
Abstract
Hydrogen is viewed as the future carbon–neutral fuel, yet hydrogen storage is a key issue for developing the hydrogen economy because current storage techniques are expensive and potentially unsafe due to pressures reaching up to 700 bar. As a consequence, research has recently designed advanced hydrogen sorbents, such as metal–organic frameworks, covalent organic frameworks, porous carbon-based adsorbents, zeolite, and advanced composites, for safer hydrogen storage. Here, we review hydrogen storage with a focus on hydrogen sources and production, advanced sorbents, and machine learning. Carbon-based sorbents include graphene, fullerene, carbon nanotubes and activated carbon. We observed that storage capacities reach up to 10 wt.% for metal–organic frameworks, 6 wt.% for covalent organic frameworks, and 3–5 wt.% for porous carbon-based adsorbents. High-entropy alloys and advanced composites exhibit improved stability and hydrogen uptake. Machine learning has allowed predicting efficient storage materials.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In recent years, hydrogen has emerged as a promising clean and sustainable energy carrier, garnering attention as a viable alternative to conventional fossil fuels across diverse sectors, including transportation, power generation, and industrial processes. The drive toward a low-carbon future has reignited interest in the role of hydrogen, with governments, businesses, and researchers investing in its potential. Its energy-rich nature makes it an appealing choice for transportation, while its storability as both gas and liquid offers application flexibility (Osman et al. 2022, 2024c).
Hydrogen fuel, with more than double the efficiency of fuels like gasoline (Balat 2008; Gutiérrez-Martín et al. 2009), is primarily produced through industrial steam reforming (accounting for 96% of hydrogen production), satisfying a large portion of the United States hydrogen demand (Gutiérrez-Martín et al. 2009; Pareek et al. 2020). Electrolysis, a significant and emerging technology, accounts for the remaining 4% of hydrogen production. This process comprises water splitting into its constituent elements, oxygen, and hydrogen, through the application of electrical energy (Mazloomi et al. 2012; Nagar et al. 2023; Osman et al. 2023a; Li et al. 2023b). While alternative methods, such as thermochemical, biomass gasification, wind, and solar radiation, are being explored, they require further experimental evidence for quantitative hydrogen production (Pareek et al. 2020; Osman et al. 2023b). This endeavor is complemented by a growing emphasis on harnessing renewable energy sources, such as solar and wind for hydrogen production. This approach can potentially create an entirely emissions-free energy cycle, aligning with the broader goals of sustainability and environmental responsibility, generating several environmental and economic benefits, as shown in Fig. 1. However, the broad implementation of hydrogen as an energy carrier is impeded by several technical obstacles, one of the most significant being the issue of hydrogen storage (Chen and Zhu 2008; Barthélémy et al. 2017).

Benefits of harnessing hydrogen energy for a sustainable future. The figure serves to highlight some of these key advantages. Firstly, hydrogen demonstrates efficiency in energy transportation, presenting a promising solution for sustainable fuel distribution networks. Moreover, its rapid refueling capabilities, akin to traditional gasoline, offer convenience and accessibility to consumers. Notably, hydrogen's zero emissions during usage significantly contribute to environmental preservation, fostering a cleaner ecosystem, and mitigating harmful air pollutants. Economically, the utilization of hydrogen presents opportunities for substantial gains, particularly through advancements in hydrogen technology and infrastructure. Additionally, hydrogen-powered vehicles hold promise for reduced long-term operational costs, potentially offering financial incentives for both consumers and industries. Furthermore, the quieter operation of hydrogen-fueled vehicles contributes to decreased noise pollution, promoting quieter and more peaceful urban environments
Hydrogen storage presents a distinctive set of challenges due to hydrogen’s inherent characteristics. Hydrogen exhibits a low volumetric energy density as the lightest element under standard conditions. This necessitates the development of storage methods that can effectively, compactly, safely, and economically accommodate the energy needs of vehicles (Ratnakar et al. 2021; Usman 2022; Navaid et al. 2023). Over time, various storage techniques have emerged as solutions. Generally, hydrogen storage technologies are divided into two main categories: physical-based and material-based.
Physical storage relies on altering storage conditions, such as pressure in compressed gas or, temperature in liquid storage, or both parameters in the case of cryo-compressed gas storage. In contrast, material-based or solid-state storage like metal hydrides, complex hydrides, and carbon-based materials involves absorption or adsorption techniques (Hassan et al. 2021). Solid-state materials have garnered significant attention as a promising alternative to compressed gas and cryogenic liquid methods due to their drawbacks. These conventional methods suffer from various limitations, including inefficiency and safety concerns during transportation. Cryogenic liquid storage, for instance, only allows for storing a relatively small amount of hydrogen per unit volume, with significant energy losses during liquefaction. Moreover, the heightened flammability of compressed hydrogen, coupled with the need for expensive high-quality cylinders for cryogenic liquid storage, renders these methods economically impractical (Gu et al. 2019; Rimza et al. 2022). In contrast, solid-state hydrogen storage is being increasingly recognized as a safer, more affordable, and compact solution. This approach involves storing hydrogen through adsorption mechanisms on the surface of solid substrates, offering a promising avenue for efficient storage. Extensive research efforts have been directed towards identifying suitable solid-state materials capable of effectively storing hydrogen (Rimza et al. 2022). However, each solution has limitations, ranging from storage efficiency and weight to considerations and operational temperature ranges. Consequently, this field continues to offer opportunities for further research and development. While the advantages and potential of hydrogen as a fuel are widely recognized, the challenges, especially concerning storage, remain substantial and multifaceted.
Hence, this review provides a comprehensive analysis to explore recent advancements and challenges in hydrogen storage, shedding light on sorbent materials and techniques that promise to overcome existing limitations. The review aims to analyze various sorbent hydrogen storage methods, including metal–organic frameworks (MOFs), covalent organic frameworks (COFs), porous carbon-based adsorbents, zeolite, high-entropy alloys, and advanced composites, and highlight their potential to enhance hydrogen storage performance. Additionally, the review emphasizes the significant role of machine learning in the exploration of hydrogen storage materials. The objective is to showcase how machine learning techniques facilitate the understanding of material properties and enable the design of improved materials, even in the face of data challenges. By addressing the limitations of current storage methods and highlighting the potential of emerging technologies, this review aims to provide valuable insights for the development of efficient and sustainable hydrogen storage systems.
Hydrogen sources and production
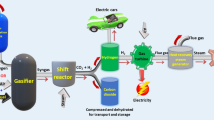
The evolution of hydrogen technology demands a comprehensive hydrogen supply chain encompassing sources, production, storage, and utilization. Figure 2 outlines these essential components and proposes hydrogen production and utilization strategies.

Essential elements of the hydrogen supply network. This figure provides a comprehensive overview of the hydrogen supply chain, illustrating the various sources and processes involved in hydrogen production, storage, delivery, and utilization. Starting with natural resources like natural gas, methane, propane, naphtha, coal, water, and renewable fuels, such as ethanol and biomass, the diagram outlines the conversion methods, including steam reforming, water gas shift, partial oxidation, pyrolysis, high-temperature solar/nuclear splitting, electrolytic processes, coal gasification, photolytic, and hydrolysis. The produced hydrogen is then subjected to liquefaction or compression and sometimes chemically bonded for storage and delivery in forms like liquid hydrogen, ammonia (NH3), metal hydride, or compressed hydrogen gas. The utilization phase spans various applications, from carbon-free chemical and steel manufacturing to heavy-duty transportation, long-distance power generation, and even aviation or spacecraft. This figure encapsulates the potential of hydrogen to serve as a versatile and clean energy carrier, contributing to a sustainable energy future
Hydrogen, the most abundant element in the universe, can be sourced from various methods. The production methods can be categorized broadly into non-renewable and renewable sources. Most global hydrogen production relies on non-renewable methods, notably natural gas reforming or gasification, which combines methane with steam under high pressure and temperature conditions to produce hydrogen, carbon monoxide, and carbon dioxide as byproducts. Steam methane reforming (SMR) has been utilized for hydrogen production for many years and is considered a mature technology. It involves the use of natural gas and steam to generate hydrogen, constituting a significant portion of 48% of global hydrogen production. Commercially available capacities for steam methane-reforming range from 130,000 to 300,000 tons per year, allowing for handling of large volumes. Moreover, steam methane reforming plants can capture a substantial portion of the carbon dioxide generated, ranging from 50 to 92%. This aspect underscores the technology's potential for contributing to carbon capture and reduction efforts (Oni et al. 2022).
Hydrogen can also be derived from coal through a gasification process. During gasification, coal undergoes partial oxidation by steam and oxygen, resulting in the production of primarily carbon monoxide and hydrogen, combined with carbon dioxide and steam, to form syngas within a high-pressure and temperature reactor (Younas et al. 2022). However, coal gasification is a significant source of carbon dioxide emissions. Kothari et al. (2008) observed a carbon dioxide emission rate of 29.33 kg per kg of hydrogen produced from a coal gasification system operating at 75% efficiency. Although carbon capture and storage (CCS) technology can be employed to capture carbon dioxide emissions, it is not considered a suitable solution on a large scale. Furthermore, the world's coal reserves are projected to last for approximately 150 years at current production rates (Younas et al. 2022). However, concerns about environmental impact and the depletion of reserves are increasingly shifting focus towards renewable feedstocks for future hydrogen production.
Currently, coal is the primary hydrogen source, accounting for about 21.5 billion tons annually (Dash et al. 2023). This significant reliance on coal must shift towards renewable alternatives. The rising demand for hydrogen is not only driven by its potential as a transport fuel or a carrier of portable energy but also by its crucial role in oil refining, petroleum desulfurization and upgrading, and ammonia production. Relying on existing technologies will increase the consumption of conventional hydrocarbons, predominantly natural gas, subsequently escalating greenhouse gas emissions.
In contrast, renewable sources offer more sustainable alternatives. For instance, water electrolysis, which uses electrical energy to split water into hydrogen and oxygen, can be considered nearly emission-free when powered by renewable sources like wind or solar energy (Hassan et al. 2024a). The electrolysis method stands out among various hydrogen production techniques for its ability to yield high-density and environmentally friendly hydrogen alongside pure oxygen through the process of water electrolysis. However, the efficiency of hydrogen production via water electrolysis remains limited due to its high power consumption and low hydrogen release rate. Globally, only a small fraction, approximately 4%, of hydrogen gas is produced through electrochemical water electrolysis, primarily due to deficiencies in this method and the prohibitively high cost of noble electrocatalysts. Currently, platinum (Pt) and ruthenium (Ru)-based compounds serve as the most effective electrocatalysts for the hydrogen evolution reaction (HER) and oxygen evolution reaction (OER), respectively. However, these materials are both exceedingly expensive and rare precious metals. Consequently, their widespread commercial application is hindered by these factors (Anwar et al. 2021).
Biomass gasification is another potentially carbon–neutral option since biomass absorbs carbon dioxide during its growth. Although still in the research stages, the biological hydrogen production by certain microorganisms holds promise (Cao et al. 2020). By the year 2050, biomass is anticipated to constitute approximately two-thirds of the total direct consumption of renewable energy. It is crucial that hydrogen production relies on renewable and sustainable resources, such as biomass to adequately address the growing global energy demand (Nguyen et al. 2024).
Solar thermochemical processes for hydrogen production face several challenges. These include high capital and operational costs due to technological development and specialized infrastructure. Additionally, these processes exhibit limited efficiency, typically below 50%, and operate at high temperatures and pressures, increasing safety risks and material expenses. Corrosion of materials is also a concern, leading to higher maintenance costs. Moreover, scalability is hindered by complex infrastructure requirements, making large-scale production costly and challenging. Overall, the complexity of solar thermochemical processes necessitates skilled personnel and process development, adding further to the overall expenses. Additionally, solar-thermal methods can yield hydrogen by utilizing concentrated solar power to split water or other compounds (Hassan et al. 2024b). These renewable approaches align with the growing emphasis on sustainable and eco-friendly hydrogen production.
Producing hydrogen from renewable resources, especially those originating from agricultural or other waste streams, offers a greener alternative. Such methods can produce hydrogen with minimal to zero net greenhouse gas emissions, primarily if carbon sequestration technologies are not utilized (Dash et al. 2023). These renewable methods also enhance the feasibility and economic viability of distributed and semi-centralized reforming (Reiter and Lindorfer 2015). Transitioning to on-site decentralized hydrogen production through electrolysis, thermocatalytic, and biological processes can eliminate the need for extensive and costly distribution networks. However, each of these methods presents its own set of technical challenges (Chakraborty et al. 2021; Pal et al. 2022). Among these challenges are optimizing conversion efficiencies, managing diverse feedstocks, and ensuring the seamless integration of hydrogen production with purification and storage technologies. Table 1 illustrates the challenges associated with hydrogen production methods.
Hydrogen storage
Hydrogen storage is crucial to the global supply chain, ensuring a consistent and timely supply to end-users. Various physical storage technologies for hydrogen have been developed. These methods, which include compression and liquefaction technologies, either individually or in combination, offer innovative solutions for storing hydrogen in dense and stable forms.
The compression of hydrogen is a well-established technology known for its high rates of hydrogen filling and release. Unlike other methods, no energy is required for the release of hydrogen. However, compressing hydrogen to high pressures consumes about 13–18% of its lower heating value, impacting the overall economics. Fortunately, the increase in pressure only marginally raises the power needed for compression. Hydrogen is typically stored in cylindrical vessels due to the difficulty in fitting spherical vessels onboard. To effectively store hydrogen, lightweight and cost-effective vessel materials capable of withstanding high pressure are essential. The chosen material must also resist hydrogen diffusion and potential embrittlement caused by stored hydrogen (Usman 2022).
Another method for physically storing hydrogen involves using cryogenic liquid. Storing hydrogen in liquid form offers a higher density compared to other methods. Liquid hydrogen, for instance, boasts a density of approximately 71 g/L at its normal boiling point of 20 K, which is roughly 1.8 times higher than hydrogen pressurized up to 70 MPa at 288 K. However, the cooling technology required for maintaining liquid hydrogen at such low temperatures consumes around 30% of its total energy content (Abe et al. 2019). However, the high costs and complex infrastructure associated with liquefaction and distribution make large-scale adoption of liquid hydrogen challenging. The liquefaction process to convert hydrogen to a liquid state demands significant capital investment to construct specialized cryogenic distillation columns and storage tanks. The distribution infrastructure also requires specialized trucks, pipelines, and dispensing equipment that can handle ultra-low temperatures without significant boil-off losses. These costs can be prohibitive, especially in the early stages of market development. Additionally, distributing a cryogenic liquid fuel on a wide scale would involve surmounting challenges related to insulation, transportation distances, dormancy losses, and safety considerations. Due to these factors, while liquid hydrogen deployment may be feasible for limited applications, expanding its use to larger markets faces critical economic and logistical barriers.
