
Abstract
Purpose
We assessed the differential effect of clarithromycin, a strong inhibitor of cytochrome P450 (CYP) 3A4 and P-glycoprotein, on the pharmacokinetics of a regular dose of edoxaban and on a microdose cocktail of factor Xa inhibitors (FXaI). Concurrently, CYP3A activity was determined with a midazolam microdose.
Methods
In an open-label fixed-sequence trial in 12 healthy volunteers, the pharmacokinetics of a microdosed FXaI cocktail (μ-FXaI; 25 μg apixaban, 50 μg edoxaban, and 25 μg rivaroxaban) and of 60 mg edoxaban before and during clarithromycin (2 x 500 mg/d) dosed to steady-state was evaluated. Plasma concentrations of study drugs were quantified using validated ultra-performance liquid chromatography–tandem mass spectrometry methods.
Results
Therapeutic clarithromycin doses increased the exposure of a therapeutic 60 mg dose of edoxaban with a geometric mean ratio (GMR) of the area under the plasma concentration-time curve (AUC) of 1.53 (90 % CI: 1.37–1.70; p < 0.0001). Clarithromycin also increased the GMR (90% CI) of the exposure of microdosed FXaI apixaban to 1.38 (1.26–1.51), edoxaban to 2.03 (1.84–2.24), and rivaroxaban to 1.44 (1.27–1.63). AUC changes observed for the therapeutic edoxaban dose were significantly smaller than those observed with the microdose (p < 0.001).
Conclusion
Clarithromycin increases FXaI exposure. However, the magnitude of this drug interaction is not expected to be clinically relevant. The edoxaban microdose overestimates the extent of the drug interaction with the therapeutic dose, whereas AUC ratios for apixaban and rivaroxaban were comparable to the interaction with therapeutic doses as reported in the literature.
Trial Registration
EudraCT Number: 2018-002490-22
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Since their approval, direct-acting oral factor Xa inhibitors (FXaI) have rapidly rising prescription rates because of at least similar effectiveness to vitamin K antagonists, less variable dosing regimens, fewer monitoring requirements, and a favorable safety profile [1,2,3]. Their effects are immediate and concentration-dependent, indicating that changes in dose or clearance will immediately translate into exposure and effect changes. Depending on the indication, FXaI maintenance doses differ, and, as shown for rivaroxaban, rather, small dose steps of 50 % or 25 % of the maximum 20 mg dose can be of importance, indicating that appropriate FXaI dose selection is critical [4,5,6].
Different pathways of transport, metabolism, and elimination are involved in the pharmacokinetics of currently available FXaI, and their relative contribution to clearance varies among different FXaI [5]. Accordingly, impairment of these pathways by drug-drug interactions (DDI) caused by co-administered drugs can result in variable exposure changes that are difficult to predict. Therefore, pharmacokinetic DDI are not a class phenomenon and need to be tested or modelled for each compound separately.
The currently marketed FXaI are substrates of drug transporters such as P-glycoprotein (P-gp, ABCB1), are metabolized by various phase-I (cytochrome P450 (CYP) 3A, CYP2J2, and carboxylesterase 1) and phase-II enzymes (e.g., UDP-glucuronosyltransferases), and are partly eliminated renally. But not all pathways are equally relevant for each compound [5, 7,8,9]; as an example, edoxaban is only minimally metabolized by CYP, whereas the CYP contribution to overall apixaban and rivaroxaban clearance is 21 and 32 %, respectively. In addition, applying results of DDI studies with FXaI to individual patient settings is complicated by the large interindividual variability in the pharmacokinetics of FXaI [10,11,12].
As different as the individual metabolic pathways of FXaI are, so is the extent of interaction with a particular perpetrator drug [5]. It is therefore important to evaluate pharmacokinetic drug interactions for each individual victim drug. An attractive way to study the interaction profile of an entire drug class is to administer these drugs simultaneously (cocktail) in microdoses, minimizing inter-day and inter-subject variability and increasing statistical power [13]. This is particularly important for narrow therapeutic index compounds such as FXaI to minimize bleeding risks. By administering all three FXaI as a microdose cocktail, it is therefore possible to reduce intraindividual and interindividual variability with negligible effects on coagulation, while simplifying study conduct and minimizing time, risk, and cost [14].
Clarithromycin is a known CYP3A inhibitor, which increased apixaban concentrations by 60 % [15], and rivaroxaban by 50–100 % [11, 16] in previous trials and which was associated with bleeding events [17, 18]. In a clinical trial in healthy volunteers, we assessed the yet unknown effect of the macrolide antibiotic clarithromycin on the pharmacokinetics of a therapeutic dose of edoxaban. In addition, we evaluated the effect of clarithromycin on the pharmacokinetics of apixaban, edoxaban, and rivaroxaban administered as a microdosed FXaI cocktail to further evaluate the utility of such a cocktail for assessing pharmacokinetic DDI with these FXaI.
