
Abstract
We have established considerable expertise in studying the role of platelets in cancer biology. From this expertise, we were keen to recognize the numerous venous-, arterial-, microvascular-, and macrovascular thrombotic events and immunologic disorders are caused by severe, acute-respiratory-syndrome coronavirus 2 (SARS-CoV-2) infections. With this offering, we explore the evolutionary connections that place platelets at the center of hemostasis, immunity, and adaptive phylogeny. Coevolutionary changes have also occurred in vertebrate viruses and their vertebrate hosts that reflect their respective evolutionary interactions. As mammals adapted from aquatic to terrestrial life and the heavy blood loss associated with placentalization-based live birth, platelets evolved phylogenetically from thrombocytes toward higher megakaryocyte-blebbing-based production rates and the lack of nuclei. With no nuclei and robust RNA synthesis, this adaptation may have influenced viral replication to become less efficient after virus particles are engulfed. Human platelets express numerous receptors that bind viral particles, which developed from archetypal origins to initiate aggregation and exocytic-release of thrombo-, immuno-, angiogenic-, growth-, and repair-stimulatory granule contents. Whether by direct, evolutionary, selective pressure, or not, these responses may help to contain virus spread, attract immune cells for eradication, and stimulate angiogenesis, growth, and wound repair after viral damage. Because mammalian and marsupial platelets became smaller and more plate-like their biophysical properties improved in function, which facilitated distribution near vessel walls in fluid-shear fields. This adaptation increased the probability that platelets could then interact with and engulf shedding virus particles. Platelets also generate circulating microvesicles that increase membrane surface-area encounters and mark viral targets. In order to match virus-production rates, billions of platelets are generated and turned over per day to continually provide active defenses and adaptation to suppress the spectrum of evolving threats like SARS-CoV-2.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Offerings from cancer expertise to understanding platelet evolution
We have acquired an informed perspective and extensive expertise in the field investigating the role of platelets in cancer biology [1,2,3,4,5,6,7,8,9,10]. One of the key discoveries that led to a better understanding of the links between coagulation and cancer was that of Trousseau’s syndrome, which is defined as a migratory thrombophlebitis found typically in patients with an underlying malignancy. Among other discoveries throughout the years, one of the key discoveries linking platelets to tumor cell interactions was made by Gasic et al. [11]. Our efforts in this field greatly expanded the knowledge base regarding platelet-tumor cell interactions and the role of prostaglandins in particular during cancer metastasis [2, 12]. Fast forward, this study is for expanding our current knowledge base in identifying unique aspects of platelet function and platelet biology in the growth and progression of ovarian cancer [4, 10]. It is from this unique cancer-centric perspective that we recognized that the numerous venous-, arterial-, microvascular-, and macrovascular thrombotic events and immunologic disorders are caused by severe, acute-respiratory syndrome coronavirus 2 (SARS-CoV-2) infections. This perspective led us to examine the evolutionary connections placing platelets at the epicenter of hemostasis, immunity, and adaptive phylogeny. All of these novel perspectives may provide new leads to better understand not only the role of platelets in vascular protection and immune function but also a keener insight into the role of platelets in cancer and metastasis.
2 Viral and platelet evolution
2.1 Single-stranded RNA virus evolution
Positive sense, single-stranded RNA viruses are thought to be among the earliest forms of life; lineages have been traced through the conservation of RNA-dependent RNA polymerases within the picornavirus super-group [13,14,15,16]. Over the past century, multiple single-stranded RNA viruses, including human immunodeficiency virus-1, influenza A, and severe, acute-respiratory-syndrome coronavirus 2 (SARS-CoV-2), have emerged to infect humans on a large scale—a modern consequence of ancient virus evolution [17, 18]. Endogenous retroviruses help to record past RNA retroviral infections and are ubiquitous in vertebrate genomes [19]. Coronaviruses are among the class of positive-strand RNA viruses that infect mammals and birds and can achieve zoonosis [20,21,22]. Evidence of a SARS-like coronavirus arising in bats shows zoonotic implications of a direct ancestor of SARS coronavirus [23, 24]. Bats can carry viruses that are deadly to other mammals without having serious symptoms, possibly involving unique immune- and rapid cellular-spreading properties [25]. During the current SARS-CoV-2 pandemic, key viral mutations may have also occurred via person-to-person transfer complicated by zoonotic passage, highlighting the rapid evolution of this virus [26,27,28,29,30,31,32]. Overall, the phylogeny of vertebrate RNA viruses reflects that of their vertebrate hosts coevolving with a transition from ocean-to-land and exhibits origins that date back to the time of early terrestrial animals [18, 33, 34].
2.2 Phylogenetic co-evolution of platelets in adapting to viral pathogens
Many aspects of platelet biology and evolution remain unrecognized, especially regarding their rapid responsiveness to viruses [35,36,37]. Platelets may have coevolved with viruses to provide speedy responses but this possibility remains unexplored. However, platelets have been shown to endocytose viral particles and are activated via toll-like receptor signaling [38]. The severity of low-platelet count (thrombocytopenia), a common complication of influenza-virus infection, predicts the clinical outcome of critically ill patients [39]. These platelet responses are biochemically and biologically rapid and may also involve integrin receptors [40]. In the case of viral diseases like COVID-19, the responses may remain unnoticed during asymptomatic and early phases [41, 42]. As there is an expression gradient of entry factors for SARS-CoV-2 along the airways, these are the likely sites of initial viral infection, spread, and clearance [43]. General symptoms can consist of cough, myalgia, loss of appetite, and gastrointestinal discomfort. Olfactory and gastrointestinal disorders are prevalent with sudden anosmia (loss of smell), ageusia (loss of taste), or dysgeusia (metallic taste), but not always fever, as informative symptoms of the COVID-19 infection [44,45,46,47,48]. Given the one-to-two, cell-layer separation between many airway passages and the microcirculation coupled with the evolutionary characteristics of platelets, this suggests that they are the first thrombocyte/immunocyte that virions such as SARS-CoV-2 may encounter and thereby rapidly respond to in the bloodstream [38, 49]. For example, with the human immunodeficiency virus, platelets have been shown to reduce viral load [50] and harbor viral particles [51].
2.3 Early evolutionary ties between innate immunity and coagulation
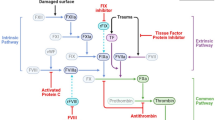
Coincident activation of innate immunity and the coagulation system after an injury is a phylogenetically ancient, adaptive response that can be traced back to the early stages of eukaryotic and chordate evolution [52,53,54,55,56,57]. Most invertebrate species like the horseshoe crab (Limulus polyphemus) lack differentiated phagocytic cells and platelets, but they do have large nucleated amoeboid granular hemocytes [58,59,60] (Fig. 1). Once their open circulatory system is breached, they respond to microbial threats via a common amoebocyte- and humoral pathway of inflammation and clotting [58,59,60]. Lysates prepared from the amebocytes of Limulus have long been known to undergo coagulation upon exposure to endotoxin and have been used to detect that endotoxin [61]. Archetypal functions of Limulus amebocytes are found in human platelets, including aggregation, adhesion, spreading, and granule-based release of coagulation factors [62,63,64]. These properties are found throughout phylogenetic evolution as common antimicrobial and proinflammatory responses to stimuli that activate both the clotting cascade and the phagocytic effector cells. Non-mammalian vertebrates—that include early chordates, fish, birds, amphibians, and reptiles—have large, nucleated, often spindle-shaped thrombocytes that mediate thrombocytic activity [36, 54,55,56,57, 65].

Platelet evolution. Thrombocyte and platelet evolution remain at the center of both hemostasis and immunity. Depicted are species and thrombocyte/platelet reproductions where transmission electron micrography-, morphological-, and functional data were available. Invertebrate thrombocytes of the horseshoe crab (Limulus polyphemus) are typically large amoeboid cells. Thrombocytes gradually became reduced in size, more elongated, and retained nuclei throughout the evolution of (1) the chordate and aquatic vertebrates such as agnatha or jawless fishes like the hagfish (Myxine glutinosa), (2) the cartilaginous or Chondrichthyes fish such as the dogfish shark (Squalus acanthias), and (3) the boney or teleost fish such as zebrafish (Danio rerio). As vertebrates adapted to terrestrial life, these trends continued in amphibians such as the green frog (Rana clamitans), reptiles like crocodiles (Crocodylus porosus), and even avian forms such as the chicken (Gallus gallus). Unique to mammals and marsupials, platelets dramatically decreased in size and developed improved biophysical properties, including the elimination of nuclei
2.4 Platelet adaptation through decreasing size and losing nuclei
Throughout evolution, the cell size and dimensions of thrombocyte nuclei have gradually decreased; this process accelerated as species became more terrestrial and began to thermoregulate (Fig. 1). In the jawless fish-like Hagfish (Myxine glutinosa) [66, 67], cartilaginous fish-like sharks (Squalus acanthias) [68, 69], boney fishes like Zebrafish (Danio rerio)[70], and other fish species, circulating nucleated thrombocytes have some phenotypic features and functional characteristics similar to those of mammalian platelets [71, 72]. As aquatic animals like the lungfish (Neoceratodus forsteri, Lepidosiren paradoxa) adapted to terrestrial life, thrombocytes remained large and retained large nuclei, while developing a sophisticated granule system to effectively regulate and maintain coagulation and immune functions, particularly during the dramatic changes associated with dry-season-based estivation or dormancy [73, 74]. Similar morphological associations found in thrombocytes of other non-mammalian vertebrates were retained in amphibians such as the green frog (Rana clamitans) [75], reptiles such as crocodiles (Crocodylus porosus) [75, 76], and birds such as the chicken (Gallus gallus) [76, 77]. In contrast, mammals [78] and marsupials have developed various essential evolutionary changes during the processes of erythroid and thrombocytic development and differentiation. Examples include megakaryocyte endoreduplication and platelet enucleation, each involving a reduction in size and improvements in functional biophysical morphology [36, 76, 79]. Mammals have also developed more intricate associations between platelets and other immune cells (neutrophils, monocytes, and macrophages) to coordinate innate immunity and the coagulation system [53]. From an evolutionary perspective directed at defending against viral threats, platelet adaptations may be an effective way to improve host responses. Platelets are important anucleate elements of the immune and hemostatic systems that have minimal nucleotide- or protein synthesis machinery to indefinitely make new gene products [80,81,82]; their ability to reproduce viral particles is limited or has been shown for only a few single-stranded-RNA viruses like Dengue virus and potentially SARS-CoV-2 [83,84,85]. Some viral replication may be excluded by restricting gene expression to a pre-existing, limited subset of ribosome-bound messenger RNAs that engage the ribosome rescue-factor pelota (PELO) in regulating messenger-RNA decay [86]. The possibility also exists that viral replication may occur in megakaryocytes within the bone marrow prior to their budding from parent cells, but this has not been fully explored [87, 88]. Although unrecognized as part of platelet evolutionary biology to date [36, 37], a more important function of platelets in mammals may be the lack of nuclei and limited replication machinery to support long-term viral replication, providing a more highly evolved defense and, potentially, a sink against these infections.
The vertebrate transition from aquatic to terrestrial environments applied significant selective pressure to the co-evolution of viruses and animals. This was accompanied by multiple geophysical, external, selective pressures, which include changes in luminosity, oxygen, carbon dioxide, and asteroid-based impacts (Fig. 2). Co-evolution is a complex interaction of co-divergence between virus and host over an extended time and includes frequent cross-species transmissions coupled with external survival pressures severe enough to elicit the change [18, 33]. In this manner, SARS-CoV-2 evolution has accelerated in conjunction with zoonotic exchanges with humans over the past 100 years (on the time scale in Fig. 2, this would be approximately the size of a period).

Evolutionary timeline. Using available transmission electron micrography- and morphological data from the literature, a nodal evolutionary tree for the species listed in Fig. 1 was constructed using the Time Tree Website, http://www.timetree.org. A species list was generated as a text file list for the following: Limulus polyphemus, Myxine glutinosa, Squalus acanthias, Danio rerio, Rana clamitans, Crocodylus porosus, Gallus gallus domesticus, and Mammals/Homo sapiens. Egg-laying mammalian evolution began 220 MYA, placental-Eutherian evolution occurred 125 MYA and human evolution began 2.5 MYA. This file was then uploaded as a list file. Time: million years ago, MYA Periods: Cambrian, C:542.0–488.3; Ordovician, O: 488.3–443.7; Silurian, S:443.70–416.0; Devonian, D: 416.0–359.2; Mississippian, MIS: 359.2–318.1; Pennsylvanian, PEN: 318.10–299.0; Permian, P: 299.0–251.0; Triassic, Tr: 251.0–199.6; Jurassic, J:199.6–145.5; Cretaceous, K:145.50–65.50; Paleogene, Pg:65.50–23.03; Neogene, N:23.03–0.0. Eras: Neo-Proterozoic: 1000.0–542.0; Paleozoic: 542.0–251.0; Mesozoic: 251.0–65.5; Cenozoic, Cz: 65.50–0.0–542.0. Eons: Proterozoic: 2500.0–542.0; Phanerozoic: 542.0–0.0. Earth impacts: Sphere size equals the diameter of the impact (e.g., Chicxulub, location: Yucatan, Mexico, coordinates: 21.333, − 89.50, diameter: 150.0 km, age: 65 MYA)
3 Adaptive characteristics
The evolution of platelets includes multiple adaptations to deal with viral infections. As one evolutionary mechanism for dealing with viral pathogens, large, multinucleated megakaryocytes evolved in the bone marrow to protect and enable the production of high numbers of platelets [89,90,91]. These mechanisms have avian roots and enabled eutherian placentalization [92, 93]. There is another significant advantage over the 2-N thrombocytes of lower vertebrates found in mammalian megakaryocytes. Significantly enlarged megakaryocytes provide a major amplification response to bleeding or viral immune threats incurred by increasing DNA content up to 128 N [93] No studies have yet determined whether these megakaryocytes serve and monoclonal source of targeted platelets responding to a specific threat. The adaptation of increasing DNA content significantly increases the capacity to produce even more platelets with increased receptor density; more organelles per unit cellular volume; and more prothrombotic proteins to reduce bleeding time or adapt to viral threats. This process culminates in mammalian platelet evolution and hemochorial placentation because mammals are the only vertebrate group that has evolved with a highly effective and unique system of hemostasis [93]. This hemostasis effectiveness is essential at parturition where even minimally invasive placentae can hemorrhage [93]. Platelet evolution has enabled the prevention of rapid blood loss and damage from massive injury or microinjury, which is particularly important when the brain is more advanced. Because microbes and viruses have a short ten-day life span, platelets must have co-evolved to create survival advantages with adaptive mechanisms to meet and sequester rapidly cycled microbial- and viral threats and pathogen response-based turnover.
The sheer number and biophysical properties of megakaryocyte-derived mammalian platelets make them more likely to be the first blood component to encounter virus particles and limit spread. Human platelet genesis is well studied and primarily occurs by membrane-bound, organelle-containing cytoplasmic extrusion into the blood stream of numerous small size- and volume blebs ranging between 9.7 and 12.8 femtoliters or in plate-like structures of 2.6 to 2.9 μm in diameter [90]. This blebbing, subcellular, genesis process occurs primarily in the bone marrow from surfaces of megakaryocytes—the largest cell in the body (ranging between 40 and 100 μm in diameter). Normal human-platelet concentrations range between 150,000 and 400,000 per microliter. The roughly 1011 platelets produced each day are responsible for coagulation—specifically for creating a fibrin plug at the site of blood vessel injury. However, emerging data suggest a greater impact on viral interdiction, pathogen surveillance, cancer biology, and immune function. As part of the anti-viral response, platelets are known to actively engage and directly phagocytose viral pathogens [94,95,96,97,98,99,100,101,102,103,104,105,106]. Note that this ability of nucleated thrombocytes to phagocytose pathogens is also present in lower vertebrates [107], which also exhibit an extensive open-canalicular system [72]. Platelets have been shown to endocytose viral particles and become activated via toll-like receptor (TLR) signaling [38]. Mammalian virus uptake may also involve the platelet open-canalicular system that exchanges factors with the surrounding microenvironment [95, 97, 108,109,110,111]. Once platelets take in virus particles, they may then undergo phagocytosis by other immune cells[106]. Platelet surface changes that occur after taking in viral particles can also mark the platelets for clearance by other organs including the liver and spleen.
3.1 Platelet formation of plate-like structures and wall shear biophysics
Other key aspects of adaptive evolution in the mammalian platelet are size decreases and improvements in biophysical shape properties to enhance encounters with virus-infected vascular surfaces. In lower vertebrates, nucleated thrombocytes are less plate-like and10 to 20 times larger than platelets causing them to primarily distribute in the center of laminar shear fields [36, 70, 72] (Fig. 1). In humans, resting platelets are plate-like discs designed to maximize planar-surface interactions[112,113,114] that, due to their small size and shape, biophysically concentrate toward the outer fluid-shear fields of flowing blood [115,116,117,118,119,120,121,122]. Taken together, these properties enhance encounters and recognition of any vascular-wall lesions, wounds, or tumors. Should platelets encounter basement membranes or their underlying matrix, the platelets undergo receptor-mediated activation [2, 123,124,125,126,127]. This activation is connected to rapid cytoskeletal- and membrane changes to form filopodia leading to adhesion [128,129,130,131,132,133,134,135]. This is a process that occurs within seconds along with a shape change from exocytic degranulation. Degranulation, in turn, encompasses the release of proteins, growth factors, cytokines, and lysozymes along with a variety of bioactive lipids, small molecules, and other factors. Successful sequential recruitment of additional platelets and immune cells coupled with thrombogenesis ultimately seals any tissue gaps and sterilizes the lesion. Since platelets segregate within shear fields near the endothelial cell surfaces, they have a high probability of encountering viral particles that enter the bloodstream. This behavior also affords platelet endothelial-cell interactions that can occur with an aggressive, influenza-like cytokine storm that triggers cell damage in lung capillaries [136].
3.2 Platelet microvesicle production and increased membrane surface area
Compared to any other form of circulating microvesicle, exocytic platelets generate the majority of microparticles and exosomes in circulation [137,138,139,140,141,142]—possibly another form of platelet evolution to deal with viral threats. In lower vertebrates, microparticle generation by thrombocytes plays a key role in thrombocyte function [70]. During laser-injury experiments in zebrafish, thrombocyte microparticles were the first cell-based entities to arrive at the injury site, even before the young thrombocytes [143, 144]. In the case of viral infections like SARS-CoV-2, platelet microparticle and exosome shedding would significantly increase the platelet-membrane surface area and possibly mitigate the threat potential through target decoration and further immune-system stimulation[145].