In addition, current infrastructure and technology have limitations, with most hydrogen pipelines located near refineries and chemical plants. Constructing new pipelines for transporting large volumes of gaseous hydrogen poses significant capital costs and safety concerns, i.e., hydrogen embrittlement and leaks. Continuous endeavors are underway to investigate alternative approaches, such as repurposing existing liquefied natural gas (LNG) networks to convey gaseous hydrogen or exploring alternative hydrogen carriers like liquid ethanol or ammonia. Additionally, advancements in storage methodologies using various means, such as trucks, railcars, ships, or barges are being pursued. Its relatively low energy density per unit volume is the chief determinant affecting the costs associated with hydrogen storage and delivery (Sazali 2020).
Hydrogen storage sorbents are materials engineered to capture hydrogen by physically adsorbing it on their surfaces or chemically integrating it into their structures. Contemporary research predominantly uses microporous sorbents, which provide extensive surface areas. The interaction between these sorbents and hydrogen is primarily governed by the dynamics of molecular hydrogen (H2), characterized by weak physisorptive attraction. Furthermore, the amount of hydrogen adsorbed is directly related to the specific surface area of the sorbent. Various high surface area materials are available, each possessing distinct physical and chemical characteristics. Figure 3 illustrates the broad categorization of hydrogen storage methods into physical and chemical storage, each characterized by distinct techniques and principles. This review primarily focuses on storing hydrogen using innovative sorbent materials. Here are several types of sorbents commonly used for hydrogen storage:

Classification of hydrogen storage methods. This figure illustrates the various methods of hydrogen storage, highlighting the versatility and adaptability of hydrogen as an energy carrier. It details two primary categories: physical storage, which includes liquid hydrogen tanks, compressed hydrogen tanks, and cryo-compressed tanks; and material-based storage, which encompasses metal hydrides (both simple and complex), adsorbents like carbon-based materials and metal–organic frameworks (MOFs), as well as chemical hydrides, such as borohydrides and ammonia borane. The figure also notes that some complex metal hydrides are not reversible on-board, while others can be regenerated off-board. This visual representation underscores the importance of ongoing research and development in enhancing hydrogen storage efficiency, which is crucial for the widespread adoption of hydrogen as a clean energy solution
Metal–organic frameworks
Metal–organic frameworks (MOFs) represent a class of highly porous materials where metal ions or clusters (referred to as secondary building units or SBUs) are interconnected by organic linkers. The versatility of metal–organic frameworks lies in their ability to be tailored using different ligands and secondary building units, resulting in a diverse range of metal–organic frameworks structures, as illustrated in Fig. 4. Moreover, the pore sizes within metal–organic frameworks can be fine-tuned by extending carbon chains, with linker length dictating pore size. Additionally, linkers can undergo modifications to enhance their chemical properties and selectivity (Butova et al. 2016).

Metal–organic framework (MOF) synthesis employing a node-and-connector approach. The figure illustrates how the careful selection of organic linkers, such as terephthalic acid, and the strategic connection of metal clusters, including Zn2+ and Zr4+, are crucial in constructing the metal–organic framework’s unique structure. These decisions directly influence the metal–organic framework’s topology, pore dimensions, and surface area, which are essential characteristics for their function. For instance, MOF-5 (Zn) features an octahedron shape with a pore size of 11 Å and a surface area of 950 m2 g−1, while UiO-66 (Zr) has a cuboctahedron shape with a pore size of 8 Å and a surface area of 881 m2 g−1. Other variations like MIL-53 (Al) and MIL-101 (Cr) demonstrate the diversity in pore sizes and surface areas, ranging from 7 and 2.1 Å to 29 and 34 Å, with corresponding surface areas of 1500 m2 g−1 and 4100 m2 g−1, respectively. This figure representation underscores the versatility and potential of metal–organic frameworks in various applications, driven by their customizable properties. MIL refers to Materials of Institute Lavoisier, and UiO refers to the University of Oslo. BET refers to Brunauer–Emmett–Teller surface area
Various research teams have established a classification system outlining seven primary categories of metal–organic frameworks based on distinct structures synthesized. These categories include isoreticular metal–organic frameworks (IRMOF), Hong Kong University of Science and Technology metal–organic frameworks (HKUST), materials of institute lavoisier (MIL), zeolitic imidazolate frameworks (ZIF), University of Oslo (UiO), porous coordination networks (PCN), and coordination pillared-layer (CPL) (Li et al. 2024). Among these, IRMOF, MIL, UiO, and ZIF are frequently utilized in solid-state hydrogen storage. IRMOFs, exemplified by IRMOF-1 and MOF-5, boast a cubic lattice structure with adjustable pore sizes through ligand modifications, making them versatile in gas storage and catalysis (Zhang and Hu 2011). UiO features a zirconium metal (Zr) core surrounded by organic ligands, offering high thermal stability and reusability (Liu 2020). MIL-type metal–organic frameworks showcase flexibility with a diamond-shaped pore structure and a unique "breathing phenomenon" (Li et al. 2021). The ZIF series, with representatives like ZIF-8 and ZIF-68, exhibit molecular sieving properties and robust thermal stability, attributed to the substitution of tetrahedral atoms by transition metal elements and the bridging of oxygen atoms by organic ligands (Tiba et al. 2019). These metal–organic frameworks hold promise for various applications due to their diverse structures and functionalities.
Metal–organic frameworks exhibit great promise for hydrogen storage due to their exceptional porosity and customizable structural attributes (Rosi et al. 2003). These microporous solids exhibit exceptional surface areas, often exceeding 3000 m2 g−1. Under cryogenic conditions (77 K), metal–organic frameworks have demonstrated impressive hydrogen storage capacities, reaching up to 9.2 wt.% hydrogen (Sumida et al. 2009). Notably, metal–organic frameworks' hydrogen storage capacity has also been enhanced through metal-supported catalyst modifications (Li and Yang 2006a, b). However, achieving high volumetric capacity remains a challenge due to their extensive porosity.
Generally, the primary mechanism for energy storage in metal–organic frameworks involves the hydrogen spill-over mechanism, which is particularly effective at room temperature when coupled with metal-based catalysts. This mechanism comprises several key steps (Guo et al. 2020): (1) Surface chemisorption through phase nucleation. (2) Hydrogen dissociation on the metal catalyst, which requires an energy of 0.8–1.8 eV for hydrogen adsorption. (3) Hydrogen migration from the metal catalyst to the surface of porous carbon materials (PCM), involving an energy barrier of 2.45–3.2 eV, that can be reduced by hole doping. (4) Diffusion and desorption on the substrate which is challenging due to the strong C–H bond, presenting a 1.05–2.16 eV barrier. Ensafi et al. (2016) developed hydrogen absorbers from layered double hydroxides (Al-M) combined with reduced graphene oxide (rGO). They used Pd#Al-M/LDH-rGO-o-phenylenediamine to study the spill-over effect. Al represents the chemical hydride, M (Ni, Cu, Zn) is the metal hydride, o-phenylenediamine enhances adsorption/desorption kinetics, and reduced graphene oxide offers a high surface area with sp2 hybrid carbon atoms. Hydrogen molecules undergo dissociation at the catalytic sites of the spill-over material and subsequently transfer to high-surface receptors. Moreover, the interaction between amoxicillin (AMX) and Zr-MOFs, notably UiO-66-NH2, enhances hydrogen absorption (Liu et al. 2020).
Metal–organic frameworks are widely recognized as a prominent category of microporous materials, according to numerous studies. They are known for their ease of assembly and modification, making them highly versatile in various applications. Metal–organic frameworks exhibit exceptional hydrogen storage abilities, particularly at cryogenic temperatures (77 K), aligning closely with the United States Department of Energy's (DOE) hydrogen storage aims (Shet et al. 2021). The hydrogen storage capacities of metal–organic frameworks using hydrogen spillover and carbon bridges have been discussed by Li and Yang (2006a). This study focused on IRMOF-1 and IRMOF-8 and found that the hydrogen uptake of metal–organic frameworks could be significantly enhanced through secondary spillover via carbon bridges. The storage capacity of IRMOF-8 was increased to 4 wt.% at 298 K and 10 MPa, which was eight times higher than pure IRMOF-8. The hydrogen adsorption measurements were performed using Sievert's apparatus, and the results showed reversible and rechargeable storage. The study suggests that spillover can be a viable technique for achieving high hydrogen storage in metal–organic frameworks, and further improvements can be made with new metal–organic frameworks having higher hydrogen uptakes. Another study conducted by Bambalaza et al. (2018) discussed the compaction of a zirconium metal–organic framework (UiO-66) for high-density hydrogen storage applications. The study reports that compaction of UiO-66 at high pressure (700 MPa) resulted in densification without compromising its total gravimetric hydrogen uptake. The densified pellets achieved a total hydrogen uptake of 5.1 wt.% at 100 bar and 77 K, compared to 5.0 wt.% for the powdered form. The volumetric capacity of the densified UiO-66 was reported to be up to 74 g/L at 77 K and 100 bar, compared to 29 g/L for the powder. These values were calculated using different methods considering packing density and crystal/skeletal densities of metal–organic frameworks. The study demonstrates the potential of compaction to improve hydrogen storage capacities in metal–organic frameworks without sacrificing their gravimetric uptake.
Metal–organic frameworks with higher structural density tend to reduce gravimetric adsorption capacity while increasing volumetric adsorption due to their structural density's impact on gas capture. Factors, such as porosity, surface area, and isosteric heat positively influence hydrogen storage capacity. According to Xia and Wang (2016), MOF-808 stands out in hydrogen adsorption, with MIL-101 achieving 6.01 wt.% and IRMOF-20 reaching 6.7 wt.% at 77 K. Shet et al. (2021) provided a comprehensive review of strategies to enhance the hydrogen absorption capabilities of metal–organic frameworks by increasing their surface area, along with an exploration of the variables influencing these characteristics. Various methods, including metal ion doping, nanoparticle inclusion, and composite formation, can elevate metal–organic frameworks' hydrogen storage potential. Understanding the effects of these strategies simplifies the optimization of metal–organic frameworks. Critical factors, such as temperature, pressure, and composition significantly impact metal–organic frameworks ' ability to adsorb hydrogen.
Enhancing the room-temperature hydrogen storage capacity of porous materials can be achieved through a spillover process, where hydrogen molecules dissociate into individual atoms and adhere to the absorbent surface. Experiments have confirmed spillover in metal nanoparticle-incorporated metal–organic frameworks, determining the depths of hydrogen penetration within these materials (Prins 2012; Zhan and Zeng 2018). Efficient hydrogen spillover hinges on two crucial factors: the distribution of metal nanoparticles and their proximity to the porous carrier. Initially, research on spillover primarily concentrated on carbon-based supports (Li and Yang 2007). In this context, a recently published article conducted by Liu et al. (2024) investigated the enhancement of room-temperature hydrogen storage capacities in metal–organic frameworks by incorporating CuNi nanoparticles into UiO-66(Zr). The study demonstrates that the CuNi@UiO-66 composite exhibits significantly improved hydrogen storage performance compared to pure UiO-66. The hydrogen storage capacity of CuNi@UiO-66 increases from 0.20 to 0.74 wt.%, as measured at 6 MPa and 298 K. The results indicate that the introduction of CuNi nanoparticles promotes the adsorption and dissociation of hydrogen molecules, favoring the hydrogen spillover effect. This research highlights the potential of designing metal–organic frameworks loaded with cost-effective metal nanoparticles for optimized room-temperature hydrogen storage, offering a promising solution for practical applications. Another study conducted by Kang et al. (2021) showcased an increase in the hydrogen storage capacity of Pt-doped UiO-66-NH2, rising from 0.08 to 0.71 wt.% under room temperature conditions and at a pressure of 30 bar. Wang et al. (2018) found that when borophene was doped with lithium (Li) metal ions, its gravimetric hydrogen capacity reached 13.96 wt.%, whereas borophene doped with sodium (Na) metal ions achieved a capacity of 10.39 wt.%. In Mg-MOF-74, the study of titanium (Ti) adsorption sites revealed that Ti atoms primarily bind to the MgO2 site, possessing a stable binding energy of + 2.92 eV, before attaching to hollow and on-O sites. At temperatures of 77 K, 150 K, and 298 K, the hydrogen uptakes are 1.81, 1.74, and 1.29 wt.%, respectively. Each Ti atom can bind with up to three hydrogen atoms (Suksaengrat et al. 2016).
Researchers have been actively involved in optimizing pore structures and expanding surface area to improve the hydrogen storage capacity of metal–organic frameworks. Kaye et al. (2007) conducted several pioneering studies aimed at achieving a large specific surface area and enhancing hydrogen storage performance using metal–organic frameworks. The paper explores the impact of preparation and handling on the hydrogen storage properties of Zn4O(1,4-benzenedicarboxylate)3 (MOF-5). The researchers aimed to determine the true hydrogen storage capacity of MOF-5 and resolve discrepancies in previous studies. They found that the highest surface area material was obtained by minimizing exposure to water and air during synthesis. The material exhibited significant differences in nitrogen and hydrogen adsorption capacities depending on exposure to air. The maximum hydrogen uptake for the prepared sample was 5.2 excess wt.% at 77 K and 40 bar. The study emphasizes the importance of minimizing exposure to moisture during metal–organic framework synthesis to achieve optimal hydrogen storage capacities. In line with this approach, Rosi et al. (2003) discussed the hydrogen storage capacities of metal–organic frameworks, focusing on MOF-5. The authors found that MOF-5, composed of Zn4O(BDC)3 (BDC: 1,4-benzenedicarboxylate), exhibited a hydrogen uptake of 4.5 wt.% at 78 K and 1.0 wt.% at room temperature and 20 bar pressure. Inelastic neutron scattering spectroscopy revealed the presence of two distinct binding sites for hydrogen within MOF-5. Preliminary studies on similar MOFs, IRMOF-6 and IRMOF-8, showed approximately double and quadruple the hydrogen uptake of MOF-5 at room temperature and 10 bar pressure. These findings demonstrate the favorable hydrogen sorption properties of metal–organic frameworks, indicating their potential as materials for hydrogen storage in applications, such as hydrogen-fueled vehicles and portable electronics.