Materials and Methods
Ethics Approval
The study protocol was approved by the competent authority (BfArM, Bonn, Germany, Vorlagen Nr. 4043377) and received a positive vote of the responsible Ethics Committee of the Medical Faculty of Heidelberg University, Germany (AFmo-144/2019), and the trial was registered in the EudraCT database (EudraCT 2018-002490-22). This phase I, investigator-initiated, monocenter trial was conducted in accordance with the Declaration of Helsinki, the principles of Good Clinical Practice, and all pertinent legal requirements at the DIN EN ISO 9001-certified early clinical trial unit KliPS of the Department of Clinical Pharmacology and Pharmacoepidemiology, Heidelberg University Hospital.
Trial Population and Design
Healthy volunteers between 18 and 65 years were eligible after full information and after having given their written informed consent. Inclusion and exclusion criteria ensured the enrolment of volunteers in good health without any relevant medical history or relevant findings in laboratory exams, electrocardiogram, and physical examination that would have put them at any risk when receiving the study drugs. They had to be willing to follow strict pregnancy prevention measures.
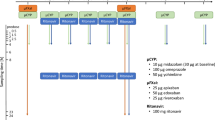
The study was an open-label, two-period, one-sequence DDI trial in healthy volunteers to assess the impact of clarithromycin on the pharmacokinetics of 60 mg edoxaban, given as a tablet, and the effect of clarithromycin on apixaban (25 μg), edoxaban (50 μg), and rivaroxaban (25 μg) administered as an oral microdose cocktail [14]. Concurrently, the effect of clarithromycin on CYP3A activity was quantified by means of an oral midazolam microdose (30 μg) (Fig. 1).

Design of a trial assessing the impact of repeated therapeutic oral doses of clarithromycin on the oral pharmacokinetics of microdosed midazolam, a microdosed factor Xa inhibitor cocktail containing apixaban, edoxaban, and rivaroxaban, and a single therapeutic dose of edoxaban in healthy volunteers. (FXaI factor Xa inhibtors)
CYP3A4 activity was quantified using a microdose of midazolam administered as an oral solution, which was administered directly before administering the FXaI [19, 20]. Clarithromycin was dosed to inhibition steady-state (7-day treatment prior to evaluation of any perpetrator effects and treatment continued until last blood sampling) and administered at a dose of 500 mg b.i.d. to ensure reliable inhibition of hepatic CYP3A4 (mechanism-based inhibition), while maximum CYP3A4 inhibition in the gut wall was expected to occur earlier [21]. Single-dose pharmacokinetics of the FXaI were assessed at baseline and on day 8 (microdosed FXaI cocktail) and day 9 (edoxaban 60 mg) of clarithromycin treatment.
Quantification of Factor Xa Inhibitors and Midazolam
Venous plasma samples for pharmacokinetic FXaI analyses were collected before and 0.25, 0.5, 0.75, 1, 1.5, 2, 2.5, 3, 3.5, 4, 5, 6, 8, 10, 12, 24 (last sampling after microdoses), and 48 h (last sampling for edoxaban 60 mg) after drug administration. Midazolam pharmacokinetics were assessed using a limited sampling strategy with sampling at 2, 2.5, 3, and 4 h post dose [19, 20]. All samples were processed within 20 min, and plasma was stored at ≤ – 20 °C until analysis. Coagulation effects (international normalized ratio (INR) and activated partial thromboplastin time (aPTT)) were assessed at expected FXaI peak plasma concentrations (3 h post dose) in the accredited central laboratory of the hospital.
Apixaban, edoxaban, midazolam, and rivaroxaban plasma concentrations were quantified using validated highly sensitive ultra-performance liquid chromatography–tandem mass spectrometry (UPLC-MS/MS) methods with accuracy and precision values of less than or equal to ± 15 % [22, 23]. The corresponding lower limits of quantification were 1 pg/ml for microdosed midazolam, 2.5 pg/ml for all microdosed FXaI, and 1 ng/ml for regular doses of edoxaban.
Pharmacokinetic and Statistical Analysis
Non-compartmental analyses of the FXaI and midazolam pharmacokinetics were performed using Phoenix WinNonlin 8.3 (Certara, Inc., Princeton, NJ, USA). Linear modelling of AUC ratios was done using the R software environment in version 4.0.4 (R Foundation for Statistical Computing, Vienna, Austria). Maximum plasma concentration (Cmax) and the time to reach Cmax (Tmax) were directly obtained from the pharmacokinetic data. The area under the concentration-time curve from 0 to infinity (AUC0-∞) was determined using the log-linear trapezoidal rule and by adding the extrapolated part until infinity. Half-life (t1/2) is calculated as \(\frac{\ln (2)}{\uplambda \textrm{z}}\), where the elimination rate constant λz was estimated using log-linear regression of the elimination phase. The apparent oral clearance (Cl/F) was calculated as \(\frac{\ \textrm{dose}}{{\textrm{AUC}}_{0\hbox{--} \infty }}\). CYP3A4 activity was determined using a limited sampling strategy using the midazolam AUC2-4 as described earlier [19, 20]. Parameters are displayed as geometric means with 95 % confidence interval (CI). Exposure changes are described by evaluating the geometric mean ratio (paired t-test on log-transformed values) of AUC0-∞ and Cmax at baseline and under the influence of steady-state clarithromycin (90 % CI). The AUC2-4 of midazolam was evaluated at multiple time points using a repeated-measures analysis of variance after logarithmic transformation.