3.3 Tissue migration
A phylogenetic adaptive loss of platelet nuclei may also enable rapid endothelial transmigration into perivascular spaces. Platelets readily undergo tissue migration because of their small size, minimal displacement volume, and highly active cytoskeletal responses linked to activation, adhesion, and aggregation [146,147,148]. Even greater is the vascular-transmigration advantage of being unencumbered by the presence of nuclei, which limit the migration of other immune cells to interdict viral threats [146,147,148,149,150]. This nuclei absence may also limit the distance that platelets can move into tissue, which limits nucleotide and protein-synthesis capabilities. Due to limited nucleotide and protein-synthesis capabilities, platelets cannot indefinitely maintain protein replacement [80, 151]. These immune-surveillance and rapid viral-response properties facilitate speedy sterilization and quickly initiate the repair process during a virus-induced, tissue-injury response. With viral infections, platelets have been shown to influence viral invasion of the central nervous system in canine distemper-virus infection [152, 153]; however, few studies exist that examine platelet invasion or extravasation as part of a viral immune response. Alternatively, because viral infection may lead to endothelial damage and platelet extravasation, activation and granule release may promote the attraction of other immune cells that produce inflammation in the affected tissues.
Various factors in response to various stimuli can contribute to platelet transmigration across leaky or inflamed vessel walls to aid in wound sterilization and tissue regeneration [154,155,156,157,158,159,160]. Platelet inflammatory responses can also cause them to undergo vascular transmigration and granule release that engages various additional immune cells [161,162,163]. Within the extravascular microenvironment, the release of the stromal cell-derived factor-1 named C-X-C chemokine-ligand 12 (CXCL12), along with other cytokines, can also provide a potent stimulus for platelet migration into extravascular spaces [10, 150, 164,165,166,167] [168,169,170,171]. Among the many response factors expressed by platelets are C-X-C chemokine-receptors 4 and 7 (CXCR-4, CXCR7), which are the cognate receptors for CXCL12. These platelet receptors may be involved in inflammatory- or allergic responses or in platelet activation in human immunodeficiency virus and other viral infections [172]. Along with those involving lymphatic biology, these thrombo-immune responses and wound healing characteristics were conserved from lower vertebrates [107, 173,174,175,176,177] [178].
4 Virus-induced platelet activation and thrombotic complications
Cyclooxygenase and prostaglandin production, associated with gene duplications and losses, have been retained throughout metazoan and vertebrate evolution [179, 180] that began in teleosts and resulted in a divergence between cyclooxygenase-1 and cyclooxygenase cyclooxygenase-2 (COX-1 and COX-2) [180]. A release of the COX-2-associated prostacyclin PGI2 from vascular endothelial cells helps to maintain the resting state of circulating platelets through G-protein-coupled receptors (GPCR) [181,182,183,184,185] (Fig. 3). Should platelets become activated during the virus uptake, responses are rapidly amplified by activating molecules such as thrombin or adenosine diphosphate (ADP). These molecules are released by platelets and trigger their cognate GPCRs at the cell surface or as a result of the release reaction. Similarly, COX-1-associated and endogenously synthesized thromboxane A2 (TXA2) amplifies the local platelet response to recruit additional platelets through TXA2-GPCRs that accelerate the thrombogenesis/clotting process (Fig. 4). Platelet TXA2 levels increase following virus infections and can be attenuated by aspirin treatment [186,187,188,189,190]. The production of TXA2 and other lipids initiates vasoconstriction [191, 192]. As a primary stimulant of platelet recruitment and aggregation, TXA2 is among the shorter-lived prostaglandins due to molecular epoxide bond strain in the active part of the molecule that is prone to rapid (< 30 s) hydrolysis [193, 194]. Because these rapid changes accelerate over a matter of minutes, a fibrin network is generated to trap and activate more platelets within the forming clot in a cyclic fashion to limit blood loss. This amplification process occurs over 20 min or less, while clot maturation and solidification continue for about an hour to initiate—over days to weeks—prompting immune-cell recruitment and inflammation. Recruitment of fibroblasts and immune cells through platelet-initiated, angiogenic repair mechanisms stimulates further wound healing and resolution [195].

Platelet responses to SARS-CoV-2. Platelet responses to viruses include activation, aggregation, and granule release. Dense δ-granules contain bioactive small molecules including thromboxane A2 (TxA2), adenosine diphosphate (ADP), serotonin (5-hydoxytryptamine [5HT]), and histamine. The α-granule contains numerous bioactive proteins involved in thrombus formation, coagulation, and aggregation including tissue factor and thrombin. It also contains numerous bioactive molecules involved in immune regulation and inflammation including Cys-X-Cys (C-X-C) motif chemokines such as CXCL1 (GRO1 oncogene [GRO-α]), CXCL4 (platelet factor 4 [PF4]), CXCL5 (ENA-78), CXCL7 (β-thromboglobulin [β-TG], platelet binding protein [PBP], CTAP-III, and NAP-2), CXCL8 (interleukin-8 [IL-8]), and CXCL12 (stromal cell-derived factor-α [SDF-1α]). Platelet α-granules also contain Cys-Cys (C–C) motif chemokines that include CCL2 (monocyte chemoattractant protein 1 [MCP-1]), CCL3 (macrophage inflammatory protein [MIP-1α]), CCL5 (regulated on activation, normal T-cell expressed and secreted [RANTES]), CCL7 (MCP-3), and CCL17 (thymus and activation regulated chemokine [TARC]), along with interleukins IL1-β and IL-6, platelet-activating factor [PAF], defensins, and complement factors C3a and C5a. Alpha-granule release factors also include growth factors such as vascular endothelial-cell growth factor (VEGF), platelet-derived growth factor (PDGF), and angiopoietin

SARS-CoV-2 replication and platelet receptor interactions. At the top, virus replication within vascular endothelial cells is initiated by SARS-CoV-2 spike-protein binding to angiotensin-converting enzyme (ACE) and enzymatic processing by transmembrane protease, serine 2 (TMPRSS2). Viral uptake results in viral-particle uncoating, viral reverse transcriptase activity of the positive single-stranded (+ ssRNA) genome, and replication. The endoplasmic reticulum of the host cell supports protein synthesis, assembly in the Golgi apparatus, and delivery to the exterior by exosomal transport or host-cell disruption, death, and retraction. The platelet depicted at the center illustrates the numerous receptors that bind to various virus types. Those that bind coronavirus species are indicated by larger-font text and include (alphabetically) coxsackie-adenovirus receptor (CAR), CCL chemokine Cys-Cys (C–C motif) ligand, cluster of differentiation (CD), C-type lectin domain family 2 and 5 (CLEC-2 and CLEC-5), complement receptor (CR 3a and CR 5a), Cys-X-Cys (C-X-C) motif chemokine receptor type (CXCR1, CXCR1-2, and CXCR1-4), dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin (DC-SIGN), Fc gamma receptor II (FcγRII), heparan sulfate proteoglycan (HSP), integrin (αvβ3), and toll-like receptor (TLR). Also depicted are the phosphate-transferring nucleotide-binding domain-like receptor protein 3 (NRLP3), the inflammasome-response switch serine/threonine-protein kinase (NEK7), and the pro-inflammatory cytokine caspase 1 that mediates the enzymatic processing of interleukin-1β (IL-1β) prior to microvesicle release. Also shown are the large numbers of G-protein-coupled receptors involved in platelet signal transduction, activation, and granule release
4.1 Granule release
The release of dense, δ-granule components influences the vascular microenvironment of the platelet (Fig. 3). The release of calcium and magnesium ions promotes platelet activation and aggregation. The release of nucleotides such as ADP activates platelets through P2Y1 and P2Y12 receptors [196, 197] and stimulates vasoconstriction [198] (Fig. 4). ADP can accumulate in the ischemic vascular microenvironment as a consequence of circulatory stasis or thrombus formation. The release of neurotransmitters such as serotonin (5-hydroxytryptamine), epinephrine, and histamine can potentiate ADP-induced platelet activation and aggregation [196, 197, 199, 200]. Platelets may serve as neuronal and innate immune cells to help mediate T cells during tissue inflammation [201]. Epinephrine is also likely to worsen COVID-19-induced stress-related effects on platelets and platelet-mediated cytokine responses [202]. By contrast, lower-vertebrate thrombocytes do not aggregate in response to ADP or epinephrine; neither do they accumulate or produce serotonin [65, 203].
Conversely, platelet release of alpha (α)-granules in serum results in substantial increases in multiple proteins responsible for most of the coagulation, growth stimulation, angiogenesis, immune function, and wound-repair factors. Adhesion molecules released from α-granules help to stimulate the rapid arrest of platelets in circulation. This is a process that is mediated by adhesion factors such as the von Willebrand factor (vWF), fibrinogen, thrombospondin, and fibronectin as well as integrins and other adhesion receptors αIIbβ3, αvβ3, and P selectin. These adhesion proteins regulate platelet interactions with other platelets, and with endothelial cells, exposed basement-membrane extracellular matrix, leukocytes, neutrophils, monocytes, and tumor cells. These same storage α-granules release the prothrombin, fibrinogen, factor V, and factor VIII that stimulate and promote coagulation and fibrin formation [3, 9, 204,205,206,207,208,209,210,211,212].
4.2 Viral infection of endothelial cells
Endothelial cells express angiotensin-converting enzyme (ACE) and transmembrane protease, serine 2 (TMPRSS2) that are involved in the SARS-CoV-2 invasion via its spike-surface protein binding and processing [213,214,215,216,217,218] (Fig. 4). After uptake, SARS-CoV-2 undergoes uncoating and viral-RNA reverse transcription and replication in the rough endoplasmic reticulum followed by new SARS-CoV-2 assembly and release (Fig. 4). Should the virus infect endothelial cells, endotheliitis, complement activation, and endothelial-cell death can occur [219,220,221,222,223,224]. Endothelial-cell retraction, whether virus- or inflammation-induced, can lead to matrix exposure that triggers platelet activation and granule release. Blood-vessel injury can amplify platelet responses and may help trigger heightened immune responses during the second phases of COVID-19 infection [219, 222, 225].
Analysis of megakaryocytes in bone marrow data from the Human Cell Atlas did not identify expression of these receptors [226, 227]; however, the detection of ACE2- and TMPRSS2 receptors in platelets requires direct protein-characterization technology, not RNA sequencing, because the platelets lack nuclei and the sequencing technique does not provide adequate sensitivity. As part of their immune-surveillance properties, platelets can recognize foreign bodies or invading pathogens [9, 228,229,230]. Along with other platelet receptors, the toll-like receptors expressed by megakaryocytes [231] may be involved in innate immune responses to viruses, may take part in endocytosis of a variety of pathogens by platelets [38, 232,233,234], and may release factors that can influence antiviral responses [157, 235,236,237,238,239,240,241,242,243,244,245,246] (Fig. 4).
4.3 SARS-CoV-2 infection and platelet recognition
Normally, platelets serve to rapidly respond during the wounding process and hemostasis. When virally damaged, blood-vessel endothelial cells die, are lost, or retract; this exposes platelets to subendothelial elements that initiate receptor-based recognition of the extracellular matrix. These interactions are driven by a variety of platelet glycoprotein receptors that bind to endothelial cells and extracellular matrix factors including proteoglycans, laminin, fibronectin, vitronectin, and various isoforms of collagen. vWF binds to exposed collagen I, collagen III, and collagen VI fibers. vWF is also recruited into matrix networks by forming tethering fiber strands [247,248,249,250,251]. Many vWF characteristics of hemostatic proteins are conserved during ancestral vertebrate evolution, but the hagfish (Myxine glutinosa) jawless vertebrate (a cyclostome) lacks functional domains required for primary hemostasis under high flow [66]. Many of the typical platelet-surface receptors also bind to viruses [34] but the role of platelet binding that may sequester viral particles in the vasculature has not been studied in detail.
Platelets contextually encounter numerous bloodstream-circulating ligands, cells, pathogens, and extracellular-matrix ligands. Immediate platelet responses are governed by a variety of integrin receptors through transmembrane glycoprotein-α and glycoprotein-β heterodimers once ligands are engaged (Fig. 4). Resting platelets express low-affinity conformation integrins that are bent to protect binding sites and to form high-affinity- or open-binding states that, once activated, efficiently interact with ligands. Rolling platelet behavior is initiated by surface-level, multimeric GPIb-complex interactions with vWF that activate a key stabilization integrin αIIbβ3 [252,253,254,255] to bind a variety of RGD (arginine-glycine-aspartic acid)-containing ligands [252,253,254,255] including fibrin, fibrinogen, fibronectin, vitronectin, thrombospondin, or vWF complexes. Platelets also interact with hanta- and adenoviruses through αIIbβ3 receptors [94, 98, 256,257,258,259,260]. Certain integrins bind to echo- and rotaviruses[261] while vitronectin is engaged by platelet αvβ3 integrin heterodimers that also bind to echovirus9; coxsackieviruses A9 and -A16; and hanta-, parecho-, and coronaviruses [98, 256, 257].
Platelet C-type lectin-like receptors CLEC-2 and CLEC-5A initiate platelet activation and trigger signal transduction through molecular multimerization [262,263,264,265,266]. CLEC-2 binds to the transmembrane sialomucin-glycoprotein podoplanin among other molecules [267,268,269]. Podoplanin is expressed on cells of the lymphatic endothelia, type I lung epithelia, choroid plexus epithelia, kidney podocytes, and lymph-node stroma to potentiate migration and invasion [267,268,269,270]. CLEC-2 expression is maintained on activated platelets and on platelet microparticles to substantially increase the probability of platelet-mediated interactions with virus particles [271]. Platelet CLEC-2 and CLEC-5A receptors bind to the human immunodeficiency- and coronaviruses [271,272,273].
Platelet P-selectin, also known as membrane glycoprotein GMP-140 [274, 275], binds to P-selectin glycoprotein ligand 1 (PSGL-1) [276], neutrophil leukocyte-endothelial cell-adhesion molecule 1 (LECAM-1) [277], endothelial cell-leukocyte adhesion-molecule 1 (ELAM-1) [278], and Sialyl Lewis (x) oligosaccharide [279]. Along with a variety of immune cells and endothelial cells, P-selectin interaction is important in mediating inflammation, autoimmunity, and wound healing, and plays a role in coxsackievirus-induced myocarditis [280,281,282,283,284]. P-, L-, and E-selectins help with the tethering and rolling of cells that flow past the vascular luminal surfaces during the initial phases of intravascular, adhesive interactions. These processes are stabilized by other receptors [285,286,287,288,289,290,291] and may contribute to interactions with virus-laden platelets.
4.4 Thrombus initiation
Platelets often serve as foci for coordinating thrombus initiation and formation. Tissue factor initiates platelet activation and aggregation [292] and may be linked to some virus-induced prothrombotic states associated with septic shock, myocarditis, and respiratory viral infections[293,294,295,296]. Thromboemboli and thrombocytopenia can also lead to COVID-19-related complications as reported in a number of publications [297,298,299,300,301,302,303,304,305,306]. In vivo, the viral threat mitigation process is likely to be rapid, as platelets aggregate in response to circulating threats. In the case of platelet interactions with virions such as SARS-CoV-2, intravital observations are unlikely due to the small, average-viral-particle size. Histologically, these changes are difficult to track microscopically due to the lack of nuclei as platelet reference points; the post-activation, morphological platelet changes; and dramatic, aggregation-related shape changes that create an amorphous mass. Rapid thrombocyte/platelet responses have been conserved phylogenetically in vertebrates[307]. Depending on the agonist, the initiation of zebrafish thrombocyte/platelet aggregation occurs within 20 s, plateaus over 1–3 min, and involves conserved αIIb receptor subunits[307]. Thrombotic plug formation is stabilized and counterbalanced by the disaggregation of platelets and fibrinolysis. Platelets migrate and help to initiate immune sterilization, tissue repair, and scar formation [195, 308,309,310,311]. In critically ill patients with a significant viral load, these platelet-mediated immune responses may be more rapid and severe.
5 Consequences of platelet-viral responses
Major and life-threatening events associated with SARS-CoV-2 infections are common. A number of coagulation/thrombotic disorders are linked to COVID-19 infections including stroke, disseminated intravascular coagulation, pulmonary embolisms, acute respiratory distress syndrome (ARDS), sepsis-induced coagulopathy, local microthrombi, venous thromboembolism, arterial thrombotic complications, and thrombo-inflammation connected to disease progress, severity, or mortality[312,313,314,315]. One tragic example in the young is the association between COVID-19-related strokes and amputations [316,317,318,319,320,321,322,323,324,325,326]. Platelets can play an integral role at any stage of coagulation/thrombotic disorders.
Like other viral infections, asymptomatic disease is present in a significant but hard-to-identify fraction of affected individuals. In most symptomatic patients, a 1-week, self-limiting, viral respiratory disease often occurs [221, 327,328,329,330,331,332,333]. This phase of disease may end when neutralizing-, anti-viral-, T-cell-, and antibody immunity is engaged. If cross-reactivity occurs with other serum-based coronavirus antigens, immunoglobulin (Ig)M-, IgA-, and IgG-type virus-specific antibody levels can be important indicators of population immunity against this disease. In the case of extreme viral load during the first infection course or repeated exposure to virus that can occur in healthcare workers, the rate and extent of this response can be an important factor for severity and localization of disease[221, 327,328,329,330,331,332]. These stage- and extent-of-disease responses will influence the development of any high-specificity and a high-accuracy serological assay that is easy to use, reliable, reproducible, and critical to the successful measurement of COVID-19 immunity in our global population [334,335,336,337,338,339,340,341,342,343,344,345,346,347,348,349,350,351,352,353].
5.1 Platelet FcγRIIA (CD32a) receptors
As COVID-19 progression becomes more severe, antibodies that platelet FcγRIIA (CD32a) receptors can recognize may be generated [259, 354] (Fig. 4). FcγRIIA—known to recognize aggregated IgG complexes [355]—can contribute to αIIbβ3 activation and aggregation [356]. Aggregated IgG complexes can trigger the release of microvesicles, as can happen with H1N1-virus (swine flu) exposure [354, 356]. FcγRIIA receptors contribute to the production of COVID-19-related autoantigens also amplified by platelet microparticle release. This may underlie the formation of potent inflammatory components: the microparticle-associated immune complexes [357]. Similarly, the complexes appear to play a role in heparin-induced thrombocytopenia, a prothrombotic disorder mediated by complexes between platelet factor 4 (PF4) and heparin or other polyanions [358]. This risk of thrombosis may extend beyond exposure to heparin implicating other PF4 partners as well [358]. Platelet FcγRIIA receptors are capable of recognizing multiple antibody subclasses that include IgM, IgA, and IgG [359,360,361,362,363,364]—all produced by patients with COVID-19 [221, 327, 339, 342, 365].
5.2 Thrombocytopenia
Thrombocytopenia initially has been associated with severe COVID-19 adverse outcomes and mortality [297, 300, 304,305,306, 366]—possibly due, in part, to the consumption of platelets during the ongoing platelet-virus interaction process. However, others have not found thrombocytopenia to be an infallible predictor of disease progression or adverse outcome [367]. Decreases in platelet numbers have been associated with poor outcomes in multiple studies [306, 368,369,370,371,372,373,374]. Platelet changes can be associated with neutrophil extracellular traps—extracellular webs of chromatin, microbicidal proteins, and oxidant enzymes that are released by neutrophils to contain infections[242, 375, 376]. Some of these conserved higher-orders in response to microorganisms can lead to clustering around pathogens that encapsulates them, properties which are also observed in thrombocytes of lower vertebrates [377,378,379].