Zhu and Zheng (2023) investigated the cryo-adsorption hydrogen storage capacity of MOF-5 using the mechanochemical method compared to the solvothermal method. The results show that the mechanochemically prepared MOF-5 (MOF-5(M)) exhibits a larger specific surface area and higher adsorption capacities for hydrogen compared to the solvothermally prepared MOF-5 (MOF-5(S)). The specific surface area of MOF-5(M) is increased by 207%, and the maximum excess adsorption capacity of hydrogen at 77 K within the pressure range of 0–10 MPa is increased by 90.5%. Grand canonical monte carlo (GCMC) simulation also supports these findings. The study suggests that MOF-5(M) holds more promise for practical applications of hydrogen storage by adsorption.
Furthermore, the effects of structural modifications on the hydrogen storage capacity of (MOF-5) have been investigated by Yang et al. (2012). Four structurally modified MOF-5s (P-MOF, C-MOF, I-MOF, and N-MOF) were prepared, and their crystal structure, pore characteristics, and hydrogen capacities were studied. The results show that the structural modifications significantly influenced the pore characteristics, leading to a decrease in specific surface areas (SSA) and an increase in ultrafine porosity. These changes correlated with an increase in the hydrogen storage capacity of MOF-5 from 1.2 to 2.0 wt.% at 469 K and 1 bar. The thermal stability of the MOF-5s was also enhanced, with the decomposition temperature increasing from 711 to 783 K. These findings provide valuable insights for designing metal–organic frameworks-based adsorbents with high hydrogen uptake and thermal stability.
In addition to using metal–organic frameworks as hydrogen storage material, they can also be used as catalysts or catalyst carriers during hydrogenation and dehydrogenation processes, owing to their large specific area, high porosity, and numerous active metal sites. In this context, a recently published article conducted by Zhang et al. (2023) investigated the catalytic effect of MOF-supported niobium pentoxide (Nb2O5) nanoparticles on the hydrogen storage behavior of magnesium hydride (MgH2). The study demonstrates that the prepared catalyst, Nb2O5@MOF, significantly enhances the hydrogen storage capacities of magnesium hydride. The composite material exhibits improved desorption kinetics, with 6.2 wt.% and 6.3 wt.% of hydrogen released within 2.6 min and 6.3 min at temperatures of 548 K and 523 K, respectively. Moreover, the fully dehydrogenated composite shows efficient hydrogenation, absorbing 4.9 wt.% and 6.5 wt.% of hydrogen within 6 min at 448 K and 423 K, respectively. The addition of Nb2O5@MOF also reduces the hydrogen capacity loss after 20 cycles compared to Nb2O5-doped magnesium hydride. The activation energies for desorption and absorption reactions are calculated to be 75.57 ± 4.16 kJ mol−1 and 51.38 ± 1.09 kJ mol−1, respectively. The study highlights the potential of metal–organic frameworks as effective catalysts for enhancing hydrogen storage capacities in Mg-based materials.
Similarly, Wang et al. (2019a) investigated the catalytic effect and mechanism of metal–organic frameworks on the hydrogen storage properties of magnesium (Mg). Three metal–organic frameworks, namely ZIF-8, ZIF-67, and MOF-74, were studied. The addition of MOFs enhanced the hydrogen storage capacities of Mg. The hydrogen release amounts from Mg, Mg/ZIF-8, Mg/ZIF-67, and Mg/MOF-74 were determined as 0.6 wt.%, 1.2 wt.%, 2.7 wt.%, and 3.7 wt.% of hydrogen, respectively, within 5000 s. The activation energy values for hydrogen release were determined as 198.9 kJmol−1, 12.8 kJmol−1, and 43.2 kJmol−1 for Mg/ZIF-8, Mg/ZIF-67, and Mg/MOF-74, respectively. The cyclic stability of Mg hydride was significantly improved with the addition of ZIF-67. The hydrogen storage capacity of the Mg/ZIF-67 nanocomposite remained unchanged even after 100 cycles of hydrogenation/dehydrogenation.
Li et al. (2014) discussed the enhanced hydrogen storage capacities of palladium (Pd) nanocrystals coated with the metal–organic framework (HKUST-1). The study reveals that the palladium nanocrystals covered with a metal–organic framework exhibit twice the storage capacity of bare palladium nanocrystals. The hydrogen storage properties were evaluated using hydrogen pressure-composition isotherms. At 101.3 kPa, the absorption of hydrogen increased from 0.5 H per Pd atom in bare palladium nanocrystals to 0.87 H per Pd atom in Pd@HKUST-1. The results suggest that the high surface area, porosity, and condensation effect of metal–organic frameworks contribute to enhanced hydrogen storage. The metal–organic framework coating also improved the reversibility and speed of the hydrogen absorption/desorption process. These findings demonstrate the potential of metal–organic frameworks as coatings for more effective hydrogen storage.
Furthermore, the inclusion of metal–organic framework structures has the potential to boost the catalytic efficacy of transition metal compounds by facilitating hydrogen diffusion within bulk MgH2 via enhanced transfer pathways. For instance, Ma et al. (2019) investigated the effects of trimesic acid-Ni MOF on the hydrogen sorption capabilities of MgH2. The composite material, Mg-(TMA-Ni MOF)-H, is prepared through ball-milling. The study finds that the addition of TMA-Ni MOF improves the absorption kinetics of hydrogen, with an onset desorption temperature 167.8 K lower than pure MgH2. The catalytic effects of nano-sized Mg2Ni/Mg2NiH4 derived from TMA-Ni MOF are attributed to enhancing the sorption kinetic properties by providing gateways for hydrogen diffusion during re/dehydrogenation processes. The hydrogenation and dehydrogenation enthalpies of the composite remain unchanged, indicating no significant alteration in thermodynamics.
In conclusion, metal–organic frameworks hold significant promise for efficient hydrogen storage due to their low production costs, minimal environmental impact, and suitability for aerospace applications owing to their lightweight nature. However, as with many emerging technologies, there is still room for improvement, and researchers are actively exploring various modifications to enhance metal–organic frameworks' hydrogen storage capabilities.
Covalent organic frameworks
Covalent organic frameworks (COFs) are organic structures formed by connecting small organic molecules into a regular and repeating pattern. These frameworks create a porous network that can store significant amounts of hydrogen. Much like metal–organic frameworks, the structure and properties of covalent organic frameworks can be adjusted by using different organic building blocks. The Yaghi research group has successfully synthesized two-dimensional (2D-COFs) (Côté et al. 2007) and three-dimensional (3D-COFs) (El-Kaderi et al. 2007; Hunt et al. 2008) using organic building units connected by strong covalent bonds like C–C, C–O, B–O, and Si–C instead of metal ions. These covalent organic frameworks exhibit high surface areas similar to metal–organic frameworks (3472 m2 g−1 for COF-102 and 4210 m2 g−1 for COF-103) and possess very low crystal densities (0.17 g cm−3 for COF-108), making them excellent candidates for hydrogen storage (Han et al. 2009).
In general, 2D-COFs typically exhibit layered structures, which can be either hexagonal or square, with the ability to adjust pore sizes by varying the size of the organic monomers (Colson and Dichtel 2013). On the other hand, 3D-COFs are typically constructed by connecting tetratopic building units, including tetrahedral and square ones, with linear or triangular organic components. The variety of available structures for 3D-COFs is somewhat more limited compared to metal–organic frameworks because their topology depends on the directionality of the organic building blocks. Common three-dimensional topologies, or nets, include carbon nitride, diamond, boracite, and platinum sulfide. Similar to their amorphous counterparts, these materials can form interpenetrated structures (Allendorf et al. 2018). Covalent organic frameworks are notable for their thermal and structural stability, allowing for the preparation of activated samples without sacrificing crystallinity. The surface areas of these activated covalent organic frameworks, as determined through nitrogen and argon adsorption studies, vary depending on their framework structural design. For instance, 2D-COFs, such as COF-6 (formed from the condensation of hexahydroxy triphenylene and 1,3,5-benzenetriboronic acid) and CTF-1 (derived from the condensation of 1,4-dicyanobenzene) possess BET (Brunauer–Emmett–Teller) surface areas of approximately 750 and 791 m2 g−1, respectively. In contrast, three-dimensional covalent organic frameworks like COF-102 and COF-103, resulting from the self-condensation of tetra(4-(dihydroxy)borylphenyl) methane and tetra(4-(dihydroxy)borylphenyl) silane, respectively, exhibit impressive BET surface areas of around 3620 and 3530 m2 g−1, respectively (Kuhn et al. 2008; Furukawa and Yaghi 2009; Allendorf et al. 2018). Regarding hydrogen absorption, COF-102 and COF-103 demonstrated gravimetric uptakes of approximately 6.8 and 6.6 wt.% at 35 bar and 77 K, respectively, despite their initial Qst (isosteric heat of adsorption) values being relatively low at 3.9 and 4.4 kJ mol−1, respectively, due to limited hydrogen binding sites (Furukawa and Yaghi 2009). In contrast, 2D-COFs like COF-10 and BLP-2(H) exhibited more modest uptakes of 3.8 and 2.5 wt.% at 77 K (Jackson et al. 2012).
Initially, the potential utilization of covalent organic frameworks for hydrogen storage underwent assessment through comprehensive multiscale theoretical methods. These analyses predominantly forecasted higher gravimetric hydrogen absorption for 3D COFs at 77 K in comparison to high surface area metal–organic frameworks (Kalidindi and Fischer 2013). In this regard, Han et al. (2008) investigated the hydrogen storage capacities of covalent organic frameworks. The researchers conducted theoretical studies and simulations to predict the hydrogen binding isotherms for various covalent organic frameworks. They found that COF-105 and COF-108 exhibited the highest hydrogen storage capacities, with predicted excess uptakes of 10.0 wt.% at 77 K. These values outperformed the highest measured values for representative metal–organic frameworks. The volumetric uptake of hydrogen was also analyzed, and COF-102 demonstrated the highest capacity of storing 40.4 g/L of H2. The study suggests that increasing both the surface area and free volume of covalent organic frameworks can enhance their maximum hydrogen uptake. The results highlight covalent organic frameworks as promising materials for practical hydrogen storage applications.
Assfour and Seifert (2010a) utilized molecular dynamics simulations to assess the stability of covalent organic frameworks under hydrogen pressure and determine the preferred adsorption sites for hydrogen in both 2D and 3D COFs. The results reveal that the most favorable adsorption sites for hydrogen are on benzene rings in the organic linkers of covalent organic frameworks, as well as near boron-oxygen networks. The adsorption interaction energy for covalent organic frameworks is found to be approximately 3.0 kJmol−1. This indicates a significant adsorption capability of covalent organic frameworks for hydrogen storage. Furthermore, the study highlights the advantages of covalent organic frameworks, such as their high thermal stability, low density, and large surface area, which make them promising candidates for hydrogen storage. The 3D COFs, particularly COF-102 and COF-103, demonstrate high hydrogen uptake, approaching the target set by the Department of Energy. The research emphasizes the importance of understanding the strength of adsorption interactions and the number of adsorption sites for efficient hydrogen storage. The findings contribute valuable insights for the rational design of covalent organic frameworks as hydrogen storage materials, supporting the development of efficient and practical hydrogen storage methods for future energy applications.
Garberoglio was the first to employ GCMC simulations to assess hydrogen absorption in covalent organic framework systems, with a particular focus on COF-102 through COF-108, at 77 K and 298 K (Assfour and Seifert 2010b; Nguyen and Gupta 2022). Among his extensive simulations, COF-105 emerged as the most promising framework for hydrogen uptake at the lower temperature of 77 K, achieving approximately 10.5 wt.%. Building on this work, Klontzas et al. (2008) applied a Lennard–Jones model to predict that COF-108 could achieve a significant gravimetric uptake of 21 wt.% at 77 K and 100 bar and 4.5 wt.% at room temperature and 100 bar. Furthermore, Klontzas et al. (2010) modified COF-102 by replacing its phenylene moieties with other extended aromatic moieties while retaining its original topology, resulting in COF-102-2, COF-102-3, COF-102-4, and COF-102-5, respectively. Among these, COF-102-3 exhibited the most promising hydrogen storage capabilities, achieving 26.7 wt.% at 77 K and 6.5 wt.% at 300 K under a pressure of 100 bar. Remarkably, these results exceeded the Department of Energy's target of 6 wt.% even at room temperature.
Enhancing the binding energy of hydrogen physisorbed on covalent organic frameworks to achieve reversible hydrogen storage at ambient temperatures is crucial. One strategy involves doping with metals, such as lithium (Li) and magnesium (Mg), which can be easily incorporated into the covalent organic frameworks, typically supported on the framework of benzene rings (Choi et al. 2008). While this approach effectively boosts hydrogen binding energy, maintaining the stability of the metal atoms within the materials and preventing metal aggregation into clusters pose significant challenges. In this context, Li et al. (2010) discussed a method to enhance the hydrogen storage properties of covalent organic frameworks through substitutional doping. The researchers proposed replacing the bridge rings in covalent organic frameworks with metal-participated rings to improve the binding energies of hydrogen molecules. First-principles calculations and Monte Carlo simulations were conducted to evaluate the effects of metal doping on hydrogen storage capacity. The results indicated that the substitutional doping strategy significantly increased the binding energy of hydrogen molecules, leading to an enhancement by a factor of four compared to undoped crystals (i.e., reaching about 10 kJmol−1). This improvement resulted in a predicted increase in the room temperature hydrogen storage capacity by a factor of two to three. The study suggests that metal doping in covalent organic frameworks can be a promising approach to achieving higher hydrogen storage capacities at ambient temperatures.
Ke et al. (2017) investigated the modification of COF-108 to enhance its hydrogen storage capacity at ambient temperature. The authors propose a two-step method involving geometric modification through C60 impregnation or aromatic ring grafting, followed by surface doping with Li atoms. The results show that the combination of Li-doping with C60 impregnation or aromatic ring grafting increases the volumetric hydrogen adsorption capacity of COF-108. One specific modified COF-108, Li6C60-impregnated COF-108, demonstrated an absolute hydrogen uptake beyond the target set by the U.S. Department of Energy (DOE). It achieved an uptake of 45.6 mg/g and 28.6 g/cm3 at 233 K and 100 bar. The impregnation and grafting techniques increased the density of doped Li and created more potential interaction sites with hydrogen, resulting in a higher number of adsorption sites. These findings indicate the potential of modified covalent organic frameworks for improving hydrogen storage capacities.