To evaluate exposure changes between the edoxaban microdose and regular dose, the following analyses were carried out: The agreement of AUC change ratios between both dose groups was evaluated by a Bland-Altman analysis plotting the difference of intraindividual AUC ratios (edoxaban 60 mg and edoxaban 50 μg) against the intraindividual mean of AUC ratios across each dose group. To evaluate P-gp activity as potential predictor for AUC change under inhibition we evaluated the linear regression of the individual baseline AUC with the AUC change.
Statistical analyses and graphical displays were carried out using Prism 9.0 (GraphPad Software Inc., La Jolla, CA, USA). A p value < 0.05 was considered significant.
Results
After having given their written informed consent, we enrolled 12 healthy volunteers (9 females), aged 20–54 years (median 26) with a mean body mass index of 22.5 kg/m2 (± standard deviation (SD) 2.36) in this trial. All participants completed the trial. Baseline characteristics are described in Supplemental Table S2.
Effect of Clarithromycin on FXaI Pharmacokinetics
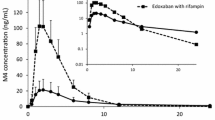
After administration of the 60 mg dose, the geometric mean ratio (GMR) of edoxaban AUC0-∞ increased to 1.53-fold. After microdosed edoxaban with a dose of 50 μg, the dose-normalized absolute exposure increased to 2.03-fold (Fig. 2, Table 1). This increase was larger than the increase observed after the 60 mg dose, which suggests that the microdose moderately overestimates the true extent of the interaction (Table 1). Clarithromycin moderately increased the exposure (Fig. 2, Table 1) and decreased clearance (Table S1) of all FXaI.

Mean values (± SD) of plasma concentration-time profiles of factor Xa inhibitors (FXaI) after administration of single oral doses before (open squares) and during clarithromycin (solid circles) to 12 healthy volunteers
At baseline, the dose-normalized AUC0-∞ of the microdose of edoxaban was similar to the AUC0-∞ of the therapeutic dose (GMR 1.08; 90 % CI: 0.97–1.20), while at steady-state of clarithromycin, the GMR of the microdose was significantly larger (1.43; 1.33–1.55) (Fig. 3). There was a significant correlation between the AUC0-∞ of edoxaban 60 mg and edoxaban 50 μg both at baseline (Pearson correlation coefficient r = 0.57, one-tailed p = 0.027) and during clarithromycin steady-state (Pearson correlation coefficient r = 0.62, one-tailed p = 0.017). In contrast, there was no correlation between the magnitude of AUC0-∞ changes after administration of the microdose and the therapeutic dose (data not shown).

Edoxaban dose-normalized AUCs at baseline (a) and during clarithromycin (b) after administration of an edoxaban microdose (50 μg) and a high regular dose (60 mg) to 12 healthy volunteers
There were no baseline characteristics (laboratory values and demographic variables) which correlated to the AUC ratio in either the microdose or the full dose setting (data not shown).
The differences of the AUC0-∞ ratios agreed only moderately between the microdose group and the regular dose in a Bland-Altman analysis (Fig. S1). In particular, the pair means of ratios of regular doses and microdose showed an upward shift, suggesting an overestimation of the effect in the microdose setting. AUC changes correlated negatively with the baseline AUC measurements both after a microdose (p = 0.010) and a therapeutic dose (p = 0.028) (Fig. S2).
Assessment of CYP3A Activity
Midazolam AUC2-4 was 6.4 min*ng/ml (90 % CI 4.9–8.3 min*ng/ml) and 40.3 (35.1–46.2) min*ng/ml at baseline and at clarithromycin steady-state, and the GMR was 6.30 (90 % CI 5.16–7.70), confirming substantial inhibition of CYP3A activity (p < 0.0001) (see Table S1). The mean increase of midazolam AUC2-4 was significantly different from the mean increase of the AUC0-∞ of individual FXaI (p < 0.0001). Tested as paired values to evaluate intraindividual correlation of changes in a participant, there was no significant linear correlation between the increase of midazolam AUC2-4 and the increase of AUC0-∞ of any of the FXaI edoxaban, apixaban, or rivaroxaban.
