-drying at different conditions (Fig. 4). Within the time (max. 1440 min) and temperature (max. 103 °C) range chosen, a pressure of 150 mbar was insufficient to reach the anhydrous state (Fig. 4a). Residual moisture contents of 0.3–2.5% were measured for beech wood even after vacuum drying at 103 °C and 150 mbar for 1440 min (24 h). In contrast, vacuum-drying at ≤ 1 mbar and 103 °C for 360 min or longer reduced the moisture content almost completely, resulting in moisture contents of 0.04, 0.02, and 0.03% for beech, pine, and MCC, respectively (Fig. 4b). Even at a pressure of ≤ 1 mbar, however, a temperature dependence was still observed after 1440 min. Residual moisture contents of 0.13–0.14% were measured after drying MCC and pine at 30 °C.
It remains unclear from our data if an insufficient drying time (max. 1440 min) caused the observed temperature dependence. However, Kollmann and Höckele (1962) vacuum-dried wood for several days at room temperature and also recorded residual moisture contents in the range of 0.2–0.3%. Although it is possible that some small quantities of absorbed water must be removed thermally, Kollmann and Höckele (1962) found similar quantities of absorbed water after oven-drying wood at 103 °C and atmospheric conditions. Hergt and Christensen (1965) observed that increasing the temperature decreased the quantity of absorbed water after vacuum-drying only slightly. Instead, they found that the sorption history before drying had a more significant effect and up to 1% residual moisture content was found if the wood was not dried rapidly from the water-saturated state. In our experiments, however, all samples reached the anhydrous state (moisture contents ≤ 0.04%) under high-temperature and low-pressure conditions, indicating that drying-resistant water molecules were not an effect of the sorption history but inadequate duration, temperature, or pressure levels.
Cellulosic materials may require a combination of minimal water contents in the surrounding atmosphere and sufficiently high temperatures to remove bound water completely due to the presence of water-attracting hydroxyl groups. However, despite clear differences in structure and chemical composition between MCC and wood samples, deuterium exchange measurements resulted in similar accessible hydroxyl group contents, with 9.4 (± 0.4) mmol g−1 measured for MCC (Väisänen et al. 2018) and 8.4–9.6 mmol g−1 for softwood samples (Thybring et al. 2017; Altgen and Rautkari 2020). Therefore, further studies are required to determine the material-inherent factors that control the residual moisture content after drying.
Nonetheless, our results demonstrate the difficulty in reaching the anhydrous state of wood and cellulose, which may be irrelevant in practical applications but has implications for scientific studies on water interactions in cellulosic materials. Incomplete drying shifts the entire sorption isotherm toward lower moisture contents (Thybring et al. 2018). This is caused by an overestimation of the reference mass (dry sample mass) and a decrease in the estimated water mass (mass difference between conditioned and dry state). Thereby, varying amounts of residual water in dried samples may limit the comparability of sorption isotherms. Based on our results, this may particularly occur when the dry sample mass is determined at different temperatures, for example when studying the effect of temperature on sorption isotherms in automated sorption balances (Hill et al. 2010; Simón et al. 2017). The observation of drying-resistant water molecules may also be related to the small number of deuterated hydroxyls that resisted re-protonation in previous studies on wood and cellulose samples (Hofstetter et al. 2006; Altgen and Rautkari 2021; Penttilä et al. 2021). Residual bound water in dried samples may cause a large error in the gravimetric quantification of accessible hydroxyl groups following the deuterium exchange approach, which has been applied to wood (Thybring et al. 2017; Uimonen et al. 2020; Altgen and Rautkari 2021) and cellulose samples (Pönni et al. 2014; Väisänen et al. 2018). When considering a typical dry mass increase of wood or cellulose samples due to deuteration in the range of 0.5–1%, an error of 11–22% is caused by a residual moisture content of 1%, and this error is reduced by one magnitude if the residual moisture content decreases to 0.1% (Thybring et al. 2018). These residual moisture contents are within the range of what we measured in the present study.
Conclusions
Dried wood and cellulose samples could contain more than 1% residual moisture content depending on the drying conditions applied. Temperature dependence of the residual moisture content was noticed during drying in dry air or nitrogen and during vacuum drying. This was not only caused by differences in the drying rate, but also by small quantities of water vapor in the surrounding atmosphere. The latter also resulted in a hysteresis effect during heating and cooling. Our results showed that variable amounts of residual water in dried wood and cellulose samples may cause significant errors in sorption isotherm and deuterium exchange measurements and should be considered when studying water interactions. However, a combination of low pressure (≤ 1 mbar) and elevated temperature (103 °C) could reduce the residual moisture content in wood and cellulose to ≤ 0.04%.