Song and Dai (2013) investigated the effect of dopants (Li, Na, Mg, and Al) on the hydrogen storage capacities of COF-108 using first-principles calculations. The binding energy of dopants in COF-108 is estimated, and their influence on the electronic structure and chemical interactions is analyzed. The study finds that Li and Na dopants exhibit positive binding energies, with the Na-doped system showing the lowest binding energy of 0.518 eV. These dopants cause the conduction band to shift and introduce weakly bonded electrons near the Fermi energy, leading to the polarization of hydrogen molecules. This polarization enhances the interaction between hydrogen and the host COF-108, thereby improving the hydrogen uptake in the doped systems. On the other hand, Mg dopant slightly reduces the band gap between the valence and conduction bands but does not significantly affect the electron distributions or chemical interactions in COF-108. Al doping requires a large amount of energy (2.692 eV) and does not show significant improvement in hydrogen uptake. The findings suggest that doping Li and Na in COF-108 can enhance its hydrogen storage capacity by promoting stronger interactions between hydrogen molecules and the material. These insights into the mechanisms of dopant influence contribute to the understanding and design of covalent organic frameworks for efficient hydrogen storage applications.
Guo et al. (2012) explored the hydrogen storage capacity of Li-doped Pc-PBBA covalent organic framework through a multiscale study. The authors combine first-principles calculations and kinetic Monte Carlo (KMC) simulations to analyze hydrogen adsorption, diffusion, and desorption processes. The first-principles calculations reveal that Li atoms can be doped on the covalent organic framework's surface with a binding energy of 1.08 eV. Each Li cation can bind three hydrogen molecules with an average adsorption energy of 0.11 eV. The maximum hydrogen uptake is found to be 24 hydrogen molecules per formula unit, corresponding to a maximum gravimetric density of 5.3 wt.% and volumetric uptake of 45.2 g/L. The diffusion barriers for hydrogen between different Li-adsorption sites range from 0.027 to 0.053 eV. The kinetic Monte Carlo simulations predict that the optimal conditions for hydrogen storage in Li-doped Pc-PBBA covalent organic framework are at temperature (250 K) and pressure (100 bar), resulting in a gravimetric density of 4.70 wt.% and volumetric uptake of 40.23 g/L. At temperature 300 K and pressure 1 bar, fast desorption kinetics are observed, with 97% of hydrogen being released from the adsorbed phase to the gas phase.
In conclusion, covalent organic frameworks exhibit remarkable potential as hydrogen storage materials due to their unique structural properties and versatile functionalities. Extensive research has highlighted their ability to achieve reversible hydrogen storage at ambient temperatures, making them promising candidates for sustainable energy applications. Covalent organic frameworks offer advantages, such as tunable pore structures, high surface areas, and customizable chemical functionalities, enabling tailored designs for enhanced hydrogen adsorption capacities. While challenges remain in optimizing stability and scalability, ongoing advancements in covalent organic framework synthesis and characterization continue to drive their development towards practical hydrogen storage solutions, showcasing covalent organic frameworks as exceptional materials in the pursuit of clean energy technologies.
Carbon-based materials
Carbon-based materials have garnered significant attention in the realm of hydrogen storage due to their unique properties. Notably, materials like graphene (Pumera 2011; Tozzini and Pellegrini 2013), fullerenes (Yoon et al. 2007; Pupysheva et al. 2008; Lan et al. 2009), carbon nanotubes (Cheng et al. 2001; Züttel et al. 2002; Oriňáková and Oriňák 2011), and activated carbon (Noh et al. 1987; Jordá-Beneyto et al. 2007; Wang et al. 2009) have undergone extensive investigations for their ability to store hydrogen with exceptional thermal stability, as depicted in Fig. 5. Matching the pore size with the kinetic diameter of the hydrogen molecule is the fundamental principle underlying the physical storage of hydrogen in these materials, with the porous structure being the primary determinant of hydrogen adsorption (Singh et al. 2023). For instance, activated carbons, such as KUA5 have exhibited significant hydrogen adsorption capabilities at varying temperatures and pressures, achieving up to 8 wt.% hydrogen storage (Jordá-Beneyto et al. 2007).

Hydrogen storage on titanium-doped fullerene, carbon nanotubes (CNTs), graphene, and activated carbons. Titanium-doped fullerene utilizes the cage-like structure of fullerene, which is doped with titanium atoms to increase its surface area. This enlarged surface area provides ample space for hydrogen adsorption. Titanium-doped fullerene demonstrates reversible hydrogen uptake, making it a promising candidate for onboard storage in fuel cell vehicles. Carbon nanotubes possess a tubular graphene lattice, which grants them exceptional strength and a large surface area. Hydrogen molecules can be stored within carbon nanotubes, enhancing their capacity for hydrogen storage. Ongoing research focuses on optimizing carbon nanotube structures to improve their practical applicability. Graphene, composed of a single layer of carbon atoms arranged in a hexagonal lattice, exhibits remarkable mechanical and electrical properties. Functionalized graphene sheets can adsorb hydrogen, making them ideal for lightweight storage systems. Activated carbons are characterized by their porous structure, consisting of micropores and mesopores. These pores effectively trap hydrogen molecules through physisorption, enabling efficient storage
Hydrogen adsorption on carbon materials is contingent upon pore size, categorized as micropores (< 2 nm), mesopores (2–50 nm), and macropores (> 50 nm). Physisorption-based hydrogen storage capacity can be computed as the aggregate of adsorption on the solid surface and compression within slit pores. Adsorption predominantly occurs in micropores, where the density of the adsorbed hydrogen phase surpasses that of the unadsorbed gaseous phase above the critical point. The hydrogen storage capacity of the adsorbent material is primarily determined by the micropore-specific surface area (SSA) (Mohan et al. 2019). Multiple studies have explored the correlation between storage capacity and specific surface area. Panella et al. (2005) concluded that hydrogen adsorption is directly proportional to specific surface area, regardless of operating temperature or carbon material type. Schimmel et al. (2003) found that the hydrogen bonding to any carbon material is weak, leading to low adsorption energy that precludes hydrogen from adsorbing in narrow interstitial channels between nanotubes. This implies that higher available surface area corresponds to higher storage capacity, as observed in activated carbons. Noh et al. (1987) demonstrated that surface modification via oxygen treatment enhances surface acidity, thereby augmenting hydrogen storage capacity without affecting specific surface area. Furthermore, Agarwal et al. (1987) also observed an increase in storage capacity with specific surface area, particularly with certain surface modifications. The hydrogen storage performance of various carbon materials under practical conditions can be summarized as follows:
Graphene
Graphene-based materials, particularly graphene oxide (GO), have been actively studied for hydrogen storage. Graphene oxide, derived from graphite, exhibits a hydrogen uptake of 1.90 wt.% (at room temperature and 80 bar pressure), while reduced graphene oxide (rGO) shows 1.34 wt.% (Rimza et al. 2022). This enhanced capacity is attributed to oxygen functional groups in graphene oxide and reduced graphene oxide, which facilitate hydrogen binding between nanosheets (Rajaura et al. 2016). Additionally, preheating graphene oxide at different temperatures (298, 523, and 723 K) results in hydrogen uptakes of 1.5, 2.0, and 2.5%, respectively, due to modifications in its interlayer spacing and improved hydrogen binding (Yadav et al. 2020). Mesoporous graphene oxide achieved a remarkable capacity of 4.65 wt.% at ambient temperature and 40 atm pressure (Kim et al. 2016).
While pure graphene cannot store significant amounts of hydrogen through physisorption due to its low binding energy, functionalization offers a promising avenue to enhance its hydrogen storage efficiency. Strategies, such as doping graphene with metals from transition, alkali, or alkaline earth metal groups, as well as introducing heteroatoms and defect engineering, have been explored to functionalize graphene (Lotfi and Saboohi 2014; Zhou et al. 2016; Singh et al. 2023). The incorporation of defects like Stone–Wales defects and single or double vacancies has been theoretically demonstrated to increase graphene's hydrogen storage capacity to up to 7.02 wt.% (Yadav et al. 2014). Additionally, adjusting the interlayer spacing of graphene using suitable spacers represents a viable approach to achieving high hydrogen storage capacity, reaching up to 6.5 wt.% (Yu et al. 2017).
The functionalization of graphene allows for the customization of material properties, such as chemical reactivity, surface area, porosity, and interlayer spacing. This tailored approach enhances adsorption capacities while simultaneously reducing the energy barrier for adsorption. In this context, Ni-doped (Ariharan et al. 2017) Al-doped (Ao et al. 2009), and Li-doped graphene (Deng et al. 2004) exhibit hydrogen storage capacities of 1.5 wt.% (at 298 K and 9 MPa), 3.84 wt.% (at 300 K and 100 MPa pressure), and a significant 6.5 wt.% (at 298 K and 2 MPa pressure), respectively. Other studies, such as the study of Zhou et al. (2016) study on Ni-graphene, found 1.18 wt.% hydrogen uptake at room temperature and 60 bar pressure, with complete desorption at 523 K. Mn-V-decorated graphene displayed even higher hydrogen uptake at 1.81 wt.% at 4 MPa, surpassing pristine graphene's 0.25 wt.% (Pei et al. 2017). This research underscores cost-effectiveness, as Mn and V are more affordable than noble metals (Pd, Pt, and Au). Notably, a significant hydrogen uptake of 3 wt.% was achieved with 20 wt.% Pd-doped graphene at 40 bar pressure (Parambhath et al. 2011), while Ni-doped graphene reached 1.1 wt.% at 2 MPa (Vinayan et al. 2013). Combining Pd–N-doped graphene resulted in 0.83 wt.% hydrogen uptake at 32 bar and 1.5–4.5 wt.% at 40 bar pressure (Vinayan et al. 2013). Additionally, Pt-nanoparticle-decorated graphene foam achieved a significant 3.19 wt.% uptake, aided by polydopamine functionalization to maintain the proper surface area, dopant dispersal, and doping amount (Jung et al. 2016). Furthermore, Vaidyanathan et al. (2024) presented the results of a study on hydrogen storage capacities of Sc-decorated Ψ-graphene. The researchers used density functional theory simulations to investigate the binding properties of Sc atoms to Ψ-graphene and its ability to store hydrogen. The results show that Sc-decorated Ψ-graphene exhibits a strong binding energy of approximately 3.02 eV and can bind seven hydrogen molecules with an average binding energy of 0.36 eV/H2. The hydrogen storage capacities were found to be 8.59 wt.% for five hydrogen molecules and 14.46 wt.% for seven hydrogen molecules. These capacities meet the requirements set by the U.S. Department of Energy for efficient hydrogen storage in light fuel cell vehicles. The study also highlights the stability of the system at room temperature and the low likelihood of Sc–Sc clustering due to the high diffusion energy barrier. Macili et al. (2023) developed a novel three-dimensional graphene structure that addresses the challenges of graphene's two-dimensional characteristics, improving its efficiency in hydrogen absorption.
Leng et al. (2020) investigated the effect of graphene on the hydrogen storage properties of MgH2. The results showed that graphene significantly enhances the kinetics of MgH2 by providing additional hydrogen diffusion channels. On the other hand, materials composed of stacked graphene layers, such as graphite, with defects or interlayer spacing ranging from 6–7 Å, demonstrate significant potential for hydrogen storage (Nair et al. 2015). However, pristine graphite possesses an interlayer spacing of 3.34 Å, which is insufficient to accommodate hydrogen molecules, requiring a minimum spacing of 4.06 Å. Therefore, modifying the structure of graphitic materials holds promise for enhancing hydrogen storage. Nair et al. (2015) investigated the hydrogen storage capacity of graphitic carbon nitride (g-C3N4) and graphitic carbon nitride doped with palladium (Pd-g-C3N4) using Sievert's apparatus. Their findings demonstrated that Pd-g-C3N4 exhibited a hydrogen storage capacity of 2.6 wt.% at 298 k and 4 MPa. Decorating Pd nanoparticles (Pd-NPs) on the g-C3N4 matrix resulted in a 66% increase in hydrogen uptake at ambient temperatures, underscoring the hydrogen spillover effect. The efficient dispersion of Pd-NPs on the g-C3N4, coupled with intensified interactions between them, catalyzed the dissociation and migration of hydrogen molecules, thereby facilitating the spillover mechanism.
It is worth noting that fully hydrogenated graphene, also known as "graphane," is a solid-state material exhibiting significant hydrogen storage capacity. With a chemical formula expressed as (CH)n, graphane can be viewed as a sp3 hybridized counterpart to graphene, comprising 2-D carbon sheets where each carbon atom in the network is covalently bonded to a hydrogen atom. Although isolating pure graphane in bulk poses challenges akin to graphene, chemical hydrogenation of graphite and other carbon forms through Birch reduction can yield hydrogenated graphene (Schäfer et al. 2013, 2016; Whitener 2018). These hydrogenated graphene materials reportedly contain covalently bound hydrogen, approaching the theoretical loading capacity of 7.7 wt.%. They remain inert under ambient conditions but irreversibly decompose to release hydrogen gas at temperatures ≥ 673 K (Subrahmanyam et al. 2011; Bouša et al. 2016; Yang et al. 2016; Eng et al. 2017). In this regard, Morse and colleagues explored the potential of chemically hydrogenated graphene as a material for hydrogen storage (Morse et al. 2021). Hydrogenated graphene has a theoretical hydrogen storage capacity of 7.7 wt.% and can release hydrogen gas upon thermal decomposition. The study aimed to characterize various properties of hydrogenated graphene related to hydrogen storage. It was found that compression of hydrogenated graphene does not affect hydrogen storage, and the fraction of hydrogen release can be controlled by temperature and heating time. The decomposition of hydrogenated graphene is the same under different environments, such as nitrogen, hydrogen, or vacuum. The activation energy for hydrogen release was calculated to be 158 kJmol−1. The authors conducted a large-scale synthesis of hydrogenated graphene using the Birch reduction method. The study provides valuable insights into the potential application of hydrogenated graphene as a hydrogen storage medium. The high energy density and promising synthesis methods make hydrogenated graphene an attractive candidate for future hydrogen storage systems. However, further research and characterization are required to fully understand and optimize the properties of hydrogenated graphene for efficient hydrogen storage.
Another study conducted by the previous team (Morse et al. 2020) presented the synthesis and evaluation of chemically hydrogenated graphene for hydrogen storage applications. The researchers successfully synthesized 75 g of hydrogenated graphene using a scaled-up Birch reduction method, representing the largest reported synthesis of this material. Characterization techniques confirmed a hydrogen loading of 3.2 wt.%. The study demonstrated the controlled release of hydrogen gas from the bulk material, revealing a bulk hydrogen storage capacity of 3.2 wt.%. The researchers also successfully operated a hydrogen fuel cell using chemically hydrogenated graphene as a power source. These findings highlight the potential of hydrogenated graphene as a high-density hydrogen storage medium, contributing to the design of prototype hydrogen storage systems.