Effect on Coagulation Markers
Regular edoxaban doses significantly increased aPTT and INR values, and anticoagulation further increased during clarithromycin (Table 2). There was also a small anticoagulant effect after the microdose cocktail but only during co-administration of clarithromycin.
Safety
Overall, 27 adverse events (AE) occurred in 11 of 12 participants, all of which were deemed possibly related to the trial. All AE were transient, none was serious, and none resulted in a withdrawal from the trial. The most frequent AE was dysgeusia reported by 9 participants while taking clarithromycin. Other AE occurring more than once were abdominal pain (n = 4), headache (n = 3), and diarrhea (n = 2), which all occurred under clarithromycin treatment.
Discussion
Influence of Clarithromycin on the Pharmacokinetics of 60 mg Edoxaban
In this first trial, examining the potential DDI of the P-gp and CYP3A4 inhibitor clarithromycin, clarithromycin moderately increased edoxaban exposure by 53 %. This is similar to the magnitude of drug interactions observed with therapeutic doses of apixaban (59 % [24]) and rivaroxaban (54 %, [16]). Overall, these exposure changes are less than those observed when therapeutic doses of FXaI were combined with the strong P-gp inhibitor ketoconazole (edoxaban, + 87 % [25]; apixaban, + 99 % [26]; rivaroxaban, + 158 % [16]). In addition to its higher potency as CYP3A inhibitor, ketoconazole also inhibits organic anion transporter 3, breast cancer resistance protein, CYP3A, and CYP2J2 [27, 28]. Because CYP2J2 appears to be the dominant hydroxylating isozyme in the metabolism of rivaroxaban in vitro [29], and CYP3A is presumably the dominant pathway of apixaban [30], it must be expected that there are differences for the effects on DDI of the perpetrator drugs ketoconazole and clarithromycin for apixaban and rivaroxaban.
Both efficacy and safety of FXaI treatment appear to be linked to FXaI exposure [31], and retrospective evidence from large cohort analyses mostly, albeit not always [32], suggests that co-medication increasing FXaI exposure is linked to major bleeding events [21, 33, 34], while combinations decreasing exposure increase the risk of thromboembolic events by approximately 60 % and more than double the risk of strokes [21, 35]. Epidemiological evidence suggests that the approximately 40 % exposure increase of apixaban and rivaroxaban caused by verapamil or diltiazem does not increase bleeding risk [36]. In contrast, in epidemiological studies evaluating patients mainly anticoagulated with FXaI, the risk of hospital admission due to major bleeding events was increased by 71 % during clarithromycin compared to azithromycin [18]. However, in this latter study, FXaI concentrations were not measured, and a significant proportion of patients had renal impairment, a comorbidity, which may have potentiated the impact of CYP3A inhibition [37]. To date, no clear therapeutic range has been defined, and it is not known what magnitude of exposure change is clinically relevant. Generally, information in the drug label approved by the authorities indicates that no dose adjustment is needed as long as exposure increases are ≤ 90 % (edoxaban [11]) or ≤ 100 % (apixaban [12], rivaroxaban [14]) but a clear threshold has not been defined.
Influence of Clarithromycin on the Pharmacokinetics of Microdoses of Edoxaban, Apixaban, and Rivaroxaban
Exposure increases observed with microdoses of apixaban and rivaroxaban agreed well with the magnitude of interaction reported with regular doses [11, 16]. In contrast, after administration of a microdose of edoxaban, the increase of exposure was significantly, albeit only moderately larger, slightly overestimating the absolute increase of exposure by the interaction of a regular high dose. The reason for these findings is not clear yet: The fact that dose-normalized AUC0-∞ values of the microdose and regular dose did not differ at baseline excludes pharmacokinetic nonlinearity in the absence of a perpetrator and formulation differences (solution of the microdose vs. tablet) as a possible cause. In addition, edoxaban does not have any perpetrator properties that could explain a potential difference of the effect of a therapeutic dose and a microdose. Therefore, the difference is unlikely due to a decrease in clarithromycin exposure under the influence of a single therapeutic dose of edoxaban. Moreover, we assume that clarithromycin exposure and thus its inhibitor effects were similar in the two phases as shown by similarly decreased CYP3A activity. We have recently shown that this FXaI microdose cocktail can predict the magnitude of the known ketoconazole-induced exposure changes observed with regular doses of FXaI [14]. This study confirms these already published findings for rivaroxaban, which was found to have an increased exposure of approximately 50–100 % with clarithromycin [11, 16] and also the corresponding findings of a 60 % increase for apixaban [15].