Data availability
The raw data to reproduce the results are available from the corresponding author upon reasonable request.
References
Altgen M, Rautkari L (2021) Humidity-dependence of the hydroxyl accessibility in Norway spruce wood. Cellulose 28:45–58. https://doi.org/10.1007/s10570-020-03535-6
Berthold J, Rinaudo M, Salmeń L (1996) Association of water to polar groups; estimations by an adsorption model for ligno-cellulosic materials. Colloids Surf Physicochem Eng Asp 112:117–129. https://doi.org/10.1016/0927-7757(95)03419-6
Chen M, Coasne B, Guyer R et al (2018) Role of hydrogen bonding in hysteresis observed in sorption-induced swelling of soft nanoporous polymers. Nat Commun 9:3507. https://doi.org/10.1038/s41467-018-05897-9
Chen Z (1997) Primary driving force in wood vacuum drying. Doctoral thesis, Virginia Polytechnic Institute and State University
EN 13183-1 (2002) Moisture content of a piece of sawn timber–Part 1: determination by oven dry method. CEN–European Comitee for Standardization, Brussels
Espino-Pérez E, Bras J, Almeida G, Relkin P, Belgacem N, Plessis C, Domenek S (2016) Cellulose nanocrystal surface functionalization for the controlled sorption of water and organic vapours. Cellulose 23:2955–2970. https://doi.org/10.1007/s10570-016-0994-y
Fischer K (1935) Neues Verfahren zur maßanalytischen Bestimmung des Wassergehaltes von Flüssigkeiten und festen Körpern. Angew Chem 48:394–396. https://doi.org/10.1002/ange.19350482605
Fredriksson M, Thybring EE (2018) Scanning or desorption isotherms? Characterising sorption hysteresis of wood. Cellulose 25:4477–4485. https://doi.org/10.1007/s10570-018-1898-9
Glass SV, Boardman CR, Zelinka SL (2017) Short hold times in dynamic vapour sorption measurements mischaracterize the equilibrium moisture content of wood. Wood Sci Technol 51:243–260. https://doi.org/10.1007/s00226-016-0883-4
Glass SV, Boardman CR, Thybring EE, Zelinka SL (2018) Quantifying and reducing errors in equilibrium moisture content measurements with dynamic vapor sorption (DVS) experiments. Wood Sci Technol 52:909–927. https://doi.org/10.1007/s00226-018-1007-0
Hatakeyama T, Nakamura K, Hatakeyama H (2000) Vaporization of bound water associated with cellulose fibers. Thermochim Acta 352–353:233–239. https://doi.org/10.1016/S0040-6031(99)00471-2
Hergt HFA, Christensen GN (1965) Variable retention of water by dry wood. J Appl Polym Sci 9:2345–2361. https://doi.org/10.1002/app.1965.070090703
Hill CAS, Norton AJ, Newman G (2010) The water vapour sorption properties of Sitka spruce determined using a dynamic vapour sorption apparatus. Wood Sci Technol 44:497–514. https://doi.org/10.1007/s00226-010-0305-y
Hofstetter K, Hinterstoisser B, Salmén L (2006) Moisture uptake in native cellulose–the roles of different hydrogen bonds: a dynamic FT-IR study using deuterium exchange. Cellulose 13:131–145. https://doi.org/10.1007/s10570-006-9055-2
Kollmann F, Höckele G (1962) Kritischer Vergleich einiger Bestimmungsverfahren der Holzfeuchtigkeit. Holz Als Roh-Werkst 20:461–473. https://doi.org/10.1007/BF02619168
McDonald AG, Dare PH, Gifford JS et al (2002) Assessment of air emissions from industrial kiln drying of Pinus radiata wood. Holz Als Roh-Werkst 60:181–190. https://doi.org/10.1007/s00107-002-0293-1
Merela M, Oven P, Serša I, Mikac U (2009) A single point NMR method for an instantaneous determination of the moisture content of wood. Holzforschung 63:348–351. https://doi.org/10.1515/HF.2009.050
Mitchell J (1940) Determination of moisture in native and processed cellulose. Ind Eng Chem Anal Ed 12:390–391. https://doi.org/10.1021/ac50147a004
Nopens M, Riegler M, Hansmann C, Krause A (2019) Simultaneous change of wood mass and dimension caused by moisture dynamics. Sci Rep 9:10309. https://doi.org/10.1038/s41598-019-46381-8
Pan Y, Zhong Z (2016) Micromechanical modeling of the wood cell wall considering moisture absorption. Compos Part B Eng 91:27–35. https://doi.org/10.1016/j.compositesb.2015.12.038