In conclusion, graphene holds promise as a material for hydrogen storage due to its unique properties. Functionalized, doped, or defected graphene variants exhibit encouraging theoretical evidence for efficient hydrogen adsorption. Transition metal decoration, nitrogen and boron doping, and combinations thereof enhance storage capacity by adjusting binding energies. However, challenges persist, particularly in achieving scalable synthesis methods and ensuring controllable adsorption/desorption kinetics. External stimuli and surface reconstruction methods offer avenues for improving kinetics. Ultimately, the commercialization of graphene-based hydrogen storage materials hinges on achieving high efficiency, low weight, reusability, low cost, and fast kinetics. With ongoing advancements in porous graphene frameworks, graphene could lead to significant breakthroughs in hydrogen storage technologies.
Fullerene
Fullerene, a highly symmetrical carbon allotrope, exhibits spherical, ellipsoidal, or tubular geometries made up of carbon atom meshes in hexagonal or pentagonal configurations (Ekpete and Orie 2023). The most common form is C60, known as Buckminster. Fullerenes possess unique properties, such as stability and the capacity to trap gas atoms, making them suitable for hydrogen storage. Metal atom-supported carbon fullerenes leverage the high electronegativity of C60 to transfer electrons from metal atoms, leaving the metal in a cationic state. This mechanism enables the trapping of molecular hydrogen through the charge polarization phenomenon. However, based on theoretical models, the metal atom coated on C60 predominantly maintains an isolated configuration (Zhao et al. 2005). In this context, Yoon et al. (2007) explored the potential of charged carbon fullerenes (Cn) as hydrogen storage materials. Through first-principles calculations, the study investigates the binding strength of molecular hydrogen to positively and negatively charged fullerenes. The results show that the binding strength can be significantly enhanced to a desirable range of 0.18–0.32 eV, suitable for near-ambient applications. This enhancement is attributed to the polarization of hydrogen molecules by the high electric field near the charged fullerene's surface. At full hydrogen coverage, the charged fullerenes can achieve storage capacities of up to 8 wt.%. These findings suggest the potential of charged carbon fullerenes as a new class of high-capacity hydrogen storage media.
Another study conducted by Pupysheva et al. (2008) examined the potential of fullerene nanocages for hydrogen storage. Using density functional theory (DFT) and ab initio molecular dynamics simulations, the researchers investigate the capacity of endohedral fullerenes (Hn@Ck) to store hydrogen. They find that for a C60 cage, the maximum stable structure can accommodate up to 58 hydrogen atoms. The breaking mechanism of this structure is also studied. The study estimates the hydrogen pressure inside the fullerene nanocage, which is shown to be close to hydrogen metallization pressure. The researchers established a general relation between hydrogen pressure and C–C bond elongation for fullerene nanocages of different radii. This provides insights into the hydrogen content achievable in larger carbon nanocages. The formation energy of Hn@C60 structures is examined, revealing that structures with more than two encapsulated hydrogen molecules are metastable. The findings of this study demonstrate the potential of fullerene nanocages for hydrogen storage. The maximum capacity of 58 hydrogen atoms within a C60 cage indicates a significant storage capability. The estimated hydrogen pressure inside the cage suggests that reasonable storage conditions can be achieved. The general relation between hydrogen pressure and C–C bond elongation provides a valuable tool for predicting hydrogen content in larger carbon nanocages. The metastable nature of structures with more than two encapsulated hydrogen molecules highlights the challenges associated with their formation. Overall, these results contribute to the understanding of hydrogen storage capacities in fullerene nanocages and offer insights for further research and development in this field. Metal-atom-supported fullerenes possess the ability to attract electrons, resulting in the metal atom being left in a cationic state, thereby enabling the trapping of molecular hydrogen. Lithium-coated fullerenes (Li2C60), the lithium atom, assume stability on the pentagonal face of the fullerene, facilitating storage of up to 120 hydrogen atoms with a binding energy of 0.075 eV/H2 (Niaz et al. 2015).
Additionally, investigations into scandium and titanium coatings have been conducted. Yildirim et al. (2005) explored the potential of titanium- and scandium-coated fullerenes and determined that a theoretical hydrogen storage capacity of 8 wt.% could be achieved, with binding energies falling within the range of 0.3–0.5 eV. Mahamiya et al. (2022) investigated the hydrogen storage capacity of scandium-decorated C24 fullerene using density functional theory simulations. The results show that the system can adsorb up to six hydrogen molecules with an average adsorption energy of 0.35 eV per H2 and an average desorption temperature of 451 K. The gravimetric weight percentage (wt.%) of hydrogen for the scandium-decorated C24 fullerene is found to be 13.02%, surpassing the Department of Energy's demand. The study also analyzes the electronic structure, orbital interactions, and charge transfer mechanisms. It reveals a total charge transfer of 1.44e from scandium to the carbon orbitals of C24 fullerene. The stability of the structure at high desorption temperatures is confirmed through ab initio molecular dynamics simulations. Overall, the results suggest that scandium-decorated C24 fullerene is a promising and thermodynamically stable material for high-capacity hydrogen storage applications.
Paul et al. (2023) explored the hydrogen storage capacity of yttrium-doped fullerene C30 using density functional theory simulations. The results indicate that a single Y atom can adsorb seven hydrogen molecules, with a binding energy falling within the range suggested by the U.S. Department of Energy. The gravimetric weight percentage for C30 loaded with five Y atoms, each adsorbing seven hydrogen molecules, is recorded to be 8.06%. This value exceeds the Department of Energy's limit of 6.5%, indicating the potential of Y-doped fullerene C30 as a hydrogen storage candidate. The study discusses the interaction between Y and C30, the Kubas interaction between the metal and hydrogen, and the elongation of the H–H bond in hydrogen molecules. These findings highlight the viability of Y-doped fullerene C30 for hydrogen storage applications.
Sahoo et al. (2021) explored the reversible hydrogen storage capacities of Li and Na-decorated C20 fullerene using density functional theory. The study reveals that Li and Na atoms bind to the C20 fullerene through non-covalent closed-shell interaction. Each Li and Na atom can adsorb up to five hydrogen molecules through the Niu-Rao-Jena interaction. The adsorption energy decreases with the addition of hydrogen molecules, with an average binding energy ranging from 0.12 to 0.13 eV. The gravimetric density of the systems can reach up to 13.08 wt.% for C20Li4 and 10.82 wt.% for C20Na4. Molecular dynamic simulations demonstrate the reversibility of adsorbed hydrogen molecules at higher temperatures. These findings indicate that Li and Na decorated C20 fullerene hold promise as hydrogen storage materials.
Jaiswal et al. (2022) reported the potential of Si-substituted and Li-decorated C20 fullerene for hydrogen storage. The study conducted density functional theory simulations to investigate the reversible hydrogen storage capacities. The newly designed Si2C18Li6 and Si4C16Li6 cages were found to exhibit stability and structural integrity at high temperatures. The adsorption energies for hydrogen molecules in Si2C18Li6-nH2 and Si4C16Li6-nH2 were determined to be in the range of 0.119–0.139 eV and 0.131–0.140 eV, respectively. The practical storage capacities of Si2C18Li6 and Si4C16Li6 cages at specific temperature and pressure ranges were found to be 16.09% and 14.77 wt.%, respectively. These capacities exceed the target set by the United States Department of Energy (5.5 wt.% by 2020). Furthermore, Zhang and Cheng (2018) explored the hydrogen storage capabilities of alkali and alkaline-earth metal atoms attached to C24 fullerene through density functional theory calculations. The study finds that the alkali and alkaline-earth metal atoms prefer to adsorb on the center of the tetragon of C24 fullerene, providing the highest binding energy. The hydrogen storage capacities of different configurations are evaluated. The 24H2/6Li/C24, 24H2/6Na/C24, and 36H2/6Ca/C24 configurations exhibit hydrogen storage gravimetric densities of 12.7 wt.%, 10.1 wt.%, and 12 wt.%, respectively. These values surpass the hydrogen storage target set by the U.S. Department of Energy for the year 2020. The average adsorption energies of hydrogen molecules in these configurations are in the desirable range for physical adsorption at ambient conditions. These findings have significant implications for designing new hydrogen storage materials in the future.
In conclusion, fullerene-based materials exhibit promising capabilities for hydrogen storage, driven by their unique structural properties and interactions with metal atoms. Metal atom-supported fullerenes demonstrate the potential to trap molecular hydrogen through charge polarization, offering significant storage capacities. Research on lithium, scandium, and yttrium-coated fullerenes reveals their ability to achieve hydrogen storage capacities exceeding the Department of Energy's targets. Additionally, studies on silicon-substituted and alkali/alkaline-earth metal-decorated fullerenes demonstrate high gravimetric densities, surpassing storage targets. The findings suggest a diverse range of fullerene-based materials with considerable potential for high-capacity hydrogen storage applications. Further research in this area holds promise for addressing energy storage challenges and advancing hydrogen fuel technologies.
Carbon nanotubes
Carbon nanotubes (CNTs), including single-walled carbon nanotubes (SWCNTs) and multi-walled carbon nanotubes (MWCNTs), are capable of storing hydrogen through physisorption. They are considered one of the best options for maximizing hydrogen storage via physisorption due to their unique properties (ullah Rather 2020). Carbon nanotubes have been explored for their hydrogen storage potential since their introduction by Dillon et al. (1997). These properties include their nanostructure, high surface area, tunable characteristics, and low mass density, which make them favorable for reversible hydrogen storage media. SWCNTs are composed of a single layer of carbon atoms arranged in a cylindrical structure, while MWCNTs consist of multiple concentric layers of graphitic filaments aligned along the same axis.
Additionally, SWCNTs can form bundles, and the length of carbon nanotubes is typically on the order of microns, while the inner diameter is thousands of times smaller (Sdanghi et al. 2020). Both theoretical and experimental evidence indicate that carbon nanotubes can adsorb hydrogen through physisorption and/or chemisorption mechanisms. Physisorption occurs via weak van der Waals forces between hydrogen molecules and carbon atoms, with a binding energy of approximately 0.1 eV. The maximum adsorption density correlates with specific surface area and pore volume. Chemisorption involves the formation of chemical bonds between hydrogen atoms and carbon, with binding energies exceeding 2–3 eV. It’s challenging to discern exclusively between physisorption and chemisorption. As the interaction strength increases, hydrogen may dissociate into atomic form and diffuse into the nanotubes, akin to metal hydrides. Modifying material chemistry or structure can alter desorption temperature, albeit within a limited range (Oriňáková and Oriňák 2011).
In order to investigate the hydrogen storage mechanism in SWCNTs, Lee et al. (2001) conducted systematic calculations at zero temperature to understand the adsorption and storage of hydrogen in SWCNTs. The results suggest that hydrogen atoms initially adsorb on the tube wall in arch-type and zigzag-type configurations up to a coverage of approximately 1. At higher coverages, hydrogen atoms are stored within the capillary of SWCNTs in the form of hydrogen molecules. The dominant storage mechanism involves breaking the C–C midbond within the tube wall while preserving the stability of the nanotube structure. The study also discusses the extraction process, where hydrogen molecules within the capillary dissociate, adsorb onto the inner wall, and are further extracted to the outer wall through the flip-out mechanism. Gu et al. (2001) investigated hydrogen storage in SWNTs using grand canonical Monte Carlo simulation. The study models hydrogen–hydrogen and hydrogen-carbon interactions with Lennard–Jones potential. The adsorption isotherms, adsorption as a function of van der Waals distance and diameter, and the influence of pressures and temperatures on adsorption are analyzed. The results provide insights into optimizing pore geometry for hydrogen storage at specific conditions. The study concludes that SWNTs show promise as a hydrogen storage system and further research is needed to develop efficient adsorbents tailored for hydrogen storage. Another study conducted by Ma et al. (2002) explored the storage capacity of hydrogen in SWCNTs through molecular dynamics simulations. The study finds that the storage capacity inside SWCNTs increases with increasing tube diameters. It is observed that for SWCNTs with diameters less than 20 Å, the storage capacity depends strongly on the helicity of the nanotube. The strain on the nanotube caused by the interaction between stored hydrogen molecules and the SWCNT is also examined. The maximum radial strain ranges from 11 to 18% and is influenced by the helicity of the nanotube. The tensile strengths of SWCNTs decrease with increasing diameters, approaching that of graphite (20 GPa) for larger-diameter tubes. The results provide insights into the hydrogen storage potential of SWCNTs and their mechanical properties, which are crucial for practical applications in energy storage.
Nijkamp et al. (2001) examined SWNTs' hydrogen sorption isotherms, obtaining maximum concentrations of 0.932 wt.% at 295 K and 2.37 wt.% at 77 K, suggesting additional sorption mechanisms beyond physisorption on carbon hexagons due to observed deviations in H/C ratios. Ye et al. (1999) investigated hydrogen adsorption on purified SWNTs, revealing capacities of up to 8 wt.% at low temperatures and pressures. Liu et al. (1999) explored pretreatments' effects on SWNT hydrogen adsorption, achieving up to 4.2 wt.% at room temperature and 10 MPa pressure through acid treatment and vacuum heating.
Furthermore, Zhou et al. (2006) investigated the hydrogen uptake mechanism on superactivated carbon and MWCTs in both powder and pellet forms, analyzing adsorption isotherms across a wide temperature and pressure range. Employing a volumetric method, hydrogen adsorption data were analyzed using advancements in supercritical adsorption theory. The study concluded that hydrogen adsorption occurs predominantly through physical means, with molecules arranged monolayerly on the carbon surface. Consequently, the storage capacity is primarily dictated by the specific surface area of the carbon material. In order to investigate the hydrogen storage capacity of MWNTs with varying diameters. Hou et al. (2003) find that the hydrogen storage capacity of MWNTs is proportional to their diameter. It is observed that hydrogen adsorbed in MWNTs cannot be completely desorbed at room temperature and ambient pressure. The researchers propose that small "carbon islands" within MWNTs serve as the main hydrogen adsorption sites.
In pursuit of meeting the Department of Energy's technical storage targets, it's evident that relying solely on hydrogen physisorption in the materials, as mentioned above, despite their unique characteristics, is insufficient. To enhance the overall hydrogen storage capacity, various proactive measures have been adopted, including activation using sodium hydroxide (NaOH) and potassium hydroxide (KOH). Chemical activation with sodium hydroxide and potassium hydroxide serves to increase surface area and pore volume, consequently improving hydrogen uptake capacity in these materials. This enhancement occurs linearly as surface area and pore volume increase (Raymundo-Piñero et al. 2005; Rather et al. 2008; ullah Rather 2020). Another significant strategy to augment hydrogen storage capacity involves the incorporation of metal or metal oxide nanoparticles into carbon nanotubes, either through decoration, embedding, or doping. This process facilitates hydrogen molecule dissociation, leading to the spillover mechanism wherein hydrogen atoms are released onto the surface. The non-classical spd hybridization and spillover phenomenon contribute to enhanced hydrogen uptake capacity in carbon nanotube composites (Zacharia et al. 2005, 2007). Various methods, including in-situ condensed phase reduction, wetness impregnation, high-energy ball milling, and sputtering, are employed for decoration, doping, or embedding purposes (Zacharia et al. 2007; Rather et al. 2007; Rather and Nahm 2014; ullah Rather 2020).