Clarithromycin is a macrolide inhibiting multiple relevant metabolic pathways including CYP3A [38], P-gp [39], and organic anion-transporting polypeptides (OATP) 1B1 and 1B3 [40]. Therefore, changes in exposure from drug interactions by clarithromycin may result from changes in either of the affected pathways. As a consequence of the very differing shares of metabolic pathways for the different FXaI [5], inhibition of individual and multiple pathways will have variable impact on exposure changes of the victim drugs, which agrees well with the fact that CYP3A is known to contribute mainly to apixaban [30, 41], less to rivaroxaban metabolism [16], whereas carboxylesterase 1 and P-gp are the dominant clearance pathways of edoxaban [42] with a contribution of CYP3A to the overall metabolic clearance of only 1 % [43]. However, despite this fact, the overall extent of interaction for edoxaban with clarithromycin was similar to the interaction observed with the other FXaI. This suggests that in the case of edoxaban, the interaction with clarithromycin is largely caused by modulation of P-gp. In theory, inhibition of carboxylesterase 1 could also cause such a phenomenon, but clarithromycin has not been reported to have such a property, and it is not converted to acyl glucuronides [44], which have been identified as carboxylesterase inhibitors [45].
Differences in the Pharmacokinetics of Microdoses and Therapeutic Doses of Edoxaban
To further evaluate the usefulness of microdose cocktail approaches, the relative divergence of the measured changes in plasma levels between microdose and 60-mg dose of edoxaban during clarithromycin was addressed. Edoxaban is actively secreted via P-gp both into urine and feces [46]. P-gp inhibition can reduce intestinal excretion during the absorption phase and biliary and renal tubular secretion during elimination, thus resulting in increased exposure. When comparing the effect of oral clarithromycin on the paradigm P-gp marker substrate digoxin given intravenously and orally, the largest perpetrator effect occurred in the absorption process in the gut with only minor reductions (15 %) of renal tubular secretion [47, 48]. While our study could not disentangle the specific mode of interaction with edoxaban, it reveals that the microdose of edoxaban is more affected by the macrolide than a regular dose, leading to overestimation of the true extent of interaction in the clinical setting when regular edoxaban doses are used. These suggest that full inhibition of pathways contributing to Cl/F by clarithromycin may be more relevant for a microdose than for a therapeutic dose, which may use alternative uptake pathways. Edoxaban is mainly transported by P-gp, CYP3A does not contribute to its clearance and F to any relevant extent [43], and selective inhibition of CYP3A (by voriconazole) does not affect its pharmacokinetics [49].
There is a significant intraindividual correlation between the AUC0-∝ of the microdose and the therapeutic dose of edoxaban. Interindividual differences of the expression and activity of P-gp (and/or similar transporters) are likely to influence edoxaban AUC0-∝ differently depending on its dose. Consistent with this, the AUC0-∝ of edoxaban shows evidence of less variability and correlation between microdose and therapeutic dose when clarithromycin is administered. This is most likely due to the elimination of variations in the activity of basic enzymes and transporters, e.g., of P-gp. The shift to overestimation of exposure has additionally been visualized via Bland-Altman analysis (Fig. S1).
These findings are further supported by the fact that the baseline AUC0-∝ of edoxaban and the factor of AUC0-∝ increase during treatment with clarithromycin were significantly correlated. Therefore, individuals with lower baseline exposure had a larger increase of AUC0-∝, which might indicate that their baseline P-gp activity was higher and that these differences in the extent of interaction are likely transporter-mediated.
Limitations
The observed inverse relationship between edoxaban exposure at baseline and extent of the interaction with clarithromycin could be the result of interindividual differences in P-gp activity or clarithromycin exposure; neither of them has been studied. Furthermore, variations in exposure could also be caused by different release characteristics of the edoxaban tablet compared to its microdose solution. We did not genotype our participants and did not power the trial for genetic differences because, in an earlier trial, P-gp haplotypes did not predict the extent of clarithromycin-induced exposure changes of rivaroxaban [11, 16]. Because clarithromycin is a mechanism-based inhibitor, which was administered until an inhibition steady-state was reached, and because CYP3A inhibition was closely followed using midazolam, it is very unlikely that small fluctuations in clarithromycin plasma concentration would have affected edoxaban pharmacokinetics.
Conclusion
This paper has shown that clarithromycin increases exposure of a therapeutic dose of edoxaban in healthy volunteers 1.53-fold, which is not considered clinically relevant. However, FXaI are often prescribed to patients with impaired renal function, which may further increase the exposure of FXaI when co-administered with clarithromycin [37]. Therefore, appropriate dose reduction for edoxaban may be needed for patients with renal impairment when clarithromycin is concomitantly used. Furthermore, our trial suggests that edoxaban microdoses might overestimate the extent of interaction with P-gp inhibitors.
Data Availability
Data will be made available by the corresponding author of this publication upon personal request.
References
Giugliano RP, Ruff CT, Braunwald E, Murphy SA, Wiviott SD, Halperin JL, et al. Edoxaban versus warfarin in patients with atrial fibrillation. N Engl J Med. 2013;369(22):2093–104.
Granger CB, Alexander JH, McMurray JJ, Lopes RD, Hylek EM, Hanna M, et al. Apixaban versus warfarin in patients with atrial fibrillation. N Engl J Med. 2011;365(11):981–92.