5.3 Thrombocytosis
Using ultrasound-guided, minimally invasive autopsy to assess COVID-19 pulmonary involvement revealed the presence of fibrinous thrombi in alveolar arterioles along with a high density of alveolar megakaryocytes that likely contribute to thrombocytosis [222] [380]. With accompanying elevations in interleukin (IL)-1 and IL-6 in patients with COVID-19 [297, 370, 381,382,383,384,385,386,387,388,389,390,391,392,393], this may lead to thrombocytosis due to inflammation-induced platelet activation, inflammasome formation, and IL-1-laden microvesicle release (Fig. 4), all of which are processes that associate with consumption-based feedback. IL-6 elevates platelet production in the metastatic ovarian-cancer setting when generated by tumors that stimulate liver-based thrombopoietin production [4, 10]. Thrombopoietin, in turn, stimulates megakaryocyte/platelet thrombocytosis in the bone marrow [4, 10]. By contrast with COVID-19, pathogenic T cells and inflammatory monocytes associated with large amounts of IL-6 secretion can incite an inflammatory (cytokine) storm, which may be curbed through treatment with tocilizumab, a monoclonal antibody treatment that targets IL-6 pathways [380].
5.4 Aspirin
Controversy exists over the use of aspirin in patients with COVID-19 particularly in those who are pregnant and display skin reactions [394,395,396]. In other studies, aspirin has shown benefit when combined with rivaroxaban (dual pathway inhibition) for the prevention of ischemic events in patients with cardiovascular- and peripheral-artery disease [397] [398]. Whether platelet consumption is the body’s way of isolating these diseases or immediately mitigating the spread of COVID-19 remains to be determined. In other studies, anti-coagulation effects were associated with improved COVID-19 survival after adjusting for mechanical ventilation.
5.5 Role of platelets in complications of viral infection
ARDS is among the most worrisome of all responses to SARS-CoV-2 infections [220, 221] (Fig. 5). Platelets can promote or contribute to all stages of ARDS development and progression, including thrombosis, inflammation, angiogenesis, and fibrosis [219,220,221, 223, 313, 366, 399,400,401,402]. Platelets can also help initiate many of the inflammatory responses in the lung. Endothelial-cell death and retraction can trigger platelet activation and granule release [219,220,221, 403], the latter of which includes many of the proinflammatory molecules that contribute to immune-cell infiltration and cytokine storming [27, 404,405,406,407,408,409,410,411,412].

SARS-CoV-2 in acute respiratory distress syndrome (ARDS). In contrast to type 1 pneumocytes, type 2 pneumocytes contain high levels of angiotensin-converting enzyme (ACE) and highly expressed transmembrane protease, serine 2 (TMPRSS2) –key targets for SARS-CoV-2 infection. The ensuing viral replication and vascular spread can trigger both vascular endothelial- and pneumocyte-cell death and matrix exposure that activates platelets. Pulmonary injury in SARS-CoV-2-induced COVID-19 can be amplified by many platelet-derived factors and bioactive processes[440]. Complete occlusion of blood vessels leads to hypoxia and additional damage. Acute respiratory distress and diffuse alveolar damage (DAD) can be induced by platelet factors and amplified by residential macrophages, neutrophils, and lymphocyte apoptosis. Platelets release cytokines and chemokines that also stimulate the production of residential, alveolar immune cells; recruitment of additional macrophages and neutrophils; lymphocyte apoptosis; formation of reactive oxygen species (ROS); and increased inflammation. Vascular endothelial-cell damage resulting from viral damage and complement activation amplifies the platelet activation and increases permeability and inflammatory thrombus formation. Fibrin formation and fibrinolysis can also be activated, releasing fibrin degradation fragments. Blood-vessel changes can dominate while damage in the alveolar space remains relatively mild. Platelet-neutrophil extracellular traps (NETs) may also play a role in severe disease [441]. Damage that accelerates to the alveolar space becomes more severe with additional immune-cell-death-related NET formation. Platelets, along with increased permeability and edema, can contribute to any cytokine storm that ensues. Platelets can also contribute to recruitment and activation of fibroblasts and myofibroblasts, formation of fibrous tissue, and scarring
Autopsies of COVID-19 patients have been difficult to achieve but they reveal many details of ARDS [220,221,222, 224, 413,414,415,416,417,418]. Certain reports suggest that in severe COVID-19 cases, platelets are centered within thrombotic responses with an absence of protective immune states [403, 419, 420]. As ARDS progresses, exudative, diffuse alveolar damage (DAD) occurs and can intensify the release of alveolar exudates to stimulate hyaline membranes, septal edema, and mild/moderate lymphocytic infiltration. This can lead to pleomorphic, alveolar, epithelial-cell alterations resulting from SARS-CoV-2 cytopathic effects, which then can cause diffuse epithelial desquamation. Epithelial cells can also develop distorted cytoplasm, large nuclei, eosinophilic nucleoli, giant cells, and squamous alveolar metaplasia. The release of platelet α-granule proteins can stimulate epithelial-cell- and endothelial proliferation and fibroblast invasion. Proliferative DAD has been characterized by disorganized fibrous tissue within alveolar septa while alveolar lumens can become severe with fibrinous thrombi[222]. These pulmonary changes are the result of severe epithelial injury and microthrombotic vascular phenomena[222].
5.6 Platelet contributions to cytokine storms
In patients with COVID-19, platelets can associate with SARS-CoV-2 RNA to become hyperactivated and can contribute to an eicosanoid storm [421] [422]. Hyperactivated platelets are characterized by proinflammatory granule release which includes C-X-C motif chemokines such as CXCL1 (GRO-α), CXCL4 (PF4), CXCL5 (ENA-78), CXCL7 (PBP, β-TG, CTAP-III, and NAP-2), CXCL8 (IL-8), and CXCL12 (also called stromal cell–derived factor-α) [171] (Fig. 4). Platelet α-granules that are C–C motif chemokines include CCL2 (MCP-1), CCL3 (MIP-1α), CCL5 (RANTES), CCL7 (MCP-3), and CCL17 (TARC); along with IL1-β, the platelet-activating factor acetylhydrolase, and lysophosphatidic acid [423]. In some cases, SARS-CoV-2-infected lung tissue may not be heavily infiltrated by CD20 + B cells, CD57 + NK cells, or lymphoid aggregates. By contrast, the number of CD4 + and CD8 + T cells may be few-to-moderate and may form small aggregates in patients with fibroproliferative DAD [222]. CD68 + macrophages populate alveolar spaces and sites of tissue remodeling in fibroproliferative areas. Some multinucleated, atypical giant-cells (CD68 + alveolar macrophages) [222] can amplify the production and release of factors that contribute to a cytokine storm [424,425,426,427]. In SARS-CoV-2-infected lung tissue ex vivo, platelets combined with resident and infiltrating immune cells led to the increased production of interferons IFNβ, IFNγ, IFNλ1, IFNλ2, and IFNλ3 as well as interleukins IL-1β, IL-6, IL-8, MCP1, MIP1α, RANTES, CXCL1, CXCL2, and CXCL5 [427].
5.7 Extrapulmonary disease
Platelets can also contribute to many of the extrapulmonary comorbidities related to ARDS [220,221,222, 402, 413,414,415,416,417]. In many cases, the extrapulmonary disease attributed to thrombotic axis comorbidities (such as hypertension and diabetes mellitus) often shows renal arteriolosclerosis, cardiomyocytes hypertrophy, myocardial fibrosis, focal glomerular sclerosis, liver steatosis, and cerebral microvascular disease [222]. COVID-19-induced shock and thrombotic lesion formation may also be associated with liver-based centrilobular congestion and acute kidney-tubular lesions. Syndromes secondary to SARS-COV-2 infection include systemic inflammation or shock linked to platelet activity associated with myositis, orchitis, lymphomononuclear myocarditis, and superficial perivascular mononuclear infiltrates in the skin.
Secondary platelet endothelial-cell-mediated changes in small vessels can include cell tumefaction, vessel wall edema, and fibrinoid alteration. Other secondary changes include fibrin microthrombi in the glomeruli, skin, testis, liver sinusoids, and heart; mesangial glomerulopathy; liver-based hyperreactive Kupffer cells; spleen-based lymphoid hypoplasia; and brain-based reactive gliosis.
6 Summary and future directions
This paper describes the importance of platelet evolution and responses secondary to SARS-CoV-2 infection along with their likely role in the thrombotic and immune function of patients with COVID-19. As primordial life forms, viruses (particularly single-stranded-RNA viruses such as SARS-CoV-2) coevolved and diverged as did vertebrates as they adapted to major environmental selective pressures while emerging from an aquatic to a terrestrial environment. Invertebrate and vertebrate thrombocytes and immunocytes retained aggregation capabilities, coagulation enzymes, and phagocytic properties involved in virus immobilization and uptake; these evolved into an active open-canalicular system. Retention of a highly active and responsive cytoskeletal system of granule release and rapid migratory capacity sustained cellular responsiveness to microbial threats. Thrombocyte/platelet evolution developed and maintained receptor-based, pathogen-directed, recognition capabilities to identify viruses. This evolution was coupled with active, membranous vesicle production to increase surface contacts and the probability of interacting with virus particles. As receptor-based response times decreased significantly and diverged over time, a finely regulated balance between endothelial COX2/PGI2- and platelet COX1/TxA2 pathway enzymes emerged. Targeting platelet adenosine receptors may also have an impact on the COVID disease state [428]. Selective serotonin reuptake inhibitors (SSRIs) are also candidates for potential intervention [429]. As vertebrates coevolved with viruses, their platelets typically remained elongated and retained morphological support by a circular ring of microtubules that ultimately formed the mammalian platelet. As they gradually became smaller and evolved into plate-like discs, this maximized planar-surface interactions and provided a biophysical advantage by concentrating the numerous platelets produced each day toward the outer fluid-shear fields of flowing blood. Management of severe COVID-19 is likely to require case-specific considerations depending on viral-load exposure, clinical presentation, and assessment of circulating platelets, megakaryocytes, cytokines, immune cells, and antibodies [430,431,432,433,434,435,436,437,438,439]. Much remains to be discovered regarding the evolution and biological function of platelets including a consideration of the many emerging selective pressures involved with SARS-CoV-2- and other viral infections. We continue to evolve in a globally connected world of rapid transit and exchange, which allows for the rapid dissemination of diseases that can outpace our ability to quickly respond with therapeutic interventions. Having learned so much from our experience studying the role of platelets in cancer and metastasis, we have tried to bring new insights into their role in immune defense mechanisms. From this perspective, we recognize that a better understanding of the platelet biology involved in the viral immune response is critical to being prepared for the rapid development of lethal viral threats such as SARS-CoV-2—and those yet to evolve.
References
Menter, D. G., Hatfield, J. S., Harkins, C., Sloane, B. F., Taylor, J. D., Crissman, J. D., et al. (1987). Tumor cell-platelet interactions in vitro and their relationship to in vivo arrest of hematogenously circulating tumor cells. Clinical and Experimental Metastasis, 5(1), 65–78.
Menter, D. G., Harkins, C., Onoda, J., Riorden, W., Sloane, B. F., Taylor, J. D., et al. (1987). Inhibition of tumor cell induced platelet aggregation by prostacyclin and carbacyclin: An ultrastructural study. Invasion and Metastasis, 7(2), 109–128.
Honn, K. V., Onoda, J. M., Menter, D. G., Cavanaugh, P. G., Taylor, J. D., Crissman, J. D., et al. (1986). Possible strategies for antimetastastic therapy. Progress in Clinical and Biological Research, 212, 217–249.
Haemmerle, M., Stone, R. L., Menter, D. G., Afshar-Kharghan, V., & Sood, A. K. (2018). The platelet lifeline to cancer: Challenges and opportunities. Cancer Cell, 33(6), 965–983.
Menter, D. G., Kopetz, S., Hawk, E., Sood, A. K., Loree, J. M., Gresele, P., et al. (2017). Platelet “first responders” in wound response, cancer, and metastasis. Cancer and Metastasis Reviews, 36(2), 199–213.
Hu, Q., Hisamatsu, T., Haemmerle, M., Cho, M. S., Pradeep, S., Rupaimoole, R., et al. (2017). Role of platelet-derived tgfbeta1 in the progression of ovarian cancer. Clinical Cancer Research, 23(18), 5611–5621.
Cho, M. S., Noh, K., Haemmerle, M., Li, D., Park, H., Hu, Q., et al. (2017). Role of ADP receptors on platelets in the growth of ovarian cancer. Blood, 130(10), 1235–1242.
Haemmerle, M., Bottsford-Miller, J., Pradeep, S., Taylor, M. L., Choi, H. J., Hansen, J. M., et al. (2016). FAK regulates platelet extravasation and tumor growth after antiangiogenic therapy withdrawal. The Journal of Clinical Investigation, 126(5), 1885–1896.
Menter, D. G., Tucker, S. C., Kopetz, S., Sood, A. K., Crissman, J. D., & Honn, K. V. (2014). Platelets and cancer: A casual or causal relationship: Revisited. Cancer and Metastasis Reviews, 33(1), 231–269.
Stone, R. L., Nick, A. M., McNeish, I. A., Balkwill, F., Han, H. D., Bottsford-Miller, J., et al. (2012). Paraneoplastic thrombocytosis in ovarian cancer. New England Journal of Medicine, 366(7), 610–618.
Gasic, G. J., Boettiger, D., Catalfamo, J. L., Gasic, T. B., & Stewart, G. J. (1978). Aggregation of platelets and cell membrane vesiculation by rat cells transformed in vitro by Rous sarcoma virus. Cancer Research, 38(9), 2950–2955.
Crissman, J. D., Hatfield, J., Schaldenbrand, M., Sloane, B. F., & Honn, K. V. (1985). Arrest and extravasation of B16 amelanotic melanoma in murine lungs. A light and electron microscopic study. Lab Invest, 53(4), 470–478.
Kazlauskas, D., Varsani, A., Koonin, E. V., & Krupovic, M. (2019). Multiple origins of prokaryotic and eukaryotic single-stranded DNA viruses from bacterial and archaeal plasmids. Nature Communications, 10(1), 3425.
Koonin, E. V., Dolja, V. V., & Krupovic, M. (2015). Origins and evolution of viruses of eukaryotes: The ultimate modularity. Virology, 479–480, 2–25.
Wolf, Y. I., Kazlauskas, D., Iranzo, J., Lucia-Sanz, A., Kuhn, J. H., Krupovic, M., et al. (2018). Origins and Evolution of the Global RNA Virome. mBio, 9(6)
Shi, M., Lin, X. D., Chen, X., Tian, J. H., Chen, L. J., Li, K., et al. (2018). The evolutionary history of vertebrate RNA viruses. Nature, 556(7700), 197–202.
Emerman, M., & Malik, H. S. (2010). Paleovirology--modern consequences of ancient viruses. PLoS Biol, 8(2), e1000301.
Zhang, Y. Z., Wu, W. C., Shi, M., & Holmes, E. C. (2018). The diversity, evolution and origins of vertebrate RNA viruses. Current Opinion in Virology, 31, 9–16.
Wang, J., Gong, Z., & Han, G. Z. (2019). Convergent co-option of the retroviral gag gene during the early evolution of mammals. J Virol, 93(14)
Gu, H., Chu, D. K. W., Peiris, M., & Poon, L. L. M. (2020). Multivariate analyses of codon usage of SARS-CoV-2 and other betacoronaviruses. Virus Evol, 6(1), veaa032.
Wong, G., Bi, Y. H., Wang, Q. H., Chen, X. W., Zhang, Z. G., & Yao, Y. G. (2020). Zoonotic origins of human coronavirus 2019 (HCoV-19 / SARS-CoV-2): Why is this work important? Zoological Research, 41(3), 213–219.
Ye, Z. W., Yuan, S., Yuen, K. S., Fung, S. Y., Chan, C. P., & Jin, D. Y. (2020). Zoonotic origins of human coronaviruses. International Journal of Biological Sciences, 16(10), 1686–1697.
Hon, C. C., Lam, T. Y., Shi, Z. L., Drummond, A. J., Yip, C. W., Zeng, F., et al. (2008). Evidence of the recombinant origin of a bat severe acute respiratory syndrome (SARS)-like coronavirus and its implications on the direct ancestor of SARS coronavirus. Journal of Virology, 82(4), 1819–1826.
Drexler, J. F., Corman, V. M., & Drosten, C. (2014). Ecology, evolution and classification of bat coronaviruses in the aftermath of SARS. Antiviral Research, 101, 45–56.
Brook, C. E., Boots, M., Chandran, K., Dobson, A. P., Drosten, C., Graham, A. L., et al. (2020). Accelerated viral dynamics in bat cell lines, with implications for zoonotic emergence. Elife, 9
Pavesi, A. (2020). New insights into the evolutionary features of viral overlapping genes by discriminant analysis. Virology, 546, 51–66.
Zhang, X., Tan, Y., Ling, Y., Lu, G., Liu, F., Yi, Z., et al. (2020). Viral and host factors related to the clinical outcome of COVID-19. Nature
Fauver, J. R., Petrone, M. E., Hodcroft, E. B., Shioda, K., Ehrlich, H. Y., Watts, A. G., et al. (2020). Coast-to-coast spread of SARS-CoV-2 during the early epidemic in the United States. Cell, 181(5), 990–996 e995.
Lu, J., du Plessis, L., Liu, Z., Hill, V., Kang, M., Lin, H., et al. (2020). Genomic epidemiology of SARS-CoV-2 in Guangdong Province, China. Cell, 181(5), 997–1003 e1009.
Vankadari, N. (2020). Overwhelming mutations or SNPs of SARS-CoV-2: A point of caution. Gene, 752, 144792.
Lam, T. T. (2020). Tracking the genomic footprints of SARS-CoV-2 transmission. Trends Genet
Li, X., Wang, W., Zhao, X., Zai, J., Zhao, Q., Li, Y., et al. (2020). Transmission dynamics and evolutionary history of 2019-nCoV. Journal of Medical Virology, 92(5), 501–511.
Geoghegan, J. L., Duchene, S., & Holmes, E. C. (2017). Comparative analysis estimates the relative frequencies of co-divergence and cross-species transmission within viral families. PLoS Pathog, 13(2), e1006215.
Pryzdial, E., Lin, B., & Sutherland, M. (2017). Virus-Platelet associations. In P. Gresele, N. Kleiman, J. Lopez, & C. Page (Eds.), Platelets in Thrombotic and Non-Thrombotic Disorders (Vol. 2, pp. 1085–1101). Springer International Publishing.
Menter, D. G. (2021). Where is Waldo? or find the platelet. Cancer and Metastasis Reviews, 40(3), 649–655.
Svoboda, O., & Bartunek, P. (2015). Origins of the vertebrate erythro/megakaryocytic system. Biomed Res Int, 2015, 632171.
Weyrich, A. S., Lindemann, S., & Zimmerman, G. A. (2003). The evolving role of platelets in inflammation. Journal of Thrombosis and Haemostasis, 1(9), 1897–1905.