In this regard, doped carbon nanotubes, such as those incorporating Co (2.62 wt.%) (Chang et al. 2014), V (0.69 wt.%) (Zacharia et al. 2005), and Pd (0.6–0.87 wt.%) (Zacharia et al. 2005; Wenelska et al. 2014), exhibit enhanced hydrogen storage potential. Liu et al. (2010) reported that carbon nanotubes' storage capacity remains below 1.7 wt.% at room temperature and moderate pressure. In terms of MWCTs, MWCNTs exhibit hydrogen uptakes of 0.3–0.9 wt.% at ambient temperatures (Rather and Hwang 2016; Hosseini et al. 2017; Han and Park 2017). The introduction of a hydrogen spillover catalyst, titanium, increases hydrogen uptake in MWCNTs by 5- and 25-fold at 298 K and an equilibrium pressure of ~ 16 atm, elevating it from 0.43 to 2.0 wt.%. Aboutalebi et al. (2012) studied the hydrogen storage performance in MWCNTs and graphene oxide, revealing adsorption capacities of approximately 0.9 wt.% and 1.4 wt.%, respectively, at 298 K and 50 bar pressure. Interestingly, their combined structures exhibited improved gravimetric storage, achieving around 2.6 wt.% in the same conditions. Similarly, MWCNTs with tin(IV) oxide (SnO2) particles exhibited a 2.62 wt.% hydrogen storage capacity (Vellingiri et al. 2018). Mehrabi et al. (2017, 2019) explored Pd- and Ni-doped MWCNTs, synthesizing them through laser ablation and chemical reduction. The results indicated that Pd nanoparticles surpassed Ni in spillover rates, with Pd-MWCNTs achieving an 8.6 wt.% uptake and Ni-MWCNTs reaching 2.5 wt.%.
Recently, Heydariyan et al. (2023) introduced a novel energetic, solid composite based on nano-sized EuMnO3/EuMn2O5 incorporated with MWCNT for the development of hydrogen storage capacity. Additionally, Alazawi et al. (2023) developed MWCNTS-ZnO (acetate) and MWCNTS-ZnO (complex) nanocomposites, with transmission electron microscopy (TEM) confirming the successful formation of irregular spherical zinc oxide nanoparticles (45–70 nm) covering carbon nanotubes with diameters up to 29 nm. The findings revealed that MWCNTS-ZnO (complex) exhibited the highest hydrogen storage capacity, achieving 4.3 wt.% H2 under conditions of 85 bar pressure and a temperature of 77 K.
Regarding SWCNTs, a recent work conducted by Verma and Jaggi (2024) investigated the use of osmium-decorated SWCNTs for hydrogen storage. The study utilizes density functional theory and explores the effects of single and dual osmium decorations on the structural, electronic, and thermodynamic properties of SWCNTs. The results indicate that osmium decoration enhances the hydrogen storage capacity through the spillover mechanism. The adsorption energy per hydrogen molecule increases with osmium decoration, reaching − 0.575 eV/H2 for single osmium decoration and − 0.681 eV/H2 for double osmium decoration. The study reveals a gravimetric hydrogen storage capacity of 1.32 wt.% and 2.53 wt.% for single and double osmium decorated SWCNTs, respectively, at 298.15 K and 10 atm.
The average Van't Hoff desorption temperature for osmium-decorated SWCNTs is also calculated. Yang et al. (2019) explored the hydrogen storage capacity of dual-Ti-doped SWCNTs using first-principles calculations. The dual-Ti-doped SWCNTs were found to stably adsorb up to six hydrogen molecules through Kubas interaction at the Ti2 active center. Additionally, the Ti-doping pattern affected the hydrogen adsorption capacity, with the dual-Ti decorated SWCNT capable of adsorbing eight hydrogen molecules with ideal adsorption energy. The synergistic effect of Ti atoms with different doping patterns enhanced the hydrogen adsorption capacity. The study emphasized the potential of Ti-doped SWCNTs as a promising material for efficient reversible hydrogen storage. Verdinelli et al. (2019) conducted a study that investigated the storage of molecular hydrogen on ruthenium (Ru) decorated SWCNTs using density functional theory. The results showed that Ru atoms act as adsorption centers for hydrogen molecules on SWCNTs, with an adsorption energy (Eads) of 0.93 eV/H2. It was found that a single Ru atom on SWCNT can hold up to four hydrogen molecules, while a uniform addition of Ru atoms allows for up to five Ru atoms without clustering. Each Ru atom in the 5Ru-decorated SWCNT system can bind up to four hydrogen molecules with an Eads of 0.83 eV/H2. The magnetic moment of Ru-decorated systems decreases as the number of hydrogen molecules increases. The study suggests that Ru-decorated SWCNTs have the potential for efficient hydrogen storage.
Modak et al. (2012) conducted research to explore the electronic structure and hydrogen adsorption characteristics of SWCNTs decorated with transition metals. The study uses density functional theory and the projector augmented wave method to analyze the ground state geometry, electronic structure, and hydrogen adsorption of various transition metal decorated SWCNTs. The results reveal a systematic change in the adsorption site of transition metal atoms with the increasing number of d electrons. Y and Zr decorated SWCNTs exhibit metallic behavior, while Nb and Mo decorated SWCNTs display semiconducting behavior. The adsorption site variation is attributed to the decreasing charge transfer from the transition metal atom to the SWCNT along the 4d series. Metallic SWCNT + TM systems are found to be more effective in hydrogen adsorption. The study emphasizes that the retention of magnetism by the system is crucial for favorable hydrogen physisorption.
In conclusion, carbon nanotubes, including SWCNTs and MWCNTs, exhibit promising potential for hydrogen storage, primarily through physisorption mechanisms. Their nanostructure, high surface area, and tunable characteristics render them favorable candidates for reversible hydrogen storage media. While SWCNTs feature a single layer of carbon atoms in cylindrical form and MWCNTs consist of multiple concentric layers, both offer avenues for hydrogen adsorption. Through various experimental and theoretical studies, it has been demonstrated that modifying carbon nanotubes with transition metals, metal oxides, or other dopants can significantly enhance their hydrogen storage capacities. These advancements, coupled with innovative approaches, such as chemical activation and nanocomposite formation, underscore the evolving landscape of hydrogen storage materials. Further research and development in this area holds promise for realizing efficient and practical solutions to meet the stringent hydrogen storage targets set by the Department of Energy.
Activated carbon
Activated carbon (AC), characterized by a substantial surface area ranging from 700 to 3300 m2 g–1, is exceptionally well-suited for both adsorption and chemical reactions (Hermosilla-Lara et al. 2007; Zhao et al. 2012). Its amorphous structure is derived from diverse materials, such as coal, wood, or coconut shells. According to IUPAC (International Union of Pure and Applied Chemistry) standards, activated carbon can be classified into microporous (< 20 Å), mesoporous (20–500 Å), and macroporous (> 500 Å) categories, with the pore size determined by the raw material and processing methods employed (Romanos et al. 2019). The synergy of pore size, volume, and a substantial surface area in activated carbon greatly enhances its hydrogen absorption capabilities. For instance, activated carbon synthesized from a biomass precursor through the potassium hydroxide activation process boasts an impressive surface area of approximately 2090 m2 g–1 and a generous pore volume of 1.44 cm3 g−1 (Samantaray et al. 2019). Its hydrogen absorption capability is considerable, achieving 1.06 wt.% at 15 bar and 298 K, highlighting its potential as a catalyst support for improved hydrogen uptake via the spill-over mechanism (Samantaray et al. 2019). Moreover, Ramesh et al. (2017) developed activated carbon materials derived from jute fibers and applied potassium hydroxide activation to enhance the sample’s porosity. The carbonization and activation processes resulted in a range of surface areas, with values ranging from 380 to 1220 m2 g−1, as listed in Table 2. Their results demonstrated that the sample exhibiting the largest surface area, 1220 m2 g−1, exhibited a high hydrogen absorption capacity of 1.2 wt.% under conditions of 303 K and a 40 bar H2 gas pressure.
Doped or pretreated activated carbons have proven effective for hydrogen adsorption (Grigorova et al. 2020). Lee and Park (2011) developed a Pt-doped AC/MOF composite with hydrogen storage of 2.3 wt.% at 298 K and 100 bar, significantly outperforming raw activated carbon. Ni nanoparticle-doped activated carbon nanofibers achieved 2.12 wt.% hydrogen adsorption at 298 K and 100 bar (Thaweelap et al. 2021). While Pd nanoparticle doping on activated carbon resulted in a relatively lower hydrogen uptake of < 0.2 wt.% at 298 K and 2–3 MPa pressure (Zhao et al. 2012), super activated carbon (Maxsorb) exhibited a hydrogen absorption capacity of 0.67 wt.% (Xu et al. 2007). Table 2 provides an overview of the characteristics of activated carbons used for hydrogen storage and their storage potential (Bosu and Rajamohan 2023).
Zeolites
Zeolites, renowned for their molecular sieve-like characteristics, facilitate hydrogen storage by effectively trapping hydrogen within their unique cavities, particularly at elevated temperatures and pressures. Zeolite variants, such as KA, Naan, RbA, and CsA, classified as microporous minerals, have been harnessed as sorbents for hydrogen storage (Chilev et al. 2012). The process involves hydrogen being driven into these cavities at higher temperatures and pressures and subsequently being retained within the zeolite structure, with release achievable upon heating. Langmi et al. (2003) conducted a comprehensive exploration of zeolites A, X, Y, and RHO for their hydrogen storage capabilities over a wide temperature range, spanning from − 196 to 573 K, and under pressures ranging from zero to 15 bar. These zeolites were synthesized using hydrothermal methods and underwent cation-exchange modifications. Their findings underscored that hydrogen uptake in zeolites is significantly influenced by variables, such as temperature, framework structure, and the type of cation present. Earlier research indicates that zeolites can store limited amounts of hydrogen (< 0.3 wt.%) when charged at room temperature or temperatures exceeding 473 K (Weitkamp et al. 1993; Langmi et al. 2003, 2005).
Weitkamp et al. (1993) delved into the potential of zeolites as hydrogen storage media, considering various zeolite structures and compositions. Their results indicated that zeolites featuring sodalite cages in their structure hold the most promise for hydrogen storage. Specifically, the zeolite with the highest concentration of sodalite cages exhibited the greatest storage capacity, reaching 9.2 cm3 g−1 when loaded at 573 K and 10 MPa. While the storage capacity of zeolites currently lags behind that of metal hydride systems, they offer distinct advantages, including robust thermal stability, cost-effectiveness, and adjustable composition, making them a promising avenue for advancing hydrogen storage technologies. Recent work by Pinjari et al. (2023) involved enhancing the hydrogen storage properties of ZIF-8 zeolite materials at room temperature. They synthesized zeolitic imidazolate frameworks (ZIF-8, ZIF-8-T) as well as their copper-doped counterparts (CuZIF-8, CuZIF-8-T) for hydrogen storage under ambient conditions. CuZIF-8-T exhibited an impressive specific surface area of 1973.7 m2 g−1 and a micropore volume of 0.72 cm3 g−1, as determined by nitrogen adsorption at 77 K. This indicated a higher number of adsorption sites compared to their counterparts. Consequently, CuZIF-8-T achieved the highest hydrogen storage of 0.70 wt.% at 298 K and 100 bar, maintaining full reversibility over ten cycles.
However, at cryogenic temperatures, the hydrogen storage capacity can surpass 1 wt.% (Kazansky et al. 1998; Langmi et al. 2003). For example, CaX zeolite demonstrated an impressive hydrogen storage capacity of 2.19 wt.% at 15 bar and 77 K (Langmi et al. 2005). Du and Wu (2006) reported a hydrogen capacity of 2.55 wt.% for NaX zeolite (with a surface area of 565 m2 g−1) at 77 K and 40 bar, though this capacity decreased to just 0.4 wt.% at 293 K and 40 MPa. Chung (2010) investigated various zeolites for hydrogen storage at 303 K and found that ultra-stable Y (USY) zeolite exhibited the highest hydrogen capacity of 0.4 wt.% at 50 bars.
Novel hydrogen storage materials
Currently, significant advancement has been achieved in hydrogen storage state-of-the-art. Researchers continue to explore various approaches to enhance hydrogen storage capacity, safety, and efficiency. Some promising materials include metal–organic frameworks, covalent organic frameworks, zeolites, and porous carbon-based adsorbents. For instance, a category of novel compounds, specifically layered transition metal carbides or nitrides, known as MXenes (e.g., Ti3C2, Nb4C3, V2C), has demonstrated substantial augmentation of the hydrogen storage capabilities of MgH2 (Liu et al. 2015, 2019, 2021, p. 2; Shen et al. 2018; Wang et al. 2019b; Lu et al. 2021; Duan et al. 2023). For example, Liu et al. (2019) found that by combining MgH2 with 5 wt.% Nb4C3 MXene, they were able to achieve a remarkable hydrogen absorption capacity of 3.5 wt.% within just two hours at 323 K. Additionally, this combination exhibited an exceptionally low initial dehydrogenation temperature of 424 K. Lu et al. (2022a) utilized V2C MXene to modify MgH2, resulting in a composite of MgH2 with 10 wt.% V2C MXene. This composite exhibited a remarkable hydrogen storage capacity of 6.4 wt.% within just 10 min at 573 K. Notably, it demonstrated a low activation energy of 87.6 kJmol−1 and a reduced dehydrogenation temperature of 463 K. These findings demonstrate the significant potential of MXenes in enhancing the hydrogen storage performance of MgH2. However, it's worth noting that the production process for MXenes often involves the use of corrosive hydrofluoric acid, which is environmentally unfriendly.
In 2023, Duan et al. (2023) investigated using Ti3AlCN MAX as an environmentally friendly alternative to improve the hydrogen storage properties of MgH2. Comparatively, Ti3AlCN MAX showed more promise in enhancing MgH2's hydrogen storage performance compared to many MXenes like Ti3C2 and V2C. The study revealed that Ti3AlCN primarily functions as an efficient catalyst for MgH2, acting as an active center for nucleation and growth of MgH2, as well as facilitating the recombination and dissociation of hydrogen molecules. These two parameters contribute to its superiority over MXenes in promoting hydrogen storage in MgH2.