Patel MR, Mahaffey KW, Garg J, Pan G, Singer DE, Hacke W, et al. Rivaroxaban versus warfarin in nonvalvular atrial fibrillation. N Engl J Med. 2011;365(10):883–91.
Vasanthamohan L, Boonyawat K, Chai-Adisaksopha C, Crowther M. Reduced-dose direct oral anticoagulants in the extended treatment of venous thromboembolism: a systematic review and meta-analysis. J Thromb Haemost. 2018;16(7):1288–95.
Foerster KI, Hermann S, Mikus G, Haefeli WE. Drug-drug interactions with direct oral anticoagulants. Clin Pharmacokinet. 2020;59(8):967–80.
Eikelboom JW, Connolly SJ, Bosch J, Dagenais GR, Hart RG, Shestakovska O, et al. Rivaroxaban with or without aspirin in stable cardiovascular disease. New Engl J Med. 2017;377(14):1319–30.
Lixiana : EPAR - Product Information. https://www.emaeuropaeu/documents/product-information/lixiana-epar-product-information_en.pdf. 25/06/2020.
Eliquis : EPAR - Product Information. https://www.emaeuropaeu/documents/product-information/eliquis-epar-product-information_en.pdf. 07/10/2020.
Xarelto : EPAR - Product Information. https://www.emaeuropaeu/documents/product-information/xarelto-epar-product-information_en.pdf. 28/11/2019.
Rosian AN, Rosian SH, Kiss B, Stefan MG, Trifa AP, Ober CD, et al. Interindividual variability of apixaban plasma concentrations: influence of clinical and genetic factors in a real-life cohort of atrial fibrillation patients. Genes (Basel). 2020;11(4).
Gouin-Thibault I, Delavenne X, Blanchard A, Siguret V, Salem JE, Narjoz C, et al. Interindividual variability in dabigatran and rivaroxaban exposure: contribution of ABCB1 genetic polymorphisms and interaction with clarithromycin. J Thromb Haemost. 2017;15(2):273–83.
Testa S, Dellanoce C, Paoletti O, Cancellieri E, Morandini R, Tala M, et al. Edoxaban plasma levels in patients with non-valvular atrial fibrillation: inter and intra-individual variability, correlation with coagulation screening test and renal function. Thromb Res. 2019;175:61–7.
Fuhr U, Jetter A, Kirchheiner J. Appropriate phenotyping procedures for drug metabolizing enzymes and transporters in humans and their simultaneous use in the "cocktail" approach. Clin Pharmacol Ther. 2007;81(2):270–83.
Mikus G, Foerster KI, Schaumaeker M, Lehmann ML, Burhenne J, Haefeli WE. Microdosed cocktail of three oral factor Xa inhibitors to evaluate drug-drug interactions with potential perpetrator drugs. Clin Pharmacokinet. 2019;58(9):1155–63.
Summary of product characteristics Eliquis. Bristol-Myers Squibb/Pfizer EEIG, United Kingdom. Availbale at https://www.ema.europa.eu/en/documents/product-information/eliquis-epar-product-information_en.pdf. Accessed 27 Feb 2023
Mueck W, Kubitza D, Becka M. Co-administration of rivaroxaban with drugs that share its elimination pathways: pharmacokinetic effects in healthy subjects. Br J Clin Pharmacol. 2013;76(3):455–66.
Fralick M, Juurlink DN, Marras T. Bleeding associated with coadministration of rivaroxaban and clarithromycin. CMAJ. 2016;188(9):669–72.
Hill K, Sucha E, Rhodes E, Carrier M, Garg AX, Harel Z, et al. Risk of hospitalization with hemorrhage among older adults taking clarithromycin vs azithromycin and direct oral anticoagulants. JAMA Intern Med. 2020;180(8):1052–60.
Katzenmaier S, Markert C, Mikus G. Proposal of a new limited sampling strategy to predict CYP3A activity using a partial AUC of midazolam. Eur J Clin Pharmacol. 2010;66(11):1137–41.
Katzenmaier S, Markert C, Riedel KD, Burhenne J, Haefeli WE, Mikus G. Determining the time course of CYP3A inhibition by potent reversible and irreversible CYP3A inhibitors using A limited sampling strategy. Clin Pharmacol Ther. 2011;90(5):666–73.
Holm J, Mannheimer B, Malmstrom RE, Eliasson E, Lindh JD. Bleeding and thromboembolism due to drug-drug interactions with non-vitamin K antagonist oral anticoagulants-a Swedish, register-based cohort study in atrial fibrillation outpatients. Eur J Clin Pharmacol. 2021;77(3):409–19.
Burhenne J, Halama B, Maurer M, Riedel KD, Hohmann N, Mikus G, et al. Quantification of femtomolar concentrations of the CYP3A substrate midazolam and its main metabolite 1'-hydroxymidazolam in human plasma using ultra performance liquid chromatography coupled to tandem mass spectrometry. Anal Bioanal Chem. 2012;402(7):2439–50.