Banerjee, M., Huang, Y., Joshi, S., Popa, G. J., Mendenhall, M. D., Wang, Q. J., et al. (2020). Platelets endocytose viral particles and are activated via TLR (toll-like receptor) signaling. Arterioscler Thromb Vasc Biol, ATVBAHA120314180.
Jansen, A. J. G., Spaan, T., Low, H. Z., Di Iorio, D., van den Brand, J., Tieke, M., et al. (2020). Influenza-induced thrombocytopenia is dependent on the subtype and sialoglycan receptor and increases with virus pathogenicity. Blood Advances, 4(13), 2967–2978.
Shimony, N., Elkin, G., Kolodkin-Gal, D., Krasny, L., Urieli-Shoval, S., & Haviv, Y. S. (2009). Analysis of adenoviral attachment to human platelets. Virol J, 6, 25.
Bai, Y., Yao, L., Wei, T., Tian, F., Jin, D. Y., Chen, L., et al. (2020). Presumed asymptomatic carrier transmission of COVID-19. JAMA
Sakurai, A., Sasaki, T., Kato, S., Hayashi, M., Tsuzuki, S. I., Ishihara, T., et al. (2020). Natural history of asymptomatic SARS-CoV-2 infection. N Engl J Med
Sungnak, W., Huang, N., Becavin, C., Berg, M., Queen, R., Litvinukova, M., et al. (2020). SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nature Medicine, 26(5), 681–687.
Bhatraju, P. K., Ghassemieh, B. J., Nichols, M., Kim, R., Jerome, K. R., Nalla, A. K., et al. (2020). Covid-19 in critically ill patients in the Seattle region - Case series. New England Journal of Medicine, 382(21), 2012–2022.
Lechien, J. R., Chiesa-Estomba, C. M., De Siati, D. R., Horoi, M., Le Bon, S. D., Rodriguez, A., et al. (2020). Olfactory and gustatory dysfunctions as a clinical presentation of mild-to-moderate forms of the coronavirus disease (COVID-19): a multicenter European study. Eur Arch Otorhinolaryngol
Mariz, B., Brandao, T. B., Ribeiro, A. C. P., Lopes, M. A., & Santos-Silva, A. R. (2020). New insights for the pathogenesis of COVID-19-related dysgeusia. J Dent Res, 22034520936638.
Poggiali, E., Ramos, P. M., Bastoni, D., Vercelli, A., & Magnacavallo, A. (2020). Abdominal pain: A real challenge in novel COVID-19 infection. Eur J Case Rep Intern Med, 7(4), 001632.
Qiu, C., Cui, C., Hautefort, C., Haehner, A., Zhao, J., Yao, Q., et al. (2020). Olfactory and gustatory dysfunction as an early identifier of COVID-19 in adults and children: An international multicenter study. Otolaryngol Head Neck Surg, 194599820934376.
Tabuchi, A., & Kuebler, W. M. (2008). Endothelium-platelet interactions in inflammatory lung disease. Vascular Pharmacology, 49(4–6), 141–150.
Solomon Tsegaye, T., Gnirss, K., Rahe-Meyer, N., Kiene, M., Kramer-Kuhl, A., Behrens, G., et al. (2013). Platelet activation suppresses HIV-1 infection of T cells. Retrovirology, 10, 48.
Real, F., Capron, C., Sennepin, A., Arrigucci, R., Zhu, A., Sannier, G., et al. (2020). Platelets from HIV-infected individuals on antiretroviral drug therapy with poor CD4(+) T cell recovery can harbor replication-competent HIV despite viral suppression. Sci Transl Med, 12(535)
Opal, S. M. (2000). Phylogenetic and functional relationships between coagulation and the innate immune response. Critical Care Medicine, 28(9 Suppl), S77-80.
Momi, S., & Wiwanitkit, V. (2017). Phylogeny of Blood Platelets. In P. Gresele, N. Kleiman, J. Lopez, & C. Page (Eds.), Platelets in Thrombotic and Non-Thrombotic Disorders (pp. 11–20). Springer.
Pascual-Anaya, J., Albuixech-Crespo, B., Somorjai, I. M., Carmona, R., Oisi, Y., Alvarez, S., et al. (2013). The evolutionary origins of chordate hematopoiesis and vertebrate endothelia. Developmental Biology, 375(2), 182–192.
Monahan-Earley, R., Dvorak, A. M., & Aird, W. C. (2013). Evolutionary origins of the blood vascular system and endothelium. Journal of Thrombosis and Haemostasis, 11(Suppl 1), 46–66.
Ponczek, M. B., Bijak, M. Z., & Nowak, P. Z. (2012). Evolution of thrombin and other hemostatic proteases by survey of protochordate, hemichordate, and echinoderm genomes. Journal of Molecular Evolution, 74(5–6), 319–331.
Han, Y., Huang, G., Zhang, Q., Yuan, S., Liu, J., Zheng, T., et al. (2010). The primitive immune system of amphioxus provides insights into the ancestral structure of the vertebrate immune system. Developmental and Comparative Immunology, 34(8), 791–796.
Levin, J., & Bang, F. B. (1964). The role of endotoxin in the extracellular coagulation of Limulus blood. Bulletin of the Johns Hopkins Hospital, 115, 265–274.
Levin, J., & Bang, F. B. (1964). A description of cellular coagulation in the Limulus. Bulletin of the Johns Hopkins Hospital, 115, 337–345.
Armstrong, P. B. (1980). Adhesion and spreading of Limulus blood cells on artificial surfaces. Journal of Cell Science, 44, 243–262.
Young, N. S., Levin, J., & Prendergast, R. A. (1972). An invertebrate coagulation system activated by endotoxin: Evidence for enzymatic mediation. The Journal of Clinical Investigation, 51(7), 1790–1797.
Armstrong, P. B., & Rickles, F. R. (1982). Endotoxin-induced degranulation of the Limulus amebocyte. Experimental Cell Research, 140(1), 15–24.
Iwanaga, S., Miyata, T., Tokunaga, F., & Muta, T. (1992). Molecular mechanism of hemolymph clotting system in Limulus. Thrombosis Research, 68(1), 1–32.
Murer, E. H., Levin, J., & Holme, R. (1975). Isolation and studies of the granules of the amebocytes of Limulus polyphemus, the horseshoe crab. Journal of Cellular Physiology, 86(3 Pt 1), 533–542.
Ratnoff, O. D. (1987). The evolution of hemostatic mechanisms. Perspectives in Biology and Medicine, 31(1), 4–33.
Grant, M. A., Beeler, D. L., Spokes, K. C., Chen, J., Dharaneeswaran, H., Sciuto, T. E., et al. (2017). Identification of extant vertebrate Myxine glutinosa VWF: Evolutionary conservation of primary hemostasis. Blood, 130(23), 2548–2558.
Mattisson, A., & Fänge, R. (1977). Light- and electronmicroscopic observations on the blood cells of the Atlantic Hagfish, Myxine glutinosa (L.). Acta Zoologica, 58, 205–221.
Hyder, S. L., Cayer, M. L., & Pettey, C. L. (1983). Cell types in peripheral blood of the nurse shark: An approach to structure and function. Tissue and Cell, 15(3), 437–455.
Shepro, D., Belamarich, F. A., & Branson, R. (1966). The fine structure of the thrombocyte in the dogfish (Mustelus canis) with special reference to microtubule orientation. Anatomical Record, 156(2), 203–214.
Khandekar, G., Kim, S., & Jagadeeswaran, P. (2012). Zebrafish thrombocytes: Functions and origins. Adv Hematol, 2012, 857058.
Rowley, A. F., Hill, D. J., Ray, C. E., & Munro, R. (1997). Haemostasis in fish–an evolutionary perspective. Thrombosis and Haemostasis, 77(2), 227–233.
Jagadeeswaran, P., Sheehan, J. P., Craig, F. E., & Troyer, D. (1999). Identification and characterization of zebrafish thrombocytes. British Journal of Haematology, 107(4), 731–738.
Ribeiro, M. L., DaMatta, R. A., Diniz, J. A., de Souza, W., & do Nascimento, J. L., & de Carvalho, T. M. (2007). Blood and inflammatory cells of the lungfish Lepidosiren paradoxa. Fish & Shellfish Immunology, 23(1), 178–187.
Hiong, K. C., Tan, X. R., Boo, M. V., Wong, W. P., Chew, S. F., & Ip, Y. K. (2015). Aestivation induces changes in transcription and translation of coagulation factor II and fibrinogen gamma chain in the liver of the African lungfish Protopterus annectens. Journal of Experimental Biology, 218(Pt 23), 3717–3728.
Lewis, J. (1996). Comparative hemostasis in vertebrates. Plenum Press.
Canfield, P. J. (1998). Comparative cell morphology in the peripheral blood film from exotic and native animals. Australian Veterinary Journal, 76(12), 793–800.
DaMatta, R. A., Manhaes, L., Lassounskaia, E., & de Souza, W. (1999). Chicken thrombocytes in culture: Lymphocyte-conditioned medium delays apoptosis. Tissue and Cell, 31(3), 255–263.
Gerrard, J. M., White, J. G., & Rao, G. H. (1974). Effects of the lonophore A23187 on the blood platelets II. Influence on ultrastructure. Am J Pathol, 77(2), 151–166.
Lewis, J. H. (1975). Comparative hematology: Studies on opossums Didelphis marsupialis (Virginianus). Comparative Biochemistry and Physiology, A: Comparative Physiology, 51(2), 275–280.
Bruce, I. J., & Kerry, R. (1987). The effect of chloramphenicol and cycloheximide on platelet aggregation and protein synthesis. Biochemical Pharmacology, 36(11), 1769–1773.
Weyrich, A. S., Schwertz, H., Kraiss, L. W., & Zimmerman, G. A. (2009). Protein synthesis by platelets: Historical and new perspectives. Journal of Thrombosis and Haemostasis, 7(2), 241–246.
Zimmerman, G. A., & Weyrich, A. S. (2008). Signal-dependent protein synthesis by activated platelets: New pathways to altered phenotype and function. Arteriosclerosis, Thrombosis, and Vascular Biology, 28(3), s17-24.
Quirino-Teixeira, A. C., Rozini, S. V., Barbosa-Lima, G., Coelho, D. R., Carneiro, P. H., Mohana-Borges, R., et al. (2020). Inflammatory signaling in dengue-infected platelets requires translation and secretion of nonstructural protein 1. Blood Advances, 4(9), 2018–2031.
Simon, A. Y., Sutherland, M. R., & Pryzdial, E. L. (2015). Dengue virus binding and replication by platelets. Blood, 126(3), 378–385.
Sutherland, M. R., Simon, A. Y., Serrano, K., Schubert, P., Acker, J. P., & Pryzdial, E. L. (2016). Dengue virus persists and replicates during storage of platelet and red blood cell units. Transfusion, 56(5), 1129–1137.
Rowley, J. W., & Weyrich, A. S. (2017). Ribosomes in platelets protect the messenger. Blood, 129(17), 2343–2345.
Vogt, M. B., Lahon, A., Arya, R. P., Spencer Clinton, J. L., & Rico-Hesse, R. (2019). Dengue viruses infect human megakaryocytes, with probable clinical consequences. PLoS Negl Trop Dis, 13(11), e0007837.
Pozner, R. G., Ure, A. E., Jaquenod de Giusti, C., D'Atri, L. P., Italiano, J. E., Torres, O., et al. (2010). Junin virus infection of human hematopoietic progenitors impairs in vitro proplatelet formation and platelet release via a bystander effect involving type I IFN signaling. PLoS Pathog, 6(4), e1000847.
Battinelli, E. M., Thon, J. N., Okazaki, R., Peters, C. G., Vijey, P., Wilkie, A. R., et al. (2019). Megakaryocytes package contents into separate alpha-granules that are differentially distributed in platelets. Blood Advances, 3(20), 3092–3098.
Thon, J. N., & Italiano, J. E. (2010). Platelet formation. Seminars in Hematology, 47(3), 220–226.
Thon, J. N., Macleod, H., Begonja, A. J., Zhu, J., Lee, K. C., Mogilner, A., et al. (2012). Microtubule and cortical forces determine platelet size during vascular platelet production. Nature Communications, 3, 852.
Brass, L. F. (2005). Did dinosaurs have megakaryocytes? New ideas about platelets and their progenitors. The Journal of Clinical Investigation, 115(12), 3329–3331.
Martin, J. F., & Wagner, G. P. (2019). The origin of platelets enabled the evolution of eutherian placentation. Biology Letters, 15(7), 20190374.
Gupalo, E., Kuk, C., Qadura, M., Buriachkovskaia, L., & Othman, M. (2013). Platelet-adenovirus vs. inert particles interaction: Effect on aggregation and the role of platelet membrane receptors. Platelets, 24(5), 383–391.
Ghosh, K., Gangodkar, S., Jain, P., Shetty, S., Ramjee, S., Poddar, P., et al. (2008). Imaging the interaction between dengue 2 virus and human blood platelets using atomic force and electron microscopy. Journal of Electron Microscopy (Tokyo), 57(3), 113–118.
Pretorius, E., Oberholzer, H. M., Smit, E., Steyn, E., Briedenhann, S., & Franz, C. R. (2008). Ultrastructural changes in platelet aggregates of HIV patients: A scanning electron microscopy study. Ultrastructural Pathology, 32(3), 75–79.
Youssefian, T., Drouin, A., Masse, J. M., Guichard, J., & Cramer, E. M. (2002). Host defense role of platelets: Engulfment of HIV and Staphylococcus aureus occurs in a specific subcellular compartment and is enhanced by platelet activation. Blood, 99(11), 4021–4029.
Rahbar, A., & Soderberg-Naucler, C. (2005). Human cytomegalovirus infection of endothelial cells triggers platelet adhesion and aggregation. Journal of Virology, 79(4), 2211–2220.
Varon, D., & Shai, E. (2009). Role of platelet-derived microparticles in angiogenesis and tumor progression. Discovery Medicine, 8(43), 237–241.
Alonzo, M. T., Lacuesta, T. L., Dimaano, E. M., Kurosu, T., Suarez, L. A., Mapua, C. A., et al. (2012). Platelet apoptosis and apoptotic platelet clearance by macrophages in secondary dengue virus infections. Journal of Infectious Diseases, 205(8), 1321–1329.
Assinger, A. (2014). Platelets and infection - an emerging role of platelets in viral infection. Frontiers in Immunology, 5, 649.
Alonso, A. L., & Cox, D. (2015). Platelet interactions with viruses and parasites. Platelets, 26(4), 317–323.
Alonso-Villaverde Lozano, C. (2009). Physiopathology of cardiovascular disease in HIV-infected patients. Enfermedades Infecciosas y Microbiologia Clinica, 27(Suppl 1), 33–39.
Hottz, E. D., Bozza, F. A., & Bozza, P. T. (2018). Platelets in Immune Response to Virus and Immunopathology of Viral Infections. Front Med (Lausanne), 5, 121.
Seyoum, M., Enawgaw, B., & Melku, M. (2018). Human blood platelets and viruses: Defense mechanism and role in the removal of viral pathogens. Thrombosis Journal, 16, 16.
Wan, S. W., Yang, Y. W., Chu, Y. T., Lin, C. F., Chang, C. P., Yeh, T. M., et al. (2016). Anti-dengue virus nonstructural protein 1 antibodies contribute to platelet phagocytosis by macrophages. Thrombosis and Haemostasis, 115(3), 646–656.
Nagasawa, T., Nakayasu, C., Rieger, A. M., Barreda, D. R., Somamoto, T., & Nakao, M. (2014). Phagocytosis by thrombocytes is a conserved innate immune mechanism in lower vertebrates. Frontiers in Immunology, 5, 445.
Liu, L. P., Shan, C. W., Liu, X. H., Xiao, H. C., & Yang, S. Q. (1998). Effect of procainamide on ultrastructure of blood platelet in rabbits. Zhongguo Yao Li Xue Bao, 19(4), 376–379.
Ludhiadch, A., Muralidharan, A., Balyan, R., & Munshi, A. (2020). The molecular basis of platelet biogenesis, activation, aggregation and implications in neurological disorders. International Journal of Neuroscience, 130(12), 1237–1249.
Nguyen, T. H., Schuster, N., Greinacher, A., & Aurich, K. (2018). Uptake pathways of protein-coated magnetic nanoparticles in platelets. ACS Applied Materials & Interfaces, 10(34), 28314–28321.
Selvadurai, M. V., & Hamilton, J. R. (2018). Structure and function of the open canalicular system - the platelet’s specialized internal membrane network. Platelets, 29(4), 319–325.
O’Brien, S., Kent, N. J., Lucitt, M., Ricco, A. J., McAtamney, C., Kenny, D., et al. (2012). Effective hydrodynamic shaping of sample streams in a microfluidic parallel-plate flow-assay device: Matching whole blood dynamic viscosity. IEEE Transactions on Biomedical Engineering, 59(2), 374–382.
Jen, C. J., & Tai, Y. W. (1992). Morphological study of platelet adhesion dynamics under whole blood flow conditions. Platelets, 3(3), 145–153.
Folie, B. J., & McIntire, L. V. (1989). Mathematical analysis of mural thrombogenesis. Concentration profiles of platelet-activating agents and effects of viscous shear flow. Biophys J, 56(6), 1121–1141.
Fedosov, D. A., Noguchi, H., & Gompper, G. (2014). Multiscale modeling of blood flow: From single cells to blood rheology. Biomechanics and Modeling in Mechanobiology, 13(2), 239–258.
Kumar, A., & Graham, M. D. (2012). Mechanism of margination in confined flows of blood and other multicomponent suspensions. Phys Rev Lett, 109(10), 108102.
Tokarev, A. A., Butylin, A. A., & Ataullakhanov, F. I. (2011). Platelet adhesion from shear blood flow is controlled by near-wall rebounding collisions with erythrocytes. Biophysical Journal, 100(4), 799–808.
Tokarev, A. A., Butylin, A. A., Ermakova, E. A., Shnol, E. E., Panasenko, G. P., & Ataullakhanov, F. I. (2011). Finite platelet size could be responsible for platelet margination effect. Biophysical Journal, 101(8), 1835–1843.
Lee, S. Y., Ferrari, M., & Decuzzi, P. (2009). Design of bio-mimetic particles with enhanced vascular interaction. Journal of Biomechanics, 42(12), 1885–1890.
Stukelj, R., Schara, K., Bedina-Zavec, A., Sustar, V., Pajnic, M., Paden, L., et al. (2017). Effect of shear stress in the flow through the sampling needle on concentration of nanovesicles isolated from blood. European Journal of Pharmaceutical Sciences, 98, 17–29.
De Gruttola, S., Boomsma, K., & Poulikakos, D. (2005). Computational simulation of a non-newtonian model of the blood separation process. Artificial Organs, 29(12), 949–959.
Nesbitt, W. S., Westein, E., Tovar-Lopez, F. J., Tolouei, E., Mitchell, A., Fu, J., et al. (2009). A shear gradient-dependent platelet aggregation mechanism drives thrombus formation. Nature Medicine, 15(6), 665–673.