Additionally, metal–organic frameworks and their composite materials have garnered considerable attention as potential candidates for hydrogen storage, as discussed earlier. They enable hydrogen storage through a combination of physisorption and chemisorption mechanisms, exemplified by hybrid materials like metal–organic frameworks (SNU-90) and Mg nanocrystals (Lim et al. 2012). Moreover, MgH2@Ni–MOF nanocomposites, synthesized using solvothermal methods and wet impregnation, exhibit efficient kinetic properties and low hydrogen absorption/desorption enthalpies (Ma et al. 2020). The outstanding performance of these materials results from the synergistic effects of nanoconfined Mg/MgH2 and in-situ catalysis from formed Mg2Ni/Mg2NiH4. This is a novel approach where the scaffold prevents Mg/MgH2 growth and also catalyzes hydrogen sorption of MgH2/Mg.
Ma et al. (2021) developed CoS-NBs (NBs: nanoparticles) as a scaffold for MgH2, achieving a high load capacity of 42.5 wt.% and reduced hydriding/dehydriding enthalpies Ren et al. (2022b) constructed a 1D N-doped hierarchically porous carbon nanofiber scaffold to facilitate the self-assembly of MgH2/Ni nanoparticles. Ren et al. (Ren et al. 2022a) demonstrated the potential of 2D transition TiO2 nanosheets as frameworks for hosting MgH2 nanoparticles, with significant contributions from the nanoconfinement effect and Mg–Ti materials. These promising materials hold great potential for stationary and on-board hydrogen storage applications.
High entropy alloys (HEAs), which include hydride-forming elements, have recently emerged as promising candidates for hydrogen storage. High entropy alloys typically contain five or more main elements within a 5–35 atomic percent range. Minor elements, less than five atomic percent, can be added to enhance specific properties (Miracle and Senkov 2017). Hydrogen storage high entropy alloys can be broadly classified into three types: BCC (body-centered cubic), lightweight, and intermetallic high entropy alloy hydrides, with body-centered cubic high entropy alloys considered the most promising for hydrogen storage. Shahi et al. (2023) conducted a comprehensive analysis of published research results, revealing that body-centered cubic high entropy alloys exhibit superior hydrogen storage capacity compared to traditional body-centered cubic alloys. However, there is a limited number of studies focusing on low-density hydrogen storage and high entropy alloys. Sahlberg et al. (2016) developed a TiVZrNbHf high-entropy alloy with a body-centered cubic structure composed of strong hydride-forming elements with a significant variance in atomic radii (δ = 6.82%). This alloy demonstrated the ability to absorb 2.7 wt.% H2 at 573 K, achieving a H/M ratio of 2.5, which is significantly higher than typical hydrides. This enhanced hydrogen storage capacity is attributed to the alloy's conversion from the body-centered cubic to the body-centered tetragonal (BCT) phase during hydrogenation, a behavior commonly observed in body-centered cubic alloys. This structural change potentially opens up new interstitial sites for hydrogen storage. In 2023, Serrano et al. (2023) developed a TiVNbCrMn high-entropy alloy system for hydrogen storage. They fabricated and tested three alloy compositions, Ti35V35Nb20Cr5Mn5, Ti27.5V27.5Nb20Cr12.5Mn12.5, and Ti32V32Nb18Cr9Mn9; which displayed high hydrogen capacities of 2.47, 3.38, and 2.09 wt.% respectively. These alloys displayed distinct activation kinetics and incubation times. Two of the alloys underwent hydrogen absorption/desorption cycles and, despite a decrease in absorption kinetics, maintained promising results by preserving their maximum hydrogen storage capacity.
Thus, developing new materials for storing hydrogen is a rapidly evolving field with immense promise for the future of renewable energy. Scientists and engineers from across the globe are engaged in this endeavor, using their collective expertise to innovate and design the next generation of hydrogen storage materials. The following section discusses some of these materials.
Machine learning in hydrogen storage
With the rapid advancements in technology, artificial intelligence (AI) is playing a pivotal role in revolutionizing the design and enhancing the efficiency of hydrogen storage models. Machine learning (ML), a subset of artificial intelligence, has emerged as a transformative tool for many industries in recent years. Machine learning models, using data from experiments or simulations, can identify materials with favorable properties, i.e., high hydrogen storage capacity and light weight with high strength. Machine learning's ability to learn from data and predict outcomes without explicit programming enhances process efficiency, reduces human error, and offers a closed-loop quality control system (Yuan et al. 2018; Osman et al. 2024a). Additionally, machine learning aids in understanding the relationships between material properties and hydrogen storage performance, guiding the design of improved and novel materials for storage. Despite challenges with data quality and availability (Osman et al. 2024b), machine learning's role in hydrogen storage exploration is anticipated to grow as the field matures.
Machine learning algorithms are mainly classified into three main categories, as presented in Fig. 6: (1) Supervised learning: This is the most common technique where the model is trained on a labeled dataset, meaning that the input data is paired with the correct output. Supervised learning primarily falls into two categories: regression and classification. In the context of hydrogen storage, regression can be used to predict continuous outcomes, such as the amount of hydrogen a material can store based on its properties. This would entail modeling the relationship between factors like surface area, porosity, or temperature and the hydrogen storage capacity. On the other hand, classification could be employed to categorize materials as either suitable or unsuitable for hydrogen storage based on predefined criteria or thresholds. This might involve assigning labels to materials based on whether they meet the required storage standards or safety criteria. Common models include the decision tree model, K-Nearest neighbor (KNN), artificial neural networks (ANN), markov model (HMM), naive bayesian model (NBM), support vector machines (SVM), etc. (Lan et al. 2020; Sunny et al. 2020; Qian et al. 2022). (2) Unlike supervised learning, unsupervised learning algorithms deal with unlabeled, open-domain data to train algorithms (Wang et al. 2022; Taherkhani et al. 2022). Unsupervised learning aims to identify hidden patterns or intrinsic structures within the data with minimal human intervention. The most commonly utilized method in this category is data clustering, which can efficiently detect grouped patterns. Common methods in unsupervised learning include the K-means clustering algorithm, density-based spatial clustering of applications with noise (DBSCAN), gaussian mixture models (GMM), hierarchical clustering, and others. The key advantage of unsupervised learning is that it does not require labeled data, making it a versatile tool in diverse data environments. Despite the differences in training datasets, the processes of testing and validation in unsupervised learning bear similarities to those in supervised machine learning. It is worth noting that both supervised and unsupervised learning leverage patterns from known data to make predictions or categorizations about new, unknown data. (3) Reinforcement learning (RL), which involves an agent that learns to make decisions by taking actions in an environment to maximize a reward signal. Reinforcement learning has effectively resolved complex optimization issues (François-Lavet et al. 2016; Dogu et al. 2022; Dreher et al. 2022; Sahar et al. 2023), as evidenced by numerous studies. Leveraging reinforcement learning could streamline the efficiency, safety, and scalability of hydrogen storage solutions, making them more viable for a range of applications.

Classification of machine learning algorithms according to their foundational operational principles. Each branch represents a distinct approach that shapes how models learn and generalize from data, contributing to the advancement of the field. Supervised learning forms the foundation of machine learning, where algorithms learn from labeled training data. By observing input–output pairs, such as in linear regression, decision trees, and neural networks, these algorithms absorb patterns from known examples and generalize to make predictions for unseen data points. Unsupervised learning, on the other hand, operates without labeled data. Its objective is to uncover hidden structures within the data. Clustering algorithms, including k-means and hierarchical clustering, group similar data points together. Reinforcement learning draws inspiration from behavioral psychology. Reinforcement learning agents learn through trial and error by interacting with an environment and receiving rewards or penalties. Algorithms like Q-learning and deep reinforcement learning have driven advancements in gaming, robotics, and finance. This taxonomy serves as a valuable compass, guiding researchers, practitioners, and enthusiasts through the vast machine-learning universe. Each algorithmic path offers unique insights and applications, contributing to the ever-evolving field of machine learning. It is important to note that while the mentioned algorithms are representative examples, there exists a wide range of other algorithms in each category that further enrich the machine-learning landscape
Each type of algorithm is used depending on the nature of the problem and the type of data available. For instance, supervised learning requires a large amount of labeled data, which can be challenging to obtain in many practical situations. Unsupervised learning, on the other hand, may not provide as accurate or direct predictions as supervised learning but can reveal useful insights from large volumes of unlabeled data.
In the context of using machine learning in hydrogen storage, Ali et al. (2022) proposed a machine learning model to design a hydrogen storage system, focusing on hydrogen storage in dibenzyl-toluene using various artificial neural networks approaches. Three artificial neural network methods were compared for prediction accuracy: levenberg marquardt (LM), scaled conjugate gradient (SCG), and bayesian regularization (BR). While levenberg marquardt showed an accuracy of 94.87%, both bayesian regularization and scaled conjugate gradient achieved an overall accuracy of 98.70%, demonstrating their superior performance in predicting hydrogen storage capacities. Similarly, Suwarno et al. (2022) utilized machine learning to analyze a database on Zr-Ti-based AB2 alloys for hydrogen storage, exploring the effect of different alloy elements on both the formation heat and storage capacity of hydrogen. They found that Ni was the most influential element in modifying the heat of formation for Zr-Ti-based AB2 alloys but simultaneously decreased hydrogen absorption. Mn was found to have a substantial effect on hydrogen storage capacity. Their findings provide insight into alloy element selection in experimental designs. Furthermore, Lu et al. (2022b) utilized machine learning to investigate the structure–property relationship of the V-Ti-Cr-Fe alloy. Their findings emphasized the significance of lattice constant, valency electron concentration, and Z/r3 in determining the hydrogen storage capacity of the alloy.
Machine learning has been employed in exploring non-metallic materials in hydrogen storage studies. Ahmed et al.(2019) employed machine learning algorithms to predict volumetric and gravimetric hydrogen capacities in metal–organic frameworks. The researchers performed a systematic assessment of real and hypothetical metal–organic frameworks by computationally screening around 500,000 compounds. The most promising metal–organic frameworks were then experimentally tested. Three metal–organic frameworks, namely SNU-70, UMCM-9, and PCN-610/NU-100, exhibited higher hydrogen capacities than the benchmark compound IRMOF-20. The study revealed a volumetric ceiling at approximately 40 g H2 L−1 and suggested surpassing this ceiling as a new capacity target for hydrogen adsorbents. Contrary to previous studies, the usable capacities in high-performing materials were found to be negatively correlated with density and volumetric surface area, emphasizing the importance of increasing gravimetric surface area and porosity. The use of machine learning techniques facilitated the identification of an optimal metal–organic framework for hydrogen storage. Furthermore, Ahmed and Siegel (2021) presented the development of machine-learning models to predict hydrogen uptake in a metal–organic framework. The machine learning models accurately predict hydrogen storage capacities using minimal input data derived from the metal–organic framework structure. The study analyzed a diverse set of 918,734 metal–organic frameworks and identified 8282 metal–organic frameworks with the potential to exceed the capacities of current materials. These promising metal–organic frameworks have low densities (< 0.31 g cm−3), high surface areas (> 5,300 m2 g−1), void fractions (> 0.90), and large pore volumes (> 3.3 cm3 g−1). The machine learning models emphasize the importance of pore volume and void fraction in predicting hydrogen uptake. The developed models are available on the web for rapid and accurate predictions of metal–organic frameworks' hydrogen capacities. The use of machine learning in this study demonstrates its potential to accelerate the discovery of high-capacity hydrogen adsorbents for efficient hydrogen storage systems.
Recently, Borja et al. (2024) explored the use of machine learning in predicting the hydrogen adsorption capabilities and moduli of metal–organic frameworks for hydrogen storage. The study evaluates 13 machine learning models and compares their predictions with simulated and experimental results. The results demonstrate that 12 out of the 13 models achieved high prediction accuracy, with a coefficient of determination (R2) greater than 0.95 for both gravimetric and volumetric hydrogen uptakes in metal–organic frameworks. Additionally, the study introduces a 4-20-1 artificial neural network model that predicts the bulk, shear, and Young's moduli for metal–organic frameworks. The use of machine learning strategies allows for efficient screening and selection of metal–organic frameworks for hydrogen storage, aiding in the development of cost-effective and time-efficient storage methods. Shekhar and Chowdhury (2024a) employed a deep learning model, specifically a feedforward neural network (FNN), and compared its performance with the extremely randomized tree (ERT) model. They utilized a dataset of 918,734 metal–organic frameworks and evaluated the hydrogen delivery capacity in terms of both gravimetric and volumetric quantities. Through their analysis, the feedforward neural network model demonstrated superior performance in predicting gravimetric capacity, while the extremely randomized tree model excelled in volumetric capacity prediction. The authors also conducted GCMC simulations to validate their model. The study highlights the potential of machine learning as a valuable tool for accelerating the discovery and prediction of optimal metal–organic frameworks for hydrogen storage applications.
To provide a more precise depiction of the geometry of metal–organic frameworks, employing feature-based representations utilizing tools from topological data analysis proves advantageous. These topological descriptors, superior to generic structural descriptors, offer a means to enhance the efficacy of machine learning methods with relative ease of implementation. In this context, a recently published article conducted by Shekhar and Chowdhury (2024b) presented a study on using machine learning and topological data analysis to predict the hydrogen storage capacity of metal–organic frameworks. The authors developed a deep learning framework that incorporates topological features along with conventional structural features to improve the prediction accuracy. The study utilized the CoRE MOF-2019 database of 4029 metal–organic frameworks and employed a temperature and pressure swing from 100 bar/77 K to 5 bar/160 K for prediction. The results show that the proposed model outperforms the baseline and demonstrates significant progress in predicting the hydrogen storage capacity of metal–organic frameworks. The topological features obtained through persistent homology analysis were found to capture additional information supplementary to the structural features. The incorporation of these topological features enhanced the predictive power of the model.
Machine learning was also used to explore the ligand effects on hydrogen storage in metal–organic frameworks. In this regard, Giappa et al. (2021) investigated the use of a combination of multi-scale calculations and machine learning to investigate hydrogen storage in metal–organic frameworks. The study focuses on the effect of ligand functionalization on the hydrogen storage profile of metal–organic frameworks. Through accurate ab-initio calculations, the binding energy of hydrogen with various functionalized benzenes was determined, revealing significant enhancements in interaction strength compared to benzene. The results indicate that functional groups, such as –OPO3H2 and –OCONH2 increase the interaction strength by 15–25%, while –OSO3H shows the most promise with an enhancement of up to 80%. GCMC calculations and machine learning analysis further support these findings. The study suggests that the functionalization strategy can be applied to enhance hydrogen storage performance in porous materials.