Foerster KI, Huppertz A, Muller OJ, Rizos T, Tilemann L, Haefeli WE, et al. Simultaneous quantification of direct oral anticoagulants currently used in anticoagulation therapy. J Pharm Biomed Anal. 2018;148:238–44.
Garonzik S, Byon W, Myers E, Li X, Marchisin D, Murthy B. The effects of clarithromycin on the pharmacokinetics of apixaban in healthy volunteers: a single-sequence crossover study. Am J Cardiovasc Drugs. 2019;19(6):561–7.
Parasrampuria DA, Mendell J, Shi M, Matsushima N, Zahir H, Truitt K. Edoxaban drug-drug interactions with ketoconazole, erythromycin, and cyclosporine. Br J Clin Pharmacol. 2016;82(6):1591–600.
Frost CE, Byon W, Song Y, Wang J, Schuster AE, Boyd RA, et al. Effect of ketoconazole and diltiazem on the pharmacokinetics of apixaban, an oral direct factor Xa inhibitor. Br J Clin Pharmacol. 2015;79(5):838–46.
Cheong EJY, Ng DZW, Chin SY, Wang Z, Chan ECY. Application of a physiologically based pharmacokinetic model of rivaroxaban to prospective simulations of drug-drug-disease interactions with protein kinase inhibitors in cancer-associated venous thromboembolism. Br J Clin Pharmacol. 2022;88(5):2267–83.
Evangelista EA, Kaspera R, Mokadam NA, Jones JP 3rd, Totah RA. Activity, inhibition, and induction of cytochrome P450 2 J2 in adult human primary cardiomyocytes. Drug Metab Dispos. 2013;41(12):2087–94.
Zhao T, Chen Y, Wang D, Wang L, Dong P, Zhao S, et al. Identifying the dominant contribution of human cytochrome P450 2 J2 to the metabolism of rivaroxaban, an oral anticoagulant. Cardiovasc Drugs Ther. 2022;36(1):121–9.
Wang L, Zhang D, Raghavan N, Yao M, Ma L, Frost CE, et al. In vitro assessment of metabolic drug-drug interaction potential of apixaban through cytochrome P450 phenotyping, inhibition, and induction studies. Drug Metab Dispos. 2010;38(3):448–58.
Herink MC, Zhuo YF, Williams CD, DeLoughery TG. Clinical management of pharmacokinetic drug interactions with direct oral anticoagulants (DOACs). Drugs. 2019;79(15):1625–34.
Chang SH, Chou IJ, Yeh YH, Chiou MJ, Wen MS, Kuo CT, et al. Association between use of non-vitamin K oral anticoagulants with and without concurrent medications and risk of major bleeding in nonvalvular atrial fibrillation. JAMA. 2017;318(13):1250–9.
Harskamp RE, Teichert M, Lucassen WAM, van Weert H, Lopes RD. Impact of polypharmacy and p-glycoprotein- and CYP3A4-modulating drugs on safety and efficacy of oral anticoagulation therapy in patients with atrial fibrillation. Cardiovasc Drugs Ther. 2019;33(5):615–23.
Li A, Li MK, Crowther M, Vazquez SR. Drug-drug interactions with direct oral anticoagulants associated with adverse events in the real world: a systematic review. Thromb Res. 2020;194:240–5.
Gronich N, Stein N, Muszkat M. Association between use of pharmacokinetic-interacting drugs and effectiveness and safety of direct acting oral anticoagulants: nested case-control study. Clin Pharmacol Ther. 2021;110(6):1526–36.
Pham P, Schmidt S, Lesko L, Lip GYH, Brown JD. Association of oral anticoagulants and verapamil or diltiazem with adverse bleeding events in patients with nonvalvular atrial fibrillation and normal kidney function. JAMA Netw Open. 2020;3(4):e203593.
Moore KT, Vaidyanathan S, Natarajan J, Ariyawansa J, Haskell L, Turner KC. An open-label study to estimate the effect of steady-state erythromycin on the pharmacokinetics, pharmacodynamics, and safety of a single dose of rivaroxaban in subjects with renal impairment and normal renal function. J Clin Pharmacol. 2014;54(12):1407–20.
Mayhew BS, Jones DR, Hall SD. An in vitro model for predicting in vivo inhibition of cytochrome P450 3A4 by metabolic intermediate complex formation. Drug Metab Dispos. 2000;28(9):1031–7.
Eberl S, Renner B, Neubert A, Reisig M, Bachmakov I, Konig J, et al. Role of p-glycoprotein inhibition for drug interactions: evidence from in vitro and pharmacoepidemiological studies. Clin Pharmacokinet. 2007;46(12):1039–49.