Penttilä PA, Zitting A, Lourençon T et al (2021) Water-accessibility of interfibrillar spaces in spruce wood cell walls. Cellulose 28:11231–11245. https://doi.org/10.1007/s10570-021-04253-3
Pönni R, Rautkari L, Hill CAS, Vuorinen T (2014) Accessibility of hydroxyl groups in birch kraft pulps quantified by deuterium exchange in D2O vapour. Cellulose 21:1217–1226. https://doi.org/10.1007/s10570-014-0166-x
Rostom L, Caré S, Courtier-Murias D (2021) Analysis of water content in wood material through 1D and 2D 1H NMR relaxometry: application to the determination of the dry mass of wood. Magn Reson Chem 59:614–627. https://doi.org/10.1002/mrc.5125
Shen Y, Zhang X, He Q et al (2020) Study of VOCs release during drying of plantation-grown Pinus sylvestris and naturally grown Russian Pinus sylvestris. J Wood Sci 66:34. https://doi.org/10.1186/s10086-020-01882-3
Simón C, Fernández FG, Esteban LG et al (2017) Comparison of the saturated salt and dynamic vapor sorption methods in obtaining the sorption properties of Pinus pinea L. Eur J Wood Wood Prod 75:919–926. https://doi.org/10.1007/s00107-016-1155-6
Steinmeier H (2004) 3. Acetate manufacturing, process and technology 3.1 chemistry of cellulose acetylation. Macromol Symp 208:49–60. https://doi.org/10.1002/masy.200450405
Thybring EE, Thygesen LG, Burgert I (2017) Hydroxyl accessibility in wood cell walls as affected by drying and re-wetting procedures. Cellulose 24:2375–2384. https://doi.org/10.1007/s10570-017-1278-x
Thybring EE, Kymäläinen M, Rautkari L (2018) Experimental techniques for characterising water in wood covering the range from dry to fully water-saturated. Wood Sci Technol 52:297–329. https://doi.org/10.1007/s00226-017-0977-7
Uimonen T, Hautamäki S, Altgen M et al (2020) Dynamic vapour sorption protocols for the quantification of accessible hydroxyl groups in wood. Holzforschung 74:412–419. https://doi.org/10.1515/hf-2019-0058
Vahtikari K, Rautkari L, Noponen T et al (2017) The influence of extractives on the sorption characteristics of Scots pine (Pinus sylvestris L.). J Mater Sci 52:10840–10852. https://doi.org/10.1007/s10853-017-1278-0
Väisänen S, Pönni R, Hämäläinen A, Vuorinen T (2018) Quantification of accessible hydroxyl groups in cellulosic pulps by dynamic vapor sorption with deuterium exchange. Cellulose 25:6923–6934. https://doi.org/10.1007/s10570-018-2064-0
Wentzel M, Altgen M, Militz H (2018) Analyzing reversible changes in hygroscopicity of thermally modified eucalypt wood from open and closed reactor systems. Wood Sci Technol 52:889–907. https://doi.org/10.1007/s00226-018-1012-3
Acknowledgments
The authors thank Prof. Dr. Gerrit A. Luinstra, Claudia Wackendorff and Dr. Ulrich Riederer from the Department of Chemistry of the Universität Hamburg for their support and the opportunity to perform the vacuum drying and Karl-Fischer titration studies at their facilities. Dr. Daniela Altgen is thanked for her help with the illustrations.
Funding
Open access funding provided by Norwegian Institute of Bioeconomy Research. The authors declare that no funds, grants, or other support were received during the preparation of this manuscript.
Author information
Authors and Affiliations
Contributions
MN designed the experiments, together with JG, AK, and MO, and prepared the material. US and MF performed the TGA-MS experiments. MN and MA recorded the dynamic mass changes in the automated sorption balances. JG conducted the vacuum-drying experiments, the coulometric Karl-Fischer titration and the multiple regression analysis. MA analysed the results, together with MN and WW, and wrote the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no relevant financial or non-financial interests to disclose.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
All authors read and approved the final manuscript.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Altgen, M., Fröba, M., Gurr, J. et al. Limits in reaching the anhydrous state of wood and cellulose. Cellulose 30, 6247–6257 (2023). https://doi.org/10.1007/s10570-023-05293-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10570-023-05293-7