Crissman, J. D., Hatfield, J. S., Menter, D. G., Sloane, B., & Honn, K. V. (1988). Morphological study of the interaction of intravascular tumor cells with endothelial cells and subendothelial matrix. Cancer Research, 48(14), 4065–4072.
Walsh, T. G., Metharom, P., & Berndt, M. C. (2015). The functional role of platelets in the regulation of angiogenesis. Platelets, 26(3), 199–211.
Kim, K. H., Barazia, A., & Cho, J. (2013). Real-time imaging of heterotypic platelet-neutrophil interactions on the activated endothelium during vascular inflammation and thrombus Formation in live mice. J Vis Exp(74)
Spectre, G., Zhu, L., Ersoy, M., Hjemdahl, P., Savion, N., Varon, D., et al. (2012). Platelets selectively enhance lymphocyte adhesion on subendothelial matrix under arterial flow conditions. Thrombosis and Haemostasis, 108(2), 328–337.
Gardiner, E., & Andrews, R. (2017). Platelet Adhesion. In P. Gresele, N. Kleiman, J. Lopez, & C. Page (Eds.), Platelets in thrombotic and non-thrombotic disorders (Vol. 2, pp. 309–319). Springer International Publishing.
Pothapragada, S., Zhang, P., Sheriff, J., Livelli, M., Slepian, M. J., Deng, Y., et al. (2015). A phenomenological particle-based platelet model for simulating filopodia formation during early activation. Int J Numer Method Biomed Eng, 31(3), e02702.
Kunert, S., Meyer, I., Fleischhauer, S., Wannack, M., Fiedler, J., Shivdasani, R. A., et al. (2009). The microtubule modulator RanBP10 plays a critical role in regulation of platelet discoid shape and degranulation. Blood, 114(27), 5532–5540.
Jackson, S. P., Nesbitt, W. S., & Westein, E. (2009). Dynamics of platelet thrombus formation. Journal of Thrombosis and Haemostasis, 7(Suppl 1), 17–20.
Italiano, J. E., Jr., Bergmeier, W., Tiwari, S., Falet, H., Hartwig, J. H., Hoffmeister, K. M., et al. (2003). Mechanisms and implications of platelet discoid shape. Blood, 101(12), 4789–4796.
Hartwig, J. H., Barkalow, K., Azim, A., & Italiano, J. (1999). The elegant platelet: Signals controlling actin assembly. Thrombosis and Haemostasis, 82(2), 392–398.
White, J. G., & Rao, G. H. (1998). Microtubule coils versus the surface membrane cytoskeleton in maintenance and restoration of platelet discoid shape. American Journal of Pathology, 152(2), 597–609.
Polanowska-Grabowska, R., Geanacopoulos, M., & Gear, A. R. (1993). Platelet adhesion to collagen via the alpha 2 beta 1 integrin under arterial flow conditions causes rapid tyrosine phosphorylation of pp125FAK. The Biochemical Journal, 296(Pt 3), 543–547.
Falet, H. (2017). Anatomy of the Platelet Cytoskeleton. In P. Gresele, N. Kleiman, J. Lopez, & C. Page (Eds.), Platelets in thrombotic and non-thrombotic disorders (Vol. 2, pp. 139–156). Springer International Publishing.
Rommel, M. G. E., Milde, C., Eberle, R., Schulze, H., & Modlich, U. (2019). Endothelial-platelet interactions in influenza-induced pneumonia: A potential therapeutic target. Anat Histol Embryol
Almeida, V. H., Rondon, A. M. R., Gomes, T., & Monteiro, R. Q. (2019). Novel aspects of extracellular vesicles as mediators of cancer-associated thrombosis. Cells, 8(7)
Enjeti, A. K., Ariyarajah, A., D’Crus, A., Seldon, M., & Lincz, L. F. (2016). Correlative analysis of nanoparticle tracking, flow cytometric and functional measurements for circulating microvesicles in normal subjects. Thrombosis Research, 145, 18–23.
Enjeti, A. K., Ariyarajah, A., D’Crus, A., Seldon, M., & Lincz, L. F. (2017). Circulating microvesicle number, function and small RNA content vary with age, gender, smoking status, lipid and hormone profiles. Thrombosis Research, 156, 65–72.
Gajos, K., Kaminska, A., Awsiuk, K., Bajor, A., Gruszczynski, K., Pawlak, A., et al. (2017). Immobilization and detection of platelet-derived extracellular vesicles on functionalized silicon substrate: Cytometric and spectrometric approach. Analytical and Bioanalytical Chemistry, 409(4), 1109–1119.
Rak, J. (2010). Microparticles in cancer. Seminars in Thrombosis and Hemostasis, 36(8), 888–906.
Kanikarla-Marie, P., Lam, M., Menter, D. G., & Kopetz, S. (2017). Platelets, circulating tumor cells, and the circulome. Cancer and Metastasis Reviews, 36(2), 235–248.
Kim, S., Carrillo, M., Radhakrishnan, U. P., & Jagadeeswaran, P. (2012). Role of zebrafish thrombocyte and non-thrombocyte microparticles in hemostasis. Blood Cells, Molecules, & Diseases, 48(3), 188–196.
Jagadeeswaran, P., Carrillo, M., Radhakrishnan, U. P., Rajpurohit, S. K., & Kim, S. (2011). Laser-induced thrombosis in zebrafish. Methods in Cell Biology, 101, 197–203.
Mauler, M., Herr, N., Schoenichen, C., Witsch, T., Marchini, T., Hardtner, C., et al. (2019). Platelet serotonin aggravates myocardial ischemia/reperfusion injury via neutrophil degranulation. Circulation, 139(7), 918–931.
Breckenridge, M. T., Egelhoff, T. T., & Baskaran, H. (2010). A microfluidic imaging chamber for the direct observation of chemotactic transmigration. Biomedical Microdevices, 12(3), 543–553.
Ellingsen, T., Storgaard, M., Moller, B. K., Buus, A., Andersen, P. L., Obel, N., et al. (2000). Migration of mononuclear cells in the modified Boyden chamber as evaluated by DNA quantification and flow cytometry. Scandinavian Journal of Immunology, 52(3), 257–263.
Friedl, P., Wolf, K., & Lammerding, J. (2011). Nuclear mechanics during cell migration. Current Opinion in Cell Biology, 23(1), 55–64.
Bdeir, K., Gollomp, K., Stasiak, M., Mei, J., Papiewska-Pajak, I., Zhao, G., et al. (2017). Platelet-specific chemokines contribute to the pathogenesis of acute lung injury. American Journal of Respiratory Cell and Molecular Biology, 56(2), 261–270.
Schmidt, E. M., Kraemer, B. F., Borst, O., Munzer, P., Schonberger, T., Schmidt, C., et al. (2012). SGK1 sensitivity of platelet migration. Cellular Physiology and Biochemistry, 30(1), 259–268.
Borisova, T. A., & Markosian, R. A. (1977). Age and biosynthesis and breakdown of thrombocyte proteins. Biulleten Eksperimentalnoi Biologii I Meditsiny, 83(1), 20–21.
Krakowka, S., Cork, L. C., Winkelstein, J. A., & Axthelm, M. K. (1987). Establishment of central nervous system infection by canine distemper virus: Breach of the blood-brain barrier and facilitation by antiviral antibody. Veterinary Immunology and Immunopathology, 17(1–4), 471–482.
Krakowka, S., Axthelm, M. K., & Gorham, J. R. (1987). Effects of induced thrombocytopenia on viral invasion of the central nervous system in canine distemper virus infection. Journal of Comparative Pathology, 97(4), 441–450.
Petito, E., Momi, S., & Gresele, P. (2017). The migration of platelets and their interaction with other migrating cells. In P. Gresele, N. Kleiman, J. Lopez, & C. Page (Eds.), Platelets in Thrombotic and Non-Thrombotic Disorders (Vol. 2, pp. 337–351). Springer International Publishing.
Sandri, G., Bonferoni, M. C., Rossi, S., Ferrari, F., Mori, M., Cervio, M., et al. (2015). Platelet lysate embedded scaffolds for skin regeneration. Expert Opinion on Drug Delivery, 12(4), 525–545.
Kurokawa, T., & Ohkohchi, N. (2017). Platelets in liver disease, cancer and regeneration. World Journal of Gastroenterology, 23(18), 3228–3239.
Mancuso, M. E., & Santagostino, E. (2017). Platelets: Much more than bricks in a breached wall. Br J Haematol
Anitua, E., Troya, M., Zalduendo, M., & Orive, G. (2017). Personalized plasma-based medicine to treat age-related diseases. Materials Science & Engineering, C: Materials for Biological Applications, 74, 459–464.
Meschi, N., Castro, A. B., Vandamme, K., Quirynen, M., & Lambrechts, P. (2016). The impact of autologous platelet concentrates on endodontic healing: A systematic review. Platelets, 27(7), 613–633.
Mlynarek, R. A., Kuhn, A. W., & Bedi, A. (2016). Platelet-Rich Plasma (PRP) in Orthopedic Sports Medicine. American Journal of Orthopedics (Belle Mead, N.J.), 45(5), 290–326.
Koupenova, M., Corkrey, H. A., Vitseva, O., Manni, G., Pang, C. J., Clancy, L., et al. (2019). The role of platelets in mediating a response to human influenza infection. Nature Communications, 10(1), 1780.
Koupenova, M., Mick, E., Corkrey, H. A., Huan, T., Clancy, L., Shah, R., et al. (2018). Micro RNAs from DNA viruses are found widely in plasma in a large observational human population. Science and Reports, 8(1), 6397.
Koupenova, M., Clancy, L., Corkrey, H. A., & Freedman, J. E. (2018). Circulating platelets as mediators of immunity, inflammation, and thrombosis. Circulation Research, 122(2), 337–351.
Goubran, H. A., Stakiw, J., Radosevic, M., & Burnouf, T. (2014). Platelets effects on tumor growth. Seminars in Oncology, 41(3), 359–369.
Unwith, S., Zhao, H., Hennah, L., & Ma, D. (2015). The potential role of HIF on tumour progression and dissemination. International Journal of Cancer, 136(11), 2491–2503.
Kraemer, B. F., Borst, O., Gehring, E. M., Schoenberger, T., Urban, B., Ninci, E., et al. (2010). PI3 kinase-dependent stimulation of platelet migration by stromal cell-derived factor 1 (SDF-1). Journal of Molecular Medicine (Berlin, Germany), 88(12), 1277–1288.
Brandt, E., Ludwig, A., Petersen, F., & Flad, H. D. (2000). Platelet-derived CXC chemokines: Old players in new games. Immunological Reviews, 177, 204–216.
Kraemer, B. F., Schmidt, C., Urban, B., Bigalke, B., Schwanitz, L., Koch, M., et al. (2011). High shear flow induces migration of adherent human platelets. Platelets, 22(6), 415–421.
Chatterjee, M., Huang, Z., Zhang, W., Jiang, L., Hultenby, K., Zhu, L., et al. (2011). Distinct platelet packaging, release, and surface expression of proangiogenic and antiangiogenic factors on different platelet stimuli. Blood, 117(14), 3907–3911.
Shenkman, B., Brill, A., Brill, G., Lider, O., Savion, N., & Varon, D. (2004). Differential response of platelets to chemokines: RANTES non-competitively inhibits stimulatory effect of SDF-1 alpha. Journal of Thrombosis and Haemostasis, 2(1), 154–160.
Gleissner, C. A., von Hundelshausen, P., & Ley, K. (2008). Platelet chemokines in vascular disease. Arteriosclerosis, Thrombosis, and Vascular Biology, 28(11), 1920–1927.
Clemetson, K. J., Clemetson, J. M., Proudfoot, A. E., Power, C. A., Baggiolini, M., & Wells, T. N. (2000). Functional expression of CCR1, CCR3, CCR4, and CXCR4 chemokine receptors on human platelets. Blood, 96(13), 4046–4054.
Stosik, M., Tokarz-Deptula, B., & Deptula, W. (2019). Characterisation of thrombocytes in osteichthyes. J Vet Res, 63(1), 123–131.
He, L., Zhang, A., Pei, Y., Chu, P., Li, Y., Huang, R., et al. (2017). Differences in responses of grass carp to different types of grass carp reovirus (GCRV) and the mechanism of hemorrhage revealed by transcriptome sequencing. BMC Genomics, 18(1), 452.
Mutoloki, S., Brudeseth, B., Reite, O. B., & Evensen, O. (2006). The contribution of Aeromonas salmonicida extracellular products to the induction of inflammation in Atlantic salmon (Salmo salar L.) following vaccination with oil-based vaccines. Fish Shellfish Immunol, 20(1), 1–11.
Mutoloki, S., Reite, O. B., Brudeseth, B., Tverdal, A., & Evensen, O. (2006). A comparative immunopathological study of injection site reactions in salmonids following intraperitoneal injection with oil-adjuvanted vaccines. Vaccine, 24(5), 578–588.
Reite, O. B., & Evensen, O. (2006). Inflammatory cells of teleostean fish: A review focusing on mast cells/eosinophilic granule cells and rodlet cells. Fish & Shellfish Immunology, 20(2), 192–208.
Chen, H., Ji, T., Chen, J., & Li, X. (2019). Matrix solid-phase dispersion combined with HPLC-DAD for simultaneous determination of nine lignans in Saururus chinensis. Journal of Chromatographic Science, 57(2), 186–193.
Yuan, D., Zou, Q., Yu, T., Song, C., Huang, S., Chen, S., et al. (2014). Ancestral genetic complexity of arachidonic acid metabolism in Metazoa. Biochimica et Biophysica Acta, 1841(9), 1272–1284.
Havird, J. C., Miyamoto, M. M., Choe, K. P., & Evans, D. H. (2008). Gene duplications and losses within the cyclooxygenase family of teleosts and other chordates. Molecular Biology and Evolution, 25(11), 2349–2359.
Dangel, O., Mergia, E., Karlisch, K., Groneberg, D., Koesling, D., & Friebe, A. (2010). Nitric oxide-sensitive guanylyl cyclase is the only nitric oxide receptor mediating platelet inhibition. Journal of Thrombosis and Haemostasis, 8(6), 1343–1352.
Koziak, K., Sevigny, J., Robson, S. C., Siegel, J. B., & Kaczmarek, E. (1999). Analysis of CD39/ATP diphosphohydrolase (ATPDase) expression in endothelial cells, platelets and leukocytes. Thrombosis and Haemostasis, 82(5), 1538–1544.
Moncada, S., Gryglewski, R., Bunting, S., & Vane, J. R. (1976). An enzyme isolated from arteries transforms prostaglandin endoperoxides to an unstable substance that inhibits platelet aggregation. Nature, 263(5579), 663–665.
Sabetkar, M., Naseem, K. M., Tullett, J. M., Friebe, A., Koesling, D., & Bruckdorfer, K. R. (2001). Synergism between nitric oxide and hydrogen peroxide in the inhibition of platelet function: The roles of soluble guanylyl cyclase and vasodilator-stimulated phosphoprotein. Nitric Oxide, 5(3), 233–242.
Zimmermann, H. (1999). Nucleotides and cd39: Principal modulatory players in hemostasis and thrombosis. Nature Medicine, 5(9), 987–988.
O'Brien, M. P., Hunt, P. W., Kitch, D. W., Klingman, K., Stein, J. H., Funderburg, N. T., et al. (2017). A randomized placebo controlled trial of aspirin effects on immune activation in chronically human immunodeficiency virus-infected adults on virologically suppressive antiretroviral therapy. Open Forum Infect Dis, 4(1), ofw278.
O’Brien, M., Montenont, E., Hu, L., Nardi, M. A., Valdes, V., Merolla, M., et al. (2013). Aspirin attenuates platelet activation and immune activation in HIV-1-infected subjects on antiretroviral therapy: A pilot study. Journal of Acquired Immune Deficiency Syndromes, 63(3), 280–288.
Loelius, S. G., Lannan, K. L., Blumberg, N., Phipps, R. P., & Spinelli, S. L. (2018). The HIV protease inhibitor, ritonavir, dysregulates human platelet function in vitro. Thrombosis Research, 169, 96–104.
Hammarstrom, S. (1982). Biosynthesis and biological actions of prostaglandins and thromboxanes. Archives of Biochemistry and Biophysics, 214(2), 431–445.
Antal, A., Gecse, A., Mojzes, L., & Foldes, V. (1985). Arachidonate cascade in the platelets of influenza virus infected mice. Acta Med Leg Soc (Liege), 35(2), 1–7.
Feletou, M., Huang, Y., & Vanhoutte, P. M. (2010). Vasoconstrictor prostanoids. Pflugers Archiv. European Journal of Physiology, 459(6), 941–950.
Kandhi, S., Zhang, B., Froogh, G., Qin, J., Alruwali, N., Le, Y., et al. (2017). EETs promote hypoxic pulmonary vasoconstriction via constrictor prostanoids. Am J Physiol Lung Cell Mol Physiol, 00038, 02017.
Bhagwat, S. S., Hamann, P. R., Still, W. C., Bunting, S., & Fitzpatrick, F. A. (1985). Synthesis and structure of the platelet aggregation factor thromboxane A2. Nature, 315(6019), 511–513.
Hamberg, M., Svensson, J., & Samuelsson, B. (1975). Thromboxanes: A new group of biologically active compounds derived from prostaglandin endoperoxides. Proc Natl Acad Sci U S A, 72(8), 2994–2998.
Gawaz, M., & Vogel, S. (2013). Platelets in tissue repair: Control of apoptosis and interactions with regenerative cells. Blood, 122(15), 2550–2554.
Fukami, M. H., & Salganicoff, L. (1977). Human platelet storage organelles. A review. Thromb Haemost, 38(4), 963–970.
Koseoglu, S., & Flaumenhaft, R. (2013). Advances in platelet granule biology. Current Opinion in Hematology, 20(5), 464–471.
Wihlborg, A. K., Wang, L., Braun, O. O., Eyjolfsson, A., Gustafsson, R., Gudbjartsson, T., et al. (2004). ADP receptor P2Y12 is expressed in vascular smooth muscle cells and stimulates contraction in human blood vessels. Arteriosclerosis, Thrombosis, and Vascular Biology, 24(10), 1810–1815.
Vanags, D. M., Rodgers, S. E., Duncan, E. M., Lloyd, J. V., & Bochner, F. (1992). Potentiation of ADP-induced aggregation in human platelet-rich plasma by 5-hydroxytryptamine and adrenaline. British Journal of Pharmacology, 106(4), 917–923.
Marketou, M. E., Kintsurashvili, E., Androulakis, N. E., Kontaraki, J., Alexandrakis, M. G., Gavras, I., et al. (2013). Blockade of platelet alpha2B-adrenergic receptors: A novel antiaggregant mechanism. International Journal of Cardiology, 168(3), 2561–2566.
Ponomarev, E. D. (2018). Fresh evidence for platelets as neuronal and innate immune cells: Their role in the activation, differentiation, and deactivation of Th1, Th17, and Tregs during tissue inflammation. Frontiers in Immunology, 9, 406.