In the case of carbon materials as promising hydrogen storage materials, Maulana Kusdhany and Lyth (2021) developed machine learning models to forecast the excess adsorption at 77 K of porous carbon materials, relying on their texture and chemical characteristics. They identified pressure and BET (Brunauer–Emmett–Teller) surface area as the key factors for redundant hydrogen uptake and surprisingly found a positive relationship between excessive absorption and oxygen content, increasing hydrogen absorption by approximately 0.6 wt.%. These findings provide a theoretical basis for designing carbon materials for storing hydrogen. Besides, Thanh et al. (2023) utilized a machine learning model that integrated with four nature-inspired approaches applied in the random forest (RF) model for predicting hydrogen storage in porous carbon-based adsorbents. Input variables include various adsorbents, activating agents, structural parameters, and operational conditions. The four applied algorithms include the particle swarm optimizer (PSO), dragonfly algorithm (DA), grey wolf optimization (GWO), and genetic algorithm (GA). The successful integration of four algorithms with random forest resulted in a superior capability for hydrogen storage prediction. Both particle swarm optimizer and grey wolf optimization within the random forest model presented equivalent results in the training and testing phases, demonstrating an R2 of ~ 0.98 and 0.91, respectively. In hydrogen prediction during the test phase, the genetic algorithm held a slightly superior lead with an R2 of ~ 0.93. Concurrently, the random forest- dragonfly algorithm exhibited precision in prediction performance with an R2 of 0.98 and 0.90 in the training and testing phases, respectively. Generally, their model showed high accuracy, and a sensitivity analysis revealed the importance of temperature, specific surface area, micropore volume, and total pore volume.
On the other hand, Rahimi et al. (2021) utilized three machine-learning models to predict hydrogen adsorption in activated carbons, leveraging structural characteristics like micropore surface area, the volume of pores, and pore size distribution. The models demonstrated high feasibility, with root mean square errors (RMSEs) ranging from 0.06 to 0.19. The support vector machines model showed the highest accuracy in their study, and optimization of microstructural properties facilitated by a support vector machines-based genetic algorithm resulted in a 2.5 wt.% increase in hydrogen uptake. The study emphasizes the significance of microstructural properties in bio-derived activated carbons for hydrogen storage and suggests that machine-learning techniques can aid in the fabrication of porous carbons for such applications. Davoodi et al. (2023) evaluated the performance of four machine learning models in predicting hydrogen uptake by porous carbon media (PCM) based on influential variables. The machine learning models tested were generalized-regression neural network (GRNN), least-squares-support-vector machine (LSSVM), adaptive-neuro-fuzzy-inference system (ANFIS), and extreme-learning machine (ELM). The study utilized a database of 2072 records with eleven independent variables and porous carbon media hydrogen uptake as the dependent variable. The LSSVM model demonstrated the best prediction performance, achieving a root mean squared error of 0.2407 wt.%. Pressure was identified as the most influential independent variable. The results indicate that the developed LSSVM model is highly generalizable for predicting porous carbon media hydrogen uptake. Machine learning shows promise in improving the efficiency of hydrogen storage for fuel-cell-powered transportation vehicles.
The advancement of clean energy solutions is dependent on novel materials for hydrogen storage and conversion. Batalović et al. (2023) applied a machine learning model to identify promising Mg-containing materials for applications like near-ambient hydrogen storage and lithium conversion electrodes. After screening over 600 compounds, 32 intermetallics were found suitable for near-ambient hydrogen storage, with MgBe13 showing high gravimetric hydrogen density. This analysis supports the strategic development of efficient hydrogen storage materials and conversion-type negative electrodes in Li-ion batteries. Recently, Dong et al. (2023) focused on the application of machine learning in the development of Mg alloys for hydrogen storage. The researchers constructed a database of Mg-based hydrogen storage materials and used supervised machine learning regression models to predict the maximum hydrogen storage (Ab max) and maximum hydrogen release (De_max) of different Mg alloys.
The machine learning models achieved high prediction accuracy, with the gradient boosting regression (GBR) model for Ab_max and the multilayer perceptron (MLP) model for De_max attaining R2 values of 0.947 and 0.922, respectively. The researchers employed the Shapley additive explanations (SHAP) algorithm to interpret the machine learning models and identify critical factors influencing the hydrogen storage property of Mg alloys. Based on the machine learning models, the study predicted the Ab_max and De_max values for various Mg-based binary and ternary alloys. Notably, the alloys 96Mg-4Sm and 95Mg-1Ni-4Sm exhibited higher Ab_max and De_max values of 6.31 wt.% and 5.69 wt.%, and 6.64 wt.% and 5.63 wt.%, respectively. These alloys met the requirement for high Ab_max/De_max at operating temperatures below 573 K. The use of machine learning in this study demonstrates its potential in accelerating the discovery and design process of hydrogen storage materials, reducing research and development cycles, and minimizing computational and experimental costs.
Regarding hydrogen storage vessel design, machine learning is gaining prominence in optimizing geometry, materials, and design parameters for increased hydrogen storage and reduced weight. Machine learning automates complex design tasks, accelerating the development of effective composite storage solutions. For this purpose, machine learning models can be sourced from data from experiments and/or simulations to identify high-performance materials for the vessels, i.e., high strength and low weight with high hydrogen storage capacity. A careful selection of training and testing datasets can ensure model accuracy. Optimizing composite pressure vessels requires an objective function reflecting material performance. While gradient-based methods are challenged by complex behaviors and can yield unreliable outcomes, metaheuristics like genetic algorithms and simulated annealing (SA) often provide more efficient optimization (Nachtane et al. 2023). Despite not always ensuring global optimization, they typically deliver better results with reduced computational effort.
For instance, Islam et al. (2018) provided a crack identification method for the pressure vessels, utilizing genetic algorithm-based feature selection paired with a deep neural network (GA + DNN) during acoustic emission examinations, achieving a 94.67% classification accuracy. Kim et al. (2005a) employed a semi-geodesic path algorithm, progressive failure analysis, and a modified genetic algorithm to optimize a Type 3 vessel against internal pressure, aiming to minimize weight without compromising structural integrity. This approach was further extended using a semi-geodesic path method, finite element analysis, and a genetic algorithm to achieve weight reduction (Kim et al. 2005b). Kaveh et al. (2021) utilized machine learning techniques, including random forest and deep learning with artificial neural networks, to predict the ultimate buckling load of variable stiffness composite cylinders. Their study aimed to determine the relationship between fiber angles and the cylinders' buckling capacity under bending-induced loads, drawing from a dataset of 11,000 cases with seven attributes (i.e., fiber angles) across 11 distinct aspect ratios (L/R). While various algorithms were tested, the deep learning model outshone others like the random forest regressor, decision tree, and multiple linear regression instability, error minimization, and generalization. In a similar vein, studies employed a multi-step design approach utilizing radial basis functions to enhance the buckling performance of laminated composite cylinders in various loading conditions, including simple (Rouhi et al. 2014, 2017) and bi-directional bending moments (Rouhi et al. 2015), compressive axial force (Rouhi et al. 2016), and bending moment (Ghayoor et al. 2017). Rikards et al. (2004) developed a method to optimize the design of composite stiffened shells, considering buckling constraints, using surrogate models based on test results and simulations, which proved accurate in predicting post-buckling behavior. Luo et al. (2021) integrated the finite element method and artificial neural networks to forecast distortion behaviors in composites, yielding efficient results even for asymmetric laminates, albeit with unavoidable shape constraints and scale limitations.
In the realm of type IV composite vessels, understanding the impact of various winding parameters on their ultimate strength holds immense industrial significance. However, the complexity of numerical models encompassing material damage in these vessels poses a considerable challenge. The extensive finite element (FE) systems governing the behavior of thin composite layers create a gap between optimization solutions and computational feasibility. When integrating machine learning into composite material response predictions, the models often necessitate simplification, typically focusing on simple geometries like cylinders or cubes, which fails to address the intricacies of geometrically complex structures, such as the dome region in hydrogen pressure vessels. Many researchers have faced challenges when aiming to enhance the performance of type IV vessels, particularly due to the significant computational resources required to consider different winding parameters (Alcántar et al. 2017). The prevalent strategy to circumvent this issue has been to either focus solely on the cylindrical portion of the vessel or employ surrogate models to simplify the dome geometry. For instance, Leh et al. (2015) utilized a genetic algorithm approach to optimize the lay-up scheme for type IV hydrogen storage vessels. Their method approximated dome responses using a polynomial function and proved successful in deriving optimal winding parameters.
Similarly, Liu and Shi (2020) analytically identified optimal winding parameters, prioritizing the mechanical performance of the vessel's cylindrical section. Recently, Li et al. (2023a) introduced a novel framework integrating machine learning with finite element analysis for a 70 MPa type IV vessel, enabling the accurate prediction of mechanical responses without simplifying the dome's geometrical intricacies and allowing for optimized winding parameters. Using high-fidelity finite element models and an integrated artificial neural network, the framework addressed material distribution challenges in vessel design. This approach drastically reduced computational time while retaining accuracy, with a damage prediction error under 2%. Experimentally, this led to a significant increase in vessel burst pressure (from 145 to 157.74 MPa), validating the model's predictions based on established failure criteria.
In conclusion, the integration of artificial intelligence, particularly machine learning, has ushered in a new era of innovation in hydrogen storage design. Machine learning models, leveraging data from experiments or simulations, offer a powerful means to identify materials with optimal properties for hydrogen storage, such as high capacity and strength-to-weight ratio. Supervised, unsupervised, and reinforcement learning algorithms provide versatile tools for predicting storage capacities, understanding material-property relationships, and optimizing design parameters. Studies have demonstrated the efficacy of machine learning in various aspects of hydrogen storage, including metal–organic frameworks, carbon materials, and Mg-based alloys. Furthermore, machine learning facilitates the exploration of ligand effects, vessel design optimization, and prediction of mechanical responses in composite vessels. Despite challenges like data quality and computational complexity, machine learning continues to evolve as a key enabler in accelerating the discovery and development of efficient hydrogen storage solutions, paving the way for cleaner energy technologies.
Challenges
Hydrogen storage remains a critical challenge for the adoption of hydrogen as a sustainable energy carrier. The primary challenges in hydrogen storage include:
-
1.
Density: Hydrogen's low volumetric storage density poses a significant challenge for practical applications, necessitating large storage systems or high-pressure conditions to store significant amounts efficiently. To achieve optimal storage capacity at room temperature, the desired enthalpy change for an adsorbent material should be at least 15.1 kJ mol−1. However, for most carbonaceous materials, this value is only around 6 kJ mol−1. Consequently, they cannot effectively adsorb hydrogen until reaching a temperature of 115 K. To address this limitation; carbonaceous materials have been doped with transition metals to promote hydrogen spillover, thereby enhancing storage capacity. Unfortunately, the driving force for hydrogen spillover is often insufficient to break the bonds between the transition metal and hydrogen. As an alternative approach, modifications to the adsorbent's pore characteristics have been made using physical and chemical activation methods. While these methods have led to improvements in storage capacity, they have not met the targets set by the Department of Energy (Meduri and Nandanavanam 2023).
-
2.
Storage safety: Hydrogen is flammable, and its storage under high pressure or at low temperatures poses risks, including leaks and explosions.
-
3.
Elevated sorption temperatures: Due to the high hydrogenation and dehydrogenation enthalpies of magnesium, its sorption temperatures are high, typically ranging from 623 to 673 K, rendering it unsuitable for room-temperature applications. To address this limitation, alloying magnesium with transition metal oxides can effectively reduce sorption temperatures. This alloying process aids in lowering the reaction enthalpies and facilitates the formation of stable compounds. However, a major challenge lies in synthesizing magnesium-based alloys. In this regard, machine learning techniques can be employed to explore various alloy combinations, offering a promising avenue for overcoming this obstacle.
-
4.
Temperature sensitivity: Many hydrogen storage systems either require very high temperatures to release the stored hydrogen or very low temperatures for efficient storage. Storing significant amounts of hydrogen at room temperature is challenging due to its low adsorption heat on porous carbon. While more hydrogen can be stored at freezing temperatures, the process is energy-consuming, and hydrogen's extremely low boiling point complicates long-term storage.
-
5.
Economic viability: Many of the materials and technologies for hydrogen storage remain expensive, making it difficult to compete with traditional fossil fuels. The cost of hydrogen largely depends on storage expenses, which include capital costs (62% of liquefaction costs), power consumption (30%), and operations and management (8%) (Broom et al. 2016). Currently, hydrogen liquefaction costs around $1.11 per kilogram, but it aims to reduce to $0.53 under the United States hydrogen program (Niaz et al. 2015; Moradi and Groth 2019). Compressed gas vessels are pricier at higher pressures, e.g., $400 per kg at 140 bars and $2100 per kg at 540 bars. Cylindrical steel tanks of 765 L at approximately 415 bars can cost around $13,000 (Broom et al. 2016). Compressing a kilogram of hydrogen costs roughly $650, compared to about $500 for compressed natural gas. Cryogenic storage vessels have varied capital costs, ranging from $20/kg to $4500/kg, depending on the storage capacity (Broom et al. 2016). For short-term storage, compressed hydrogen tanks are more affordable than liquid hydrogen tanks. However, liquid hydrogen might be more cost-effective for long-term storage.
-
6.
Material challenges: Some storage materials can degrade over time, especially if they undergo repeated hydrogen absorption and desorption cycles. For instance, in some carbon-based porous materials, the effectiveness of hydrogen storage is closely linked to pore dimensions and overall pore volume. As such, developing well-distributed and uniform porous structures is essential for optimizing hydrogen uptake in ambient conditions.
-
7.
Infrastructure: There is a lack of widespread infrastructure for refueling hydrogen, especially in comparison to gasoline or electric charging stations.
-
8.
Onboard vehicle storage: For vehicles, the storage system must be compact, lightweight, and able to quickly release hydrogen to meet acceleration demands.
-
9.
Purity requirements: Fuel cells, a common use for stored hydrogen, often require very pure hydrogen. The presence of contaminants can degrade fuel cell performance and lifespan.
-
10.
Energy loss: Certain hydrogen storage methods, such as liquefaction, can consume a significant portion of the hydrogen's energy content.
Addressing these challenges requires a combination of materials science, engineering, and economic solutions to make hydrogen storage viable for widespread application.
Conclusion
In conclusion, this comprehensive review of hydrogen storage technologies has highlighted major findings that pave the way for more efficient and sustainable energy system. The study revealed that solid-state porous materials, such as metal–organic frameworks (MOFs), covalent organic frameworks (COFs), and porous