Seithel A, Eberl S, Singer K, Auge D, Heinkele G, Wolf NB, et al. The influence of macrolide antibiotics on the uptake of organic anions and drugs mediated by OATP1B1 and OATP1B3. Drug Metab Dispos. 2007;35(5):779–86.
Zhang D, He K, Raghavan N, Wang L, Mitroka J, Maxwell BD, et al. Comparative metabolism of 14C-labeled apixaban in mice, rats, rabbits, dogs, and humans. Drug Metab Dispos. 2009;37(8):1738–48.
Bathala MS, Masumoto H, Oguma T, He L, Lowrie C, Mendell J. Pharmacokinetics, biotransformation, and mass balance of edoxaban, a selective, direct factor Xa inhibitor, in humans. Drug Metab Dispos. 2012;40(12):2250–5.
Parasrampuria DA, Truitt KE. Pharmacokinetics and pharmacodynamics of edoxaban, a non-vitamin K antagonist oral anticoagulant that inhibits clotting factor Xa. Clin Pharmacokinet. 2016;55(6):641–55.
Ferrero JL, Bopp BA, Marsh KC, Quigley SC, Johnson MJ, Anderson DJ, et al. Metabolism and disposition of clarithromycin in man. Drug Metab Dispos. 1990;18(4):441–6.
McClaine RJ, Husted TL, Hebbeler-Clark RS, Solomkin JS. Meta-analysis of trials evaluating parenteral antimicrobial therapy for skin and soft tissue infections. Clin Infect Dis. 2010;50(8):1120–6.
Mikkaichi T, Yoshigae Y, Masumoto H, Imaoka T, Rozehnal V, Fischer T, et al. Edoxaban transport via P-glycoprotein is a key factor for the drug's disposition. Drug Metab Dispos. 2014;42(4):520–8.
Rengelshausen J, Goggelmann C, Burhenne J, Riedel KD, Ludwig J, Weiss J, et al. Contribution of increased oral bioavailability and reduced nonglomerular renal clearance of digoxin to the digoxin-clarithromycin interaction. Br J Clin Pharmacol. 2003;56(1):32–8.
Fenner KS, Troutman MD, Kempshall S, Cook JA, Ware JA, Smith DA, et al. Drug-drug interactions mediated through P-glycoprotein: clinical relevance and in vitro-in vivo correlation using digoxin as a probe drug. Clin Pharmacol Ther. 2009;85(2):173–81.
Mikus G, Foerster KI, Schaumaeker M, Lehmann ML, Burhenne J, Haefeli WE. Application of a microdosed cocktail of 3 oral factor Xa inhibitors to study drug-drug interactions with different perpetrator drugs. Br J Clin Pharmacol. 2020;86(8):1632–41.
Funding
Open Access funding enabled and organized by Projekt DEAL. The Department of Clinical Pharmacology and Pharmacoepidemiology received financial research support of Daiichi Sankyo Europe GmbH to conduct this trial.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. The study protocol was written by Alexander Lenard, Antje Blank, and Walter E. Haefeli. The study was conducted by Alexander Lenard, Simon A. Hermann, Felicitas Stoll, and Antje Blank. Measurements of drug plasma concentrations were performed by Kathrin Foerster and Juergen Burhenne. Statistical analysis was performed by Alexander Lenard, Antje Blank, Gerd Mikus, and Andreas D. Meid. The first draft of the manuscript was written by Alexander Lenard, and all authors substantially contributed to the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics Approval
The study protocol was approved by the competent authority (BfArM, Bonn, Germany, Vorlagen Nr. 4043377) and received a positive vote of the responsible Ethics Committee of the Medical Faculty of Heidelberg University, Germany (AFmo-144/2019), and the trial was registered in the EudraCT database (EudraCT 2018-002490-22). This phase I, investigator-initiated, monocenter trial was conducted in accordance with the Declaration of Helsinki, the principles of Good Clinical Practice, and all pertinent legal requirements at the DIN EN ISO 9001-certified early clinical trial unit KliPS of the Department of Clinical Pharmacology and Pharmacoepidemiology, Heidelberg University Hospital.
Consent to Participate
Written informed consent was obtained from all individual participants included in the study.
Consent for Publication
No individual patient data is published in this manuscript.
Competing Interests
Walter E. Haefeli declares to have received grants, travel support, and lecture fees from Daiichi Sankyo not related to this work. All other authors declare they have no financial interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lenard, A., Hermann, S.A., Stoll, F. et al. Effect of Clarithromycin, a Strong CYP3A and P-glycoprotein Inhibitor, on the Pharmacokinetics of Edoxaban in Healthy Volunteers and the Evaluation of the Drug Interaction with Other Oral Factor Xa Inhibitors by a Microdose Cocktail Approach. Cardiovasc Drugs Ther 38, 747–756 (2024). https://doi.org/10.1007/s10557-023-07443-2
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10557-023-07443-2