Luo, P., Liu, D., & Li, J. (2020). Epinephrine use in COVID-19: Friend or foe? Eur J Hosp Pharm
Belamarich, F. A., Fusari, M. H., Shepro, D., & Kien, M. (1966). In vitro studies of aggregation of non-mammalian thrombocytes. Nature, 212(5070), 1579–1580.
Chen, W., Tang, D., Xu, Y., Zou, Y., Sui, W., Dai, Y., et al. (2018). Comprehensive analysis of lysine crotonylation in proteome of maintenance hemodialysis patients. Medicine (Baltimore), 97(37), e12035.
Stritt, S., Beck, S., Becker, I. C., Vogtle, T., Hakala, M., Heinze, K. G., et al. (2017). Twinfilin 2a regulates platelet reactivity and turnover in mice. Blood, 130(15), 1746–1756.
Hui, H., Fuller, K. A., Erber, W. N., & Linden, M. D. (2017). Imaging flow cytometry in the assessment of leukocyte-platelet aggregates. Methods, 112, 46–54.
Lowe, K. L., Finney, B. A., Deppermann, C., Hagerling, R., Gazit, S. L., Frampton, J., et al. (2015). Podoplanin and CLEC-2 drive cerebrovascular patterning and integrity during development. Blood, 125(24), 3769–3777.
Qiu, Y., Brown, A. C., Myers, D. R., Sakurai, Y., Mannino, R. G., Tran, R., et al. (2014). Platelet mechanosensing of substrate stiffness during clot formation mediates adhesion, spreading, and activation. Proc Natl Acad Sci U S A, 111(40), 14430–14435.
Peerschke, E. I., Reid, K. B., & Ghebrehiwet, B. (1993). Platelet activation by C1q results in the induction of alpha IIb/beta 3 integrins (GPIIb-IIIa) and the expression of P-selectin and procoagulant activity. Journal of Experimental Medicine, 178(2), 579–587.
Bevilacqua, M. P., Stengelin, S., Gimbrone, M. A., Jr., & Seed, B. (1989). Endothelial leukocyte adhesion molecule 1: An inducible receptor for neutrophils related to complement regulatory proteins and lectins. Science, 243(4895), 1160–1165.
Menter, D. G., Steinert, B. W., Sloane, B. F., Gundlach, N., O’Gara, C. Y., Marnett, L. J., et al. (1987). Role of platelet membrane in enhancement of tumor cell adhesion to endothelial cell extracellular matrix. Cancer Research, 47(24 Pt 1), 6751–6762.
Menter, D. G., Sloane, B. F., Steinert, B. W., Onoda, J., Craig, R., Harkins, C., et al. (1987). Platelet enhancement of tumor cell adhesion to subendothelial matrix: Role of platelet cytoskeleton and platelet membrane. Journal of the National Cancer Institute, 79(5), 1077–1090.
Hoffmann, M., Kleine-Weber, H., Schroeder, S., Kruger, N., Herrler, T., Erichsen, S., et al. (2020). SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell, 181(2), 271–280 e278.
Lan, J., Ge, J., Yu, J., Shan, S., Zhou, H., Fan, S., et al. (2020). Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature
Shang, J., Ye, G., Shi, K., Wan, Y., Luo, C., Aihara, H., et al. (2020). Structural basis of receptor recognition by SARS-CoV-2. Nature
Sun, S., Cai, X., Wang, H., He, G., Lin, Y., Lu, B., et al. (2020). Abnormalities of peripheral blood system in patients with COVID-19 in Wenzhou, China. Clinica Chimica Acta, 507, 174–180.
Wrapp, D., Wang, N., Corbett, K. S., Goldsmith, J. A., Hsieh, C. L., Abiona, O., et al. (2020). Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science, 367(6483), 1260–1263.
Yan, R., Zhang, Y., Li, Y., Xia, L., Guo, Y., & Zhou, Q. (2020). Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science, 367(6485), 1444–1448.
Varga, Z., Flammer, A. J., Steiger, P., Haberecker, M., Andermatt, R., Zinkernagel, A. S., et al. (2020). Endothelial cell infection and endotheliitis in COVID-19. Lancet, 395(10234), 1417–1418.
Ackermann, M., Verleden, S. E., Kuehnel, M., Haverich, A., Welte, T., Laenger, F., et al. (2020). Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N Engl J Med
Azkur, A. K., Akdis, M., Azkur, D., Sokolowska, M., van de Veen, W., Bruggen, M. C., et al. (2020). Immune response to SARS-CoV-2 and mechanisms of immunopathological changes in COVID-19. Allergy
Nunes Duarte-Neto, A., de Almeida Monteiro, R. A., da Silva, L. F. F., Malheiros, D., de Oliveira, E. P., Theodoro Filho, J., et al. (2020). Pulmonary and systemic involvement of COVID-19 assessed by ultrasound-guided minimally invasive autopsy. Histopathology
O’Sullivan, J., Mc Gonagle, D., Ward, S., Preston, R., & O’Donnell, J. (2020). Endothelial cells orchestrate COVID-19 coagulopathy. Lancet Haematol, 30215
Barton, L., Duval, E., Stroberg, E., Ghosh, S., & Mukhopadhyay, S. (2020). COVID-19 Autopsies, Oklahoma, USA. American Journal of Clinical Pathology, 153, 725–733.
Matacic, C. (2020). Blood vessel injury may spur disease’s fatal second phase. Science, 368(6495), 1039–1040.
Hay, S. B., Ferchen, K., Chetal, K., Grimes, H. L., & Salomonis, N. (2018). The Human Cell Atlas bone marrow single-cell interactive web portal. Experimental Hematology, 68, 51–61.
Tikhonova, A. N., Dolgalev, I., Hu, H., Sivaraj, K. K., Hoxha, E., Cuesta-Dominguez, A., et al. (2019). The bone marrow microenvironment at single-cell resolution. Nature, 569(7755), 222–228.
Rossaint, J., & Zarbock, A. (2015). Platelets in leucocyte recruitment and function. Cardiovasc Res
Garraud, O., Berthet, J., Hamzeh-Cognasse, H., & Cognasse, F. (2011). Pathogen sensing, subsequent signalling, and signalosome in human platelets. Thrombosis Research, 127(4), 283–286.
von Hundelshausen, P., & Weber, C. (2007). Platelets as immune cells: Bridging inflammation and cardiovascular disease. Circulation Research, 100(1), 27–40.
D’Atri, L. P., Etulain, J., Rivadeneyra, L., Lapponi, M. J., Centurion, M., Cheng, K., et al. (2015). Expression and functionality of Toll-like receptor 3 in the megakaryocytic lineage. Journal of Thrombosis and Haemostasis, 13(5), 839–850.
Kullaya, V. I., de Mast, Q., van der Ven, A., elMoussaoui, H., Kibiki, G., Simonetti, E., et al. (2017). Platelets modulate innate immune response against human respiratory syncytial virus in vitro. Viral Immunology, 30(8), 576–581.
LP, D. A., & Schattner, M. (2017). Platelet toll-like receptors in thromboinflammation. Front Biosci (Landmark Ed), 22, 1867–1883.
Thon, J. N., Peters, C. G., Machlus, K. R., Aslam, R., Rowley, J., Macleod, H., et al. (2012). T granules in human platelets function in TLR9 organization and signaling. Journal of Cell Biology, 198(4), 561–574.
Rath, D., Chatterjee, M., Borst, O., Muller, K., Langer, H., Mack, A. F., et al. (2015). Platelet surface expression of stromal cell-derived factor-1 receptors CXCR4 and CXCR7 is associated with clinical outcomes in patients with coronary artery disease. Journal of Thrombosis and Haemostasis, 13(5), 719–728.
Rafii, S., Cao, Z., Lis, R., Siempos, I. I., Chavez, D., Shido, K., et al. (2015). Platelet-derived SDF-1 primes the pulmonary capillary vascular niche to drive lung alveolar regeneration. Nature Cell Biology, 17(2), 123–136.
Chatterjee, M., Seizer, P., Borst, O., Schonberger, T., Mack, A., Geisler, T., et al. (2014). SDF-1alpha induces differential trafficking of CXCR4-CXCR7 involving cyclophilin A, CXCR7 ubiquitination and promotes platelet survival. The FASEB Journal, 28(7), 2864–2878.
Rath, D., Chatterjee, M., Borst, O., Muller, K., Stellos, K., Mack, A. F., et al. (2014). Expression of stromal cell-derived factor-1 receptors CXCR4 and CXCR7 on circulating platelets of patients with acute coronary syndrome and association with left ventricular functional recovery. European Heart Journal, 35(6), 386–394.
Iannacone, M. (2016). Platelet-mediated modulation of adaptive immunity. Seminars in Immunology, 28(6), 555–560.
Danese, S., & Fiocchi, C. (2016). Endothelial cell-immune cell interaction in IBD. Digestive Diseases, 34(1–2), 43–50.
Chatterjee, M., & Geisler, T. (2016). Inflammatory contribution of platelets revisited: New players in the arena of inflammation. Seminars in Thrombosis and Hemostasis, 42(3), 205–214.
Carestia, A., Kaufman, T., & Schattner, M. (2016). Platelets: New bricks in the building of neutrophil extracellular traps. Frontiers in Immunology, 7, 271.
Lam, F. W., Vijayan, K. V., & Rumbaut, R. E. (2015). Platelets and their interactions with other immune cells. Comprehensive Physiology, 5(3), 1265–1280.
Kapur, R., Zufferey, A., Boilard, E., & Semple, J. W. (2015). Nouvelle cuisine: Platelets served with inflammation. The Journal of Immunology, 194(12), 5579–5587.
Cognasse, F., Nguyen, K. A., Damien, P., McNicol, A., Pozzetto, B., Hamzeh-Cognasse, H., et al. (2015). The inflammatory role of platelets via their TLRs and Siglec receptors. Frontiers in Immunology, 6, 83.
Chatterjee, M., Rath, D., & Gawaz, M. (2015). Role of chemokine receptors CXCR4 and CXCR7 for platelet function. Biochemical Society Transactions, 43(4), 720–726.
Coburn, L. A., Damaraju, V. S., Dozic, S., Eskin, S. G., Cruz, M. A., & McIntire, L. V. (2011). GPIbalpha-vWF rolling under shear stress shows differences between type 2B and 2M von Willebrand disease. Biophysical Journal, 100(2), 304–312.
Colace, T. V., & Diamond, S. L. (2013). Direct observation of von Willebrand factor elongation and fiber formation on collagen during acute whole blood exposure to pathological flow. Arteriosclerosis, Thrombosis, and Vascular Biology, 33(1), 105–113.
Fredrickson, B. J., Dong, J. F., McIntire, L. V., & Lopez, J. A. (1998). Shear-dependent rolling on von Willebrand factor of mammalian cells expressing the platelet glycoprotein Ib-IX-V complex. Blood, 92(10), 3684–3693.
Jackson, S. P., Mistry, N., & Yuan, Y. (2000). Platelets and the injured vessel wall– “rolling into action”: Focus on glycoprotein Ib/V/IX and the platelet cytoskeleton. Trends in Cardiovascular Medicine, 10(5), 192–197.
Yago, T., Lou, J., Wu, T., Yang, J., Miner, J. J., Coburn, L., et al. (2008). Platelet glycoprotein Ibalpha forms catch bonds with human WT vWF but not with type 2B von Willebrand disease vWF. The Journal of Clinical Investigation, 118(9), 3195–3207.
Coller, B. S., & Shattil, S. J. (2008). The GPIIb/IIIa (integrin alphaIIbbeta3) odyssey: A technology-driven saga of a receptor with twists, turns, and even a bend. Blood, 112(8), 3011–3025.
Kim, C., & Kim, M. C. (2013). Differences in alpha-beta transmembrane domain interactions among integrins enable diverging integrin signaling. Biochemical and Biophysical Research Communications, 436(3), 406–412.
Kim, C., Lau, T. L., Ulmer, T. S., & Ginsberg, M. H. (2009). Interactions of platelet integrin alphaIIb and beta3 transmembrane domains in mammalian cell membranes and their role in integrin activation. Blood, 113(19), 4747–4753.
Shattil, S. J. (2009). The beta3 integrin cytoplasmic tail: Protein scaffold and control freak. Journal of Thrombosis and Haemostasis, 7(Suppl 1), 210–213.
Assinger, A., Kral, J. B., Yaiw, K. C., Schrottmaier, W. C., Kurzejamska, E., Wang, Y., et al. (2014). Human cytomegalovirus-platelet interaction triggers toll-like receptor 2-dependent proinflammatory and proangiogenic responses. Arteriosclerosis, Thrombosis, and Vascular Biology, 34(4), 801–809.
Nelsen-Salz, B., Eggers, H. J., & Zimmermann, H. (1999). Integrin alpha(v)beta3 (vitronectin receptor) is a candidate receptor for the virulent echovirus 9 strain Barty. Journal of General Virology, 80(Pt 9), 2311–2313.
Bruning, A., Kohler, T., Quist, S., Wang-Gohrke, S., Moebus, V. J., Kreienberg, R., et al. (2001). Adenoviral transduction efficiency of ovarian cancer cells can be limited by loss of integrin beta3 subunit expression and increased by reconstitution of integrin alphavbeta3. Human Gene Therapy, 12(4), 391–399.
Chabert, A., Hamzeh-Cognasse, H., Pozzetto, B., Cognasse, F., Schattner, M., Gomez, R. M., et al. (2015). Human platelets and their capacity of binding viruses: Meaning and challenges? BMC Immunology, 16, 26.
Vilela, M. C., Lima, G. K., Rodrigues, D. H., Lacerda-Queiroz, N., Pedroso, V. S., de Miranda, A. S., et al. (2016). Platelet Activating Factor (PAF) Receptor Deletion or Antagonism Attenuates Severe HSV-1 Meningoencephalitis. Journal of Neuroimmune Pharmacology, 11(4), 613–621.
Fleming, F. E., Graham, K. L., Takada, Y., & Coulson, B. S. (2011). Determinants of the specificity of rotavirus interactions with the alpha2beta1 integrin. Journal of Biological Chemistry, 286(8), 6165–6174.
Bergmeier, W., & Stefanini, L. (2013). Platelet ITAM signaling. Current Opinion in Hematology, 20(5), 445–450.
Ozaki, Y., Suzuki-Inoue, K., & Inoue, O. (2013). Platelet receptors activated via mulitmerization: Glycoprotein VI, GPIb-IX-V, and CLEC-2. Journal of Thrombosis and Haemostasis, 11(Suppl 1), 330–339.
Zahid, M., Mangin, P., Loyau, S., Hechler, B., Billiald, P., Gachet, C., et al. (2012). The future of glycoprotein VI as an antithrombotic target. Journal of Thrombosis and Haemostasis, 10(12), 2418–2427.
Takemoto, A., Okitaka, M., Takagi, S., Takami, M., Sato, S., Nishio, M., et al. (2017). A critical role of platelet TGF-beta release in podoplanin-mediated tumour invasion and metastasis. Science and Reports, 7, 42186.
Nakazawa, Y., Sato, S., Naito, M., Kato, Y., Mishima, K., Arai, H., et al. (2008). Tetraspanin family member CD9 inhibits Aggrus/podoplanin-induced platelet aggregation and suppresses pulmonary metastasis. Blood, 112(5), 1730–1739.
Navarro-Nunez, L., Langan, S. A., Nash, G. B., & Watson, S. P. (2013). The physiological and pathophysiological roles of platelet CLEC-2. Thrombosis and Haemostasis, 109(6), 991–998.
Suzuki-Inoue, K., Fuller, G. L., Garcia, A., Eble, J. A., Pohlmann, S., Inoue, O., et al. (2006). A novel Syk-dependent mechanism of platelet activation by the C-type lectin receptor CLEC-2. Blood, 107(2), 542–549.
Suzuki-Inoue, K., Kato, Y., Inoue, O., Kaneko, M. K., Mishima, K., Yatomi, Y., et al. (2007). Involvement of the snake toxin receptor CLEC-2, in podoplanin-mediated platelet activation, by cancer cells. Journal of Biological Chemistry, 282(36), 25993–26001.
Pula, B., Witkiewicz, W., Dziegiel, P., & Podhorska-Okolow, M. (2013). Significance of podoplanin expression in cancer-associated fibroblasts: A comprehensive review. International Journal of Oncology, 42(6), 1849–1857.
Gitz, E., Pollitt, A. Y., Gitz-Francois, J. J., Alshehri, O., Mori, J., Montague, S., et al. (2014). CLEC-2 expression is maintained on activated platelets and on platelet microparticles. Blood, 124(14), 2262–2270.
Golda, A., Malek, N., Dudek, B., Zeglen, S., Wojarski, J., Ochman, M., et al. (2011). Infection with human coronavirus NL63 enhances streptococcal adherence to epithelial cells. Journal of General Virology, 92(Pt 6), 1358–1368.
Sung, P. S., & Hsieh, S. L. (2019). CLEC2 and CLEC5A: Pathogenic host factors in acute viral infections. Frontiers in Immunology, 10, 2867.
Johnston, G. I., Cook, R. G., & McEver, R. P. (1989). Cloning of GMP-140, a granule membrane protein of platelets and endothelium: Sequence similarity to proteins involved in cell adhesion and inflammation. Cell, 56(6), 1033–1044.
Stenberg, P. E., McEver, R. P., Shuman, M. A., Jacques, Y. V., & Bainton, D. F. (1985). A platelet alpha-granule membrane protein (GMP-140) is expressed on the plasma membrane after activation. Journal of Cell Biology, 101(3), 880–886.
Zarbock, A., Muller, H., Kuwano, Y., & Ley, K. (2009). PSGL-1-dependent myeloid leukocyte activation. Journal of Leukocyte Biology, 86(5), 1119–1124.
Picker, L. J., Warnock, R. A., Burns, A. R., Doerschuk, C. M., Berg, E. L., & Butcher, E. C. (1991). The neutrophil selectin LECAM-1 presents carbohydrate ligands to the vascular selectins ELAM-1 and GMP-140. Cell, 66(5), 921–933.
Polley, M. J., Phillips, M. L., Wayner, E., Nudelman, E., Singhal, A. K., Hakomori, S., et al. (1991). CD62 and endothelial cell-leukocyte adhesion molecule 1 (ELAM-1) recognize the same carbohydrate ligand, sialyl-Lewis x. Proc Natl Acad Sci U S A, 88(14), 6224–6228.
Foxall, C., Watson, S. R., Dowbenko, D., Fennie, C., Lasky, L. A., Kiso, M., et al. (1992). The three members of the selectin receptor family recognize a common carbohydrate epitope, the sialyl Lewis(x) oligosaccharide. Journal of Cell Biology, 117(4), 895–902.
Habets, K. L., Huizinga, T. W., & Toes, R. E. (2013). Platelets and autoimmunity. European Journal of Clinical Investigation, 43(7), 746–757.
Kazmi, R. S., Cooper, A. J., & Lwaleed, B. A. (2011). Platelet function in pre-eclampsia. Seminars in Thrombosis and Hemostasis, 37(2), 131–136.
Nurden, A. T. (2011). Platelets, inflammation and tissue regeneration. Thrombosis and Haemostasis, 105(Suppl 1), S13-33.
Negrotto, S., Jaquenod de Giusti, C., Rivadeneyra, L., Ure, A. E., Mena, H. A., Schattner, M., et al. (2015). Platelets interact with Coxsackieviruses B and have a critical role in the pathogenesis of virus-induced myocarditis. Journal of Thrombosis and Haemostasis, 13(2), 271–282.
Moore, K. L., Varki, A., & McEver, R. P. (1991). GMP-140 binds to a glycoprotein receptor on human neutrophils: Evidence for a lectin-like interaction. Journal of Cell Biology, 112(3), 491–499.
Borsig, L. (2008). The role of platelet activation in tumor metastasis. Expert Review of Anticancer Therapy, 8(8), 1247–1255.
Dammacco, F., Vacca, A., Procaccio, P., Ria, R., Marech, I., & Racanelli, V. (2013). Cancer-related coagulopathy (Trousseau’s syndrome): Review of the literature and experience of a single center of internal medicine. Clinical and Experimental Medicine, 13(2), 85–97.
Erpenbeck, L., & Schon, M. P. (2010). Deadly allies: The fatal interplay between platelets and metastasizing cancer cells. Blood, 115(17), 3427–3436.
Gay, L. J., & Felding-Habermann, B. (2011). Contribution of platelets to tumour metastasis. Nature Reviews Cancer, 11(2), 123–134.
Gay, L. J., & Felding-Habermann, B. (2011). Platelets alter tumor cell attributes to propel metastasis: Programming in transit. Cancer Cell, 20(5), 553–554.
Kyriazi, V., & Theodoulou, E. (2013). Assessing the risk and prognosis of thrombotic complications in cancer patients. Archives of Pathology and Laboratory Medicine, 137(9), 1286–1295.
McEver, R. P. (1997). Selectin-carbohydrate interactions during inflammation and metastasis. Glycoconjugate Journal, 14(5), 585–591.
Arnout, J., Hoylaerts, M. F., & Lijnen, H. R. (2006). Haemostasis. Handb Exp Pharmacol(176 Pt 2), 1–41.
Gaitzsch, E., Czermak, T., Ribeiro, A., Heun, Y., Bohmer, M., Merkle, M., et al. (2017). Double-stranded DNA induces a prothrombotic phenotype in the vascular endothelium. Science and Reports, 7(1), 1112.
Antoniak, S., Boltzen, U., Riad, A., Kallwellis-Opara, A., Rohde, M., Dorner, A., et al. (2008). Viral myocarditis and coagulopathy: Increased tissue factor expression and plasma thrombogenicity. Journal of Molecular and Cellular Cardiology, 45(1), 118–126.
Dang, O., Navarro, L., & David, M. (2004). Inhibition of lipopolysaccharide-induced interferon regulatory factor 3 activation and protection from septic shock by hydroxystilbenes. Shock, 21(5), 470–475.
Visseren, F. L., Bouwman, J. J., Bouter, K. P., Diepersloot, R. J., de Groot, P. H., & Erkelens, D. W. (2000). Procoagulant activity of endothelial cells after infection with respiratory viruses. Thrombosis and Haemostasis, 84(2), 319–324.
Amgalan, A., & Othman, M. (2020). Exploring possible mechanisms for COVID-19 induced thrombocytopenia: Unanswered questions. Journal of Thrombosis and Haemostasis, 18(6), 1514–1516.
Chan, J. F., Yuan, S., Kok, K. H., To, K. K., Chu, H., Yang, J., et al. (2020). A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: A study of a family cluster. Lancet, 395(10223), 514–523.
Giannis, D., Ziogas, I. A., & Gianni, P. (2020). Coagulation disorders in coronavirus infected patients: COVID-19, SARS-CoV-1, MERS-CoV and lessons from the past. J Clin Virol, 127, 104362.
Lippi, G., Plebani, M., & Henry, B. M. (2020). Thrombocytopenia is associated with severe coronavirus disease 2019 (COVID-19) infections: A meta-analysis. Clinica Chimica Acta, 506, 145–148.
Misra, D. P., Agarwal, V., Gasparyan, A. Y., & Zimba, O. (2020). Rheumatologists' perspective on coronavirus disease 19 (COVID-19) and potential therapeutic targets. Clin Rheumatol
Rasmussen, S. A., Smulian, J. C., Lednicky, J. A., Wen, T. S., & Jamieson, D. J. (2020). Coronavirus disease 2019 (COVID-19) and pregnancy: What obstetricians need to know. Am J Obstet Gynecol
Sri Santosh, T., Parmar, R., Anand, H., Srikanth, K., & Saritha, M. (2020). A review of salivary diagnostics and its potential implication in detection of Covid-19. Cureus, 12(4), e7708.
Terpos, E., Ntanasis-Stathopoulos, I., Elalamy, I., Kastritis, E., Sergentanis, T. N., Politou, M., et al. (2020). Hematological findings and complications of COVID-19. Am J Hematol
Xu, P., Zhou, Q., & Xu, J. (2020). Mechanism of thrombocytopenia in COVID-19 patients. Ann Hematol
Yang, X., Yang, Q., Wang, Y., Wu, Y., Xu, J., Yu, Y., et al. (2020). Thrombocytopenia and its association with mortality in patients with COVID-19. J Thromb Haemost
Sundaramoorthi, H., Panapakam, R., & Jagadeeswaran, P. (2015). Zebrafish thrombocyte aggregation by whole blood aggregometry and flow cytometry. Platelets, 26(7), 613–619.
Feng, D., Nagy, J. A., Pyne, K., Dvorak, H. F., & Dvorak, A. M. (1998). Platelets exit venules by a transcellular pathway at sites of F-met peptide-induced acute inflammation in guinea pigs. International Archives of Allergy and Immunology, 116(3), 188–195.
Lowenhaupt, R. W., Glueck, H. I., Miller, M. A., & Kline, D. L. (1977). Factors which influence blood platelet migration. Journal of Laboratory and Clinical Medicine, 90(1), 37–45.
Nathan, P. (1973). The migration of human platelets in vitro. Thrombosis et Diathesis Haemorrhagica, 30(1), 173–177.
Schmidt, E. M., Munzer, P., Borst, O., Kraemer, B. F., Schmid, E., Urban, B., et al. (2011). Ion channels in the regulation of platelet migration. Biochemical and Biophysical Research Communications, 415(1), 54–60.
Iba, T., Levy, J. H., Levi, M., Connors, J. M., & Thachil, J. (2020). Coagulopathy of Coronavirus Disease 2019. Crit Care Med
Levi, M., Thachil, J., Iba, T., & Levy, J. H. (2020). Coagulation abnormalities and thrombosis in patients with COVID-19. Lancet Haematol, 7(6), e438–e440.
The Lancet, H. (2020). COVID-19 coagulopathy: an evolving story. Lancet Haematol, 7(6), e425.
Huang, C., Wang, Y., Li, X., Ren, L., Zhao, J., Hu, Y., et al. (2020). Clinical features of patients infected with 2019 novel coronavirus in Wuhan. China. Lancet, 395(10223), 497–506.
Bellosta, R., Luzzani, L., Natalini, G., Pegorer, M. A., Attisani, L., Cossu, L. G., et al. (2020). Acute limb ischemia in patients with COVID-19 pneumonia. J Vasc Surg
Rogers, L. C., Lavery, L. A., Joseph, W. S., & Armstrong, D. G. (2020). All Feet On Deck-The Role of Podiatry During the COVID-19 Pandemic: Preventing hospitalizations in an overburdened healthcare system, reducing amputation and death in people with diabetes. J Am Podiatr Med Assoc
Baracchini, C., Pieroni, A., Viaro, F., Cianci, V., Cattelan, A. M., Tiberio, I., et al. (2020). Acute stroke management pathway during Coronavirus-19 pandemic. Neurological Sciences, 41(5), 1003–1005.
Beyrouti, R., Adams, M. E., Benjamin, L., Cohen, H., Farmer, S. F., Goh, Y. Y., et al. (2020). Characteristics of ischaemic stroke associated with COVID-19. J Neurol Neurosurg Psychiatry
Calcagno, N., Colombo, E., Maranzano, A., Pasquini, J., Keller Sarmiento, I. J., Trogu, F., et al. (2020). Rising evidence for neurological involvement in COVID-19 pandemic. Neurol Sci
Kansagra, A. P., Goyal, M. S., Hamilton, S., & Albers, G. W. (2020). Collateral effect of Covid-19 on stroke evaluation in the United States. N Engl J Med
Larson, A. S., Savastano, L., Kadirvel, R., Kallmes, D. F., Hassan, A. E., & Brinjikji, W. (2020). COVID-19 and the cerebro-cardiovascular systems: What do we know so far? J Am Heart Assoc, e016793.
Oxley, T. J., Mocco, J., Majidi, S., Kellner, C. P., Shoirah, H., Singh, I. P., et al. (2020). Large-vessel stroke as a presenting feature of Covid-19 in the young. N Engl J Med, 382(20), e60.
Valderrama, E. V., Humbert, K., Lord, A., Frontera, J., & Yaghi, S. (2020). Severe acute respiratory syndrome coronavirus 2 infection and ischemic stroke. Stroke, STROKEAHA120030153.
Viguier, A., Delamarre, L., Duplantier, J., Olivot, J. M., & Bonneville, F. (2020). Acute ischemic stroke complicating common carotid artery thrombosis during a severe COVID-19 infection. J Neuroradiol
Zhou, Y., Yasumoto, A., Lei, C., Huang, C. J., Kobayashi, H., Wu, Y., et al. (2020). Intelligent classification of platelet aggregates by agonist type. Elife, 9
Dong, X., Cao, Y. Y., Lu, X. X., Zhang, J. J., Du, H., Yan, Y. Q., et al. (2020). Eleven faces of coronavirus disease 2019. Allergy
Li, N., Wang, X., & Lv, T. (2020). Prolonged SARS-CoV-2 RNA shedding: Not a rare phenomenon. J Med Virol
Long, Q. X., Liu, B. Z., Deng, H. J., Wu, G. C., Deng, K., Chen, Y. K., et al. (2020). Antibody responses to SARS-CoV-2 in patients with COVID-19. Nat Med
Qu, J., Wu, C., Li, X., Zhang, G., Jiang, Z., Li, X., et al. (2020). Profile of IgG and IgM antibodies against severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Clin Infect Dis
Xiang, F., Wang, X., He, X., Peng, Z., Yang, B., Zhang, J., et al. (2020). Antibody detection and dynamic characteristics in patients with COVID-19. Clin Infect Dis
Zhang, G., Nie, S., Zhang, Z., & Zhang, Z. (2020). Longitudinal change of SARS-Cov2 antibodies in patients with COVID-19. J Infect Dis
Payne, D. C., Smith-Jeffcoat, S. E., Nowak, G., Chukwuma, U., Geibe, J. R., Hawkins, R. J., et al. (2020). SARS-CoV-2 infections and serologic responses from a sample of U.S. Navy Service Members - USS Theodore Roosevelt, April 2020. MMWR Morb Mortal Wkly Rep, 69(23), 714–721.
Cai, X. F., Chen, J., Hu, J. L., Long, Q. X., Deng, H. J., Fan, K., et al. (2020). A peptide-based magnetic chemiluminescence enzyme immunoassay for serological diagnosis of coronavirus disease 2019 (COVID-19). J Infect Dis
Cassaniti, I., Novazzi, F., Giardina, F., Salinaro, F., Sachs, M., Perlini, S., et al. (2020). Performance of VivaDiag COVID-19 IgM/IgG rapid test is inadequate for diagnosis of COVID-19 in acute patients referring to emergency room department. J Med Virol
Castro, R., Luz, P. M., Wakimoto, M. D., Veloso, V. G., Grinsztejn, B., & Perazzo, H. (2020). COVID-19: A meta-analysis of diagnostic test accuracy of commercial assays registered in Brazil. Braz J Infect Dis
di Mauro, G., Cristina, S., Concetta, R., Francesco, R., & Annalisa, C. (2020). SARS-Cov-2 infection: Response of human immune system and possible implications for the rapid test and treatment. Int Immunopharmacol, 84, 106519.
Dohla, M., Boesecke, C., Schulte, B., Diegmann, C., Sib, E., Richter, E., et al. (2020). Rapid point-of-care testing for SARS-CoV-2 in a community screening setting shows low sensitivity. Public Health, 182, 170–172.
Hou, H., Wang, T., Zhang, B., Luo, Y., Mao, L., Wang, F., et al. (2020). Detection of IgM and IgG antibodies in patients with coronavirus disease 2019. Clin Transl Immunology, 9(5), e01136.
Infantino, M., Grossi, V., Lari, B., Bambi, R., Perri, A., Manneschi, M., et al. (2020). Diagnostic accuracy of an automated chemiluminescent immunoassay for anti-SARS-CoV-2 IgM and IgG antibodies: an Italian experience. J Med Virol
Jin, Y., Wang, M., Zuo, Z., Fan, C., Ye, F., Cai, Z., et al. (2020). Diagnostic value and dynamic variance of serum antibody in coronavirus disease 2019. International Journal of Infectious Diseases, 94, 49–52.
Li, Z., Yi, Y., Luo, X., Xiong, N., Liu, Y., Li, S., et al. (2020). Development and clinical application of a rapid IgM-IgG combined antibody test for SARS-CoV-2 infection diagnosis. J Med Virol
Lippi, G., Salvagno, G. L., Pegoraro, M., Militello, V., Caloi, C., Peretti, A., et al. (2020). Assessment of immune response to SARS-CoV-2 with fully automated MAGLUMI 2019-nCoV IgG and IgM chemiluminescence immunoassays. Clin Chem Lab Med
Liu, W., Liu, L., Kou, G., Zheng, Y., Ding, Y., Ni, W., et al. (2020). Evaluation of nucleocapsid and spike protein-based ELISAs for detecting antibodies against SARS-CoV-2. J Clin Microbiol
Padoan, A., Cosma, C., Sciacovelli, L., Faggian, D., & Plebani, M. (2020). Analytical performances of a chemiluminescence immunoassay for SARS-CoV-2 IgM/IgG and antibody kinetics. Clin Chem Lab Med
Shen, B., Zheng, Y., Zhang, X., Zhang, W., Wang, D., Jin, J., et al. (2020). Clinical evaluation of a rapid colloidal gold immunochromatography assay for SARS-Cov-2 IgM/IgG. Am J Transl Res, 12(4), 1348–1354.
Spicuzza, L., Montineri, A., Manuele, R., Crimi, C., Pistorio, M. P., Campisi, R., et al. (2020). Reliability and usefulness of a rapid IgM-IgG antibody test for the diagnosis of SARS-CoV-2 infection: A preliminary report. J Infect
Vashist, S. K. (2020). In Vitro Diagnostic Assays for COVID-19: Recent Advances and Emerging Trends. Diagnostics (Basel), 10(4)
Wang, Q., Du, Q., Guo, B., Mu, D., Lu, X., Ma, Q., et al. (2020). A method to prevent SARS-CoV-2 IgM false positives in gold immunochromatography and enzyme-linked immunosorbent assays. J Clin Microbiol
Xie, J., Ding, C., Li, J., Wang, Y., Guo, H., Lu, Z., et al. (2020). Characteristics of patients with coronavirus disease (COVID-19) confirmed using an IgM-IgG antibody test. J Med Virol
Yong, G., Yi, Y., Tuantuan, L., Xiaowu, W., Xiuyong, L., Ang, L., et al. (2020). Evaluation of the auxiliary diagnostic value of antibody assays for the detection of novel coronavirus (SARS-CoV-2). J Med Virol
Zainol Rashid, Z., Othman, S. N., Abdul Samat, M. N., Ali, U. K., & Wong, K. K. (2020). Diagnostic performance of COVID-19 serology assays. Malaysian Journal of Pathology, 42(1), 13–21.
Zhong, L., Chuan, J., Gong, B., Shuai, P., Zhou, Y., Zhang, Y., et al. (2020). Detection of serum IgM and IgG for COVID-19 diagnosis. Sci China Life Sci, 63(5), 777–780.
Arman, M., & Krauel, K. (2015). Human platelet IgG Fc receptor FcgammaRIIA in immunity and thrombosis. Journal of Thrombosis and Haemostasis, 13(6), 893–908.
Worth, R. G., Chien, C. D., Chien, P., Reilly, M. P., McKenzie, S. E., & Schreiber, A. D. (2006). Platelet FcgammaRIIA binds and internalizes IgG-containing complexes. Experimental Hematology, 34(11), 1490–1495.
Boilard, E., Pare, G., Rousseau, M., Cloutier, N., Dubuc, I., Levesque, T., et al. (2014). Influenza virus H1N1 activates platelets through FcgammaRIIA signaling and thrombin generation. Blood, 123(18), 2854–2863.
Cloutier, N., Tan, S., Boudreau, L. H., Cramb, C., Subbaiah, R., Lahey, L., et al. (2013). The exposure of autoantigens by microparticles underlies the formation of potent inflammatory components: The microparticle-associated immune complexes. EMBO Molecular Medicine, 5(2), 235–249.
Johnston, I., Sarkar, A., Hayes, V., Koma, G. T., Arepally, G. M., Chen, J., et al. (2020). Recognition of PF4-VWF complexes by heparin-induced thrombocytopenia antibodies contributes to thrombus propagation. Blood, 135(15), 1270–1280.
Bossuyt, X., Moens, L., Van Hoeyveld, E., Jeurissen, A., Bogaert, G., Sauer, K., et al. (2007). Coexistence of (partial) immune defects and risk of recurrent respiratory infections. Clinical Chemistry, 53(1), 124–130.
Kyaw, A. K., Ngwe Tun, M. M., Moi, M. L., Nabeshima, T., Soe, K. T., Thwe, S. M., et al. (2017). Clinical, virological and epidemiological characterization of dengue outbreak in Myanmar, 2015. Epidemiology and Infection, 145(9), 1886–1897.
Payeras, A., Martinez, P., Mila, J., Riera, M., Pareja, A., Casal, J., et al. (2002). Risk factors in HIV-1-infected patients developing repetitive bacterial infections: Toxicological, clinical, specific antibody class responses, opsonophagocytosis and Fc(gamma) RIIa polymorphism characteristics. Clinical and Experimental Immunology, 130(2), 271–278.
Pereira, A., de Siqueira, T. R., de Oliveira Prado, A. A., da Silva, C. A. V., de Fatima Silva Moraes, T., Aleixo, A. A., et al. (2018). High prevalence of dengue antibodies and the arginine variant of the FcgammaRIIa polymorphism in asymptomatic individuals in a population of Minas Gerais State Southeast Brazil. Immunogenetics, 70(6), 355–362.
Sanchez Guiu, I. M., Martinez-Martinez, I., Martinez, C., Navarro-Fernandez, J., Garcia-Candel, F., Ferrer-Marin, F., et al. (2015). An atypical IgM class platelet cold agglutinin induces GPVI-dependent aggregation of human platelets. Thrombosis and Haemostasis, 114(2), 313–324.
Schmitz, H., Gabriel, M., & Emmerich, P. (2011). Specific detection of antibodies to different flaviviruses using a new immune complex ELISA. Medical Microbiology and Immunology, 200(4), 233–239.
Zhao, J., Yuan, Q., Wang, H., Liu, W., Liao, X., Su, Y., et al. (2020). Antibody responses to SARS-CoV-2 in patients of novel coronavirus disease 2019. Clin Infect Dis
Larsen, J. B., Pasalic, L., & Hvas, A. M. (2020). Platelets in Coronavirus Disease 2019. Semin Thromb Hemost
Guan, W. J., Ni, Z. Y., Hu, Y., Liang, W. H., Ou, C. Q., He, J. X., et al. (2020). Clinical characteristics of coronavirus disease 2019 in China. New England Journal of Medicine, 382(18), 1708–1720.
Zheng, Y., Zhang, Y., Chi, H., Chen, S., Peng, M., Luo, L., et al. (2020). The hemocyte counts as a potential biomarker for predicting disease progression in COVID-19: A retrospective study. Clin Chem Lab Med
Chen, R., Sang, L., Jiang, M., Yang, Z., Jia, N., Fu, W., et al. (2020). Longitudinal hematologic and immunologic variations associated with the progression of COVID-19 patients in China. J Allergy Clin Immunol
Henry, B. M., de Oliveira, M. H. S., Benoit, S., Plebani, M., & Lippi, G. (2020). Hematologic, biochemical and immune biomarker abnormalities associated with severe illness and mortality in coronavirus disease 2019 (COVID-19): A meta-analysis. Clin Chem Lab Med
Connors, J. M., & Levy, J. H. (2020). COVID-19 and its implications for thrombosis and anticoagulation. Blood
Cheng, Z., Lu, Y., Cao, Q., Qin, L., Pan, Z., Yan, F., et al. (2020). Clinical features and chest CT manifestations of coronavirus disease 2019 (COVID-19) in a single-center study in Shanghai, China. AJR Am J Roentgenol, 1–6.
Hwang, S. M., Na, B. J., Jung, Y., Lim, H. S., Seo, J. E., Park, S. A., et al. (2019). Clinical and laboratory findings of middle east respiratory syndrome coronavirus infection. Japanese Journal of Infectious Diseases, 72(3), 160–167.
Alfaraj, S. H., Al-Tawfiq, J. A., Altuwaijri, T. A., & Memish, Z. A. (2019). Middle East respiratory syndrome coronavirus in pediatrics: A report of seven cases from Saudi Arabia. Frontiers in Medicine, 13(1), 126–130.
Zuo, Y., Yalavarthi, S., Shi, H., Gockman, K., Zuo, M., Madison, J. A., et al. (2020). Neutrophil extracellular traps in COVID-19. JCI Insight
Clark, S. R., Ma, A. C., Tavener, S. A., McDonald, B., Goodarzi, Z., Kelly, M. M., et al. (2007). Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nature Medicine, 13(4), 463–469.
Peters, M. J., Dixon, G., Kotowicz, K. T., Hatch, D. J., Heyderman, R. S., & Klein, N. J. (1999). Circulating platelet-neutrophil complexes represent a subpopulation of activated neutrophils primed for adhesion, phagocytosis and intracellular killing. British Journal of Haematology, 106(2), 391–399.
de Gaetano, G., Cerletti, C., & Evangelista, V. (1999). Recent advances in platelet-polymorphonuclear leukocyte interaction. Haemostasis, 29(1), 41–49.
Koppang, E. O., Fischer, U., Satoh, M., & Jirillo, E. (2007). Inflammation in fish as seen from a morphological point of view with special reference to the vascular compartment. Current Pharmaceutical Design, 13(36), 3649–3655.
Xu, X., Han, M., Li, T., Sun, W., Wang, D., Fu, B., et al. (2020). {Luo, 2020 #20;Morrison, 2020 #11}Effective treatment of severe COVID-19 patients with tocilizumab. Proc Natl Acad Sci U S A
Aziz, M., Fatima, R., & Assaly, R. (2020). Elevated Interleukin-6 and Severe COVID-19: A Meta-Analysis. J Med Virol
Chen, X., Zhao, B., Qu, Y., Chen, Y., Xiong, J., Feng, Y., et al. (2020). Detectable serum SARS-CoV-2 viral load (RNAaemia) is closely correlated with drastically elevated interleukin 6 (IL-6) level in critically ill COVID-19 patients. Clin Infect Dis
Guzik, T. J., Mohiddin, S. A., Dimarco, A., Patel, V., Savvatis, K., Marelli-Berg, F. M., et al. (2020). COVID-19 and the cardiovascular system: Implications for risk assessment, diagnosis, and treatment options. Cardiovasc Res
Ke, C., Wang, Y., Zeng, X., Yang, C., & Hu, Z. (2020). 2019 novel coronavirus disease (COVID-19) in hemodialysis patients: A report of two cases. Clin Biochem
Liu, B., Li, M., Zhou, Z., Guan, X., & Xiang, Y. (2020). Can we use interleukin-6 (IL-6) blockade for coronavirus disease 2019 (COVID-19)-induced cytokine release syndrome (CRS)? J Autoimmun, 102452.
Wei, X. S., Wang, X. R., Zhang, J. C., Yang, W. B., Ma, W. L., Yang, B. H., et al. (2020). A cluster of health care workers with COVID-19 pneumonia caused by SARS-CoV-2. J Microbiol Immunol Infect
Yao, Z., Zheng, Z., Wu, K., & Junhua, Z. (2020). Immune environment modulation in pneumonia patients caused by coronavirus: SARS-CoV, MERS-CoV and SARS-CoV-2. Aging (Albany NY), 12
Zeng, J. H., Liu, Y. X., Yuan, J., Wang, F. X., Wu, W. B., Li, J. X., et al. (2020). First case of COVID-19 complicated with fulminant myocarditis: A case report and insights. Infection
Zhang, J., Yu, M., Tong, S., Liu, L. Y., & Tang, L. V. (2020). Predictive factors for disease progression in hospitalized patients with coronavirus disease 2019 in Wuhan, China. J Clin Virol, 127, 104392.
Zhou, Y., Han, T., Chen, J., Hou, C., Hua, L., He, S., et al. (2020). Clinical and autoimmune characteristics of severe and critical cases with COVID-19. Clin Transl Sci
Conti, P., Gallenga, C. E., Tete, G., Caraffa, A., Ronconi, G., Younes, A., et al. (2020). How to reduce the likelihood of coronavirus-19 (CoV-19 or SARS-CoV-2) infection and lung inflammation mediated by IL-1. J Biol Regul Homeost Agents, 34(2)
Conti, P., Ronconi, G., Caraffa, A., Gallenga, C. E., Ross, R., Frydas, I., et al. (2020). Induction of pro-inflammatory cytokines (IL-1 and IL-6) and lung inflammation by Coronavirus-19 (COVI-19 or SARS-CoV-2): Anti-inflammatory strategies. J Biol Regul Homeost Agents, 34(2)
Amgalan, A., & Othman, M. (2020). Hemostatic laboratory derangements in COVID-19 with a focus on platelet count. Platelets, 1–6.
Salah, H. M., & Mehta, J. L. (2021). Meta-analysis of the effect of aspirin on mortality in COVID-19. American Journal of Cardiology, 142, 158–159.
Meizlish, M. L., Goshua, G., Liu, Y., Fine, R., Amin, K., Chang, E., et al. (2021). Intermediate-dose anticoagulation, aspirin, and in-hospital mortality in COVID-19: A propensity score-matched analysis. Am J Hematol
Kwiatkowski, S., Borowski, D., Kajdy, A., Poon, L. C., Rokita, W., & Wielgos, M. (2020). Why we should not stop giving aspirin to pregnant women during the COVID-19 pandemic. Ultrasound in Obstetrics and Gynecology, 55(6), 841–843.
Rauch, B. (2020). Cost-effectiveness of rivaroxaban plus aspirin (dual pathway inhibition) for prevention of ischaemic events in patients with cardiovascular disease: on top optimisation of secondary prevention medication in the context of COVID-19 pandemia. Eur J Prev Cardiol, 2047487320920754.
McClure, G. R., Kaplovitch, E., Narula, S., Bhagirath, V. C., & Anand, S. S. (2019). Rivaroxaban and aspirin in peripheral vascular disease: A review of implementation strategies and management of common clinical scenarios. Current Cardiology Reports, 21(10), 115.
Casey, K., Iteen, A., Nicolini, R., & Auten, J. (2020). COVID-19 pneumonia with hemoptysis: Acute segmental pulmonary emboli associated with novel coronavirus infection. Am J Emerg Med
Yin, S., Huang, M., Li, D., & Tang, N. (2020). Difference of coagulation features between severe pneumonia induced by SARS-CoV2 and non-SARS-CoV2. J Thromb Thrombolysis
Willyard, C. (2020). Coronavirus blood-clot mystery intensifies. Nature, 581(7808), 250.
Li, H., Liu, L., Zhang, D., Xu, J., Dai, H., Tang, N., et al. (2020). SARS-CoV-2 and viral sepsis: Observations and hypotheses. Lancet, 395(10235), 1517–1520.
Canzano, P., Brambilla, M., Porro, B., Cosentino, N., Tortorici, E., Vicini, S., et al. (2021). Platelet and endothelial activation as potential mechanisms behind the thrombotic complications of COVID-19 patients. JACC Basic Transl Sci
Costela-Ruiz, V. J., Illescas-Montes, R., Puerta-Puerta, J. M., Ruiz, C., & Melguizo-Rodriguez, L. (2020). SARS-CoV-2 infection: The role of cytokines in COVID-19 disease. Cytokine Growth Factor Rev
Debnath, M., Banerjee, M., & Berk, M. (2020). Genetic gateways to COVID-19 infection: Implications for risk, severity, and outcomes. FASEB J
Mehta, P., McAuley, D. F., Brown, M., Sanchez, E., Tattersall, R. S., Manson, J. J., et al. (2020). COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet, 395(10229), 1033–1034.
Pain, C. E., Felsenstein, S., Cleary, G., Mayell, S., Conrad, K., Harave, S., et al. (2020). Novel paediatric presentation of COVID-19 with ARDS and cytokine storm syndrome without respiratory symptoms. Lancet Rheumatol
Pedersen, S. F., & Ho, Y. C. (2020). SARS-CoV-2: A storm is raging. The Journal of Clinical Investigation, 130(5), 2202–2205.
Song, P., Li, W., Xie, J., Hou, Y., & You, C. (2020). Cytokine storm induced by SARS-CoV-2. Clin Chim Acta
Tomar, B., Anders, H. J., Desai, J., & Mulay, S. R. (2020). Neutrophils and neutrophil extracellular traps drive necroinflammation in COVID-19. Cells, 9(6)
Vaninov, N. (2020). In the eye of the COVID-19 cytokine storm. Nature Reviews Immunology, 20(5), 277.
Wright, D. J. M. (2020). Prevention of the cytokine storm in COVID-19. Lancet Infect Dis
Barnes, B. J., Adrover, J. M., Baxter-Stoltzfus, A., Borczuk, A., Cools-Lartigue, J., Crawford, J. M., et al. (2020). Targeting potential drivers of COVID-19: Neutrophil extracellular traps. J Exp Med, 217(6)
Konopka, K. E., Wilson, A., & Myers, J. L. (2020). Postmortem lung findings in an asthmatic patient with coronavirus disease 2019. Chest
Suess, C., & Hausmann, R. (2020). Gross and histopathological pulmonary findings in a COVID-19 associated death during self-isolation. International Journal of Legal Medicine, 134(4), 1285–1290.
Wang, J., Hajizadeh, N., Moore, E. E., McIntyre, R. C., Moore, P. K., Veress, L. A., et al. (2020). Tissue plasminogen activator (tPA) treatment for COVID-19 associated acute respiratory distress syndrome (ARDS): A case series. J Thromb Haemost
Yan, L., Mir, M., Sanchez, P., Beg, M., Peters, J., Enriquez, O., et al. (2020). Autopsy report with clinical pathological correlation. Arch Pathol Lab Med
Youd, E., & Moore, L. (2020). COVID-19 autopsy in people who died in community settings: The first series. Journal of Clinical Pathology, 206710, 1–5.
Dwiputra Hernugrahanto, K., Novembri Utomo, D., Hariman, H., Budhiparama, N. C., Medika Hertanto, D., Santoso, D., et al. (2021). Thromboembolic involvement and its possible pathogenesis in COVID-19 mortality: Lesson from post-mortem reports. European Review for Medical and Pharmacological Sciences, 25(3), 1670–1679.
Combes, A. J., Courau, T., Kuhn, N. F., Hu, K. H., Ray, A., Chen, W. S., et al. (2021). Global absence and targeting of protective immune states in severe COVID-19. Nature
Zaid, Y., Puhm, F., Allaeys, I., Naya, A., Oudghiri, M., Khalki, L., et al. (2020). Platelets Can Associate With SARS-CoV-2 RNA and Are Hyperactivated in COVID-19. Circulation Research, 127, 1404–1418.
Panigrahy, D., Gilligan, M. M., Huang, S., Gartung, A., Cortes-Puch, I., Sime, P. J., et al. (2020). Inflammation resolution: A dual-pronged approach to averting cytokine storms in COVID-19? Cancer Metastasis Rev
Blair, P., & Flaumenhaft, R. (2009). Platelet alpha-granules: Basic biology and clinical correlates. Blood Reviews, 23(4), 177–189.
Chang, Y. S., Ko, B. H., Ju, J. C., Chang, H. H., Huang, S. H., & Lin, C. W. (2020). SARS unique domain (SUD) of severe acute respiratory syndrome coronavirus induces NLRP3 inflammasome-dependent CXCL10-mediated pulmonary inflammation. Int J Mol Sci, 21(9)
Hui, K. P. Y., Cheung, M. C., Perera, R., Ng, K. C., Bui, C. H. T., Ho, J. C. W., et al. (2020). Tropism, replication competence, and innate immune responses of the coronavirus SARS-CoV-2 in human respiratory tract and conjunctiva: an analysis in ex-vivo and in-vitro cultures. Lancet Respir Med
Magrone, T., Magrone, M., & Jirillo, E. (2020). Focus on receptors for coronaviruses with special reference to angiotensin-converting enzyme 2 as a potential drug target - A perspective. Endocr Metab Immune Disord Drug Targets
Chu, H., Chan, J. F., Wang, Y., Yuen, T. T., Chai, Y., Hou, Y., et al. (2020). Comparative replication and immune activation profiles of SARS-CoV-2 and SARS-CoV in human lungs: an ex vivo study with implications for the pathogenesis of COVID-19. Clin Infect Dis
DiNicolantonio, J. J., & Barroso-Aranda, J. (2020). Harnessing adenosine A2A receptors as a strategy for suppressing the lung inflammation and thrombotic complications of COVID-19: Potential of pentoxifylline and dipyridamole. Med Hypotheses, 143, 110051.
Meikle, C. K. S., Creeden, J. F., McCullumsmith, C., & Worth, R. G. (2021). SSRIs: Applications in inflammatory lung disease and implications for COVID-19. Neuropsychopharmacol Rep, 41(3), 325–335.
Morris, G., Bortolasci, C. C., Puri, B. K., Olive, L., Marx, W., O'Neil, A., et al. (2021). Preventing the development of severe COVID-19 by modifying immunothrombosis. Life Sci, 264, 118617.
Mir, N., D'Amico, A., Dasher, J., Tolwani, A., & Valentine, V. (2021). Understanding the andromeda strain - The role of cytokine release, coagulopathy and antithrombin III in SARS-CoV2 critical illness. Blood Rev, 45, 100731.
Theoharides, T. C., Antonopoulou, S., & Demopoulos, C. A. (2020). Coronavirus 2019, Microthromboses, and Platelet Activating Factor. Clinical Therapeutics, 42(10), 1850–1852.
Salamanna, F., Maglio, M., Landini, M. P., & Fini, M. (2020). Platelet functions and activities as potential hematologic parameters related to Coronavirus Disease 2019 (Covid-19). Platelets, 1–6.
Pavord, S., Thachil, J., Hunt, B. J., Murphy, M., Lowe, G., Laffan, M., et al. (2020). Practical guidance for the management of adults with immune thrombocytopenia during the COVID-19 pandemic. British Journal of Haematology, 189(6), 1038–1043.
Crispin, P. J., & Montague, S. J. (2020). Megakaryocytes: Masters of innate immunity? Arteriosclerosis, Thrombosis, and Vascular Biology, 40(12), 2812–2814.
Colling, M. E., & Kanthi, Y. (2020). COVID-19-associated coagulopathy: An exploration of mechanisms. Vascular Medicine, 25(5), 471–478.
Bautista-Vargas, M., Bonilla-Abadia, F., & Canas, C. A. (2020). Potential role for tissue factor in the pathogenesis of hypercoagulability associated with in COVID-19. Journal of Thrombosis and Thrombolysis, 50(3), 479–483.
Bao, C., Tao, X., Cui, W., Hao, Y., Zheng, S., Yi, B., et al. (2021). Natural killer cells associated with SARS-CoV-2 viral RNA shedding, antibody response and mortality in COVID-19 patients. Experimental Hematology & Oncology, 10(1), 5.
Althaus, K., Marini, I., Zlamal, J., Pelzl, L., Singh, A., Haberle, H., et al. (2021). Antibody-induced procoagulant platelets in severe COVID-19 infection. Blood, 137(8), 1061–1071.
McMullen, P. D., Cho, J. H., Miller, J. L., Husain, A. N., Pytel, P., & Krausz, T. (2021). A descriptive and quantitative immunohistochemical study demonstrating a spectrum of platelet recruitment patterns across pulmonary infections including COVID-19. American Journal of Clinical Pathology, 155(3), 354–363.
Middleton, E. A., He, X. Y., Denorme, F., Campbell, R. A., Ng, D., Salvatore, S. P., et al. (2020). Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood, 136(10), 1169–1179.
Funding
DGM is supported by Institutional Research Grant from UT MD Anderson Cancer Center. SK, DGM, JPS, and SK are supported by the Colorectal Cancer Moon Shot at the UT MD Anderson Cancer Center. SK, DGM, and JPS are supported by 3P30CA016672, P30CA0166723, P50CA221707, R01CA218230 and R01CA184843. AM is supported by the Pancreatic Cancer Moon Shot at the UT MD Anderson Cancer Center, R01CA218230, and P01CA117969. AKS is supported by the Ovarian Cancer Moon Shot at the UT MD Anderson Cancer Center, R01CA177909, P50 CA098258, P50CA217685, R35 CA209904, the American Cancer Society, and the Frank McGraw Memorial Chair in Cancer Research at the UT MD Anderson Cancer Center. VAV is supported by R01CA177909 and R01CA231141.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
AKS: Consulting (Merck, Kiyatec), research funding (MTrap), shareholder (BioPath).
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Menter, D.G., Afshar-Kharghan, V., Shen, J.P. et al. Of vascular defense, hemostasis, cancer, and platelet biology: an evolutionary perspective. Cancer Metastasis Rev 41, 147–172 (2022). https://doi.org/10.1007/s10555-022-10019-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-022-10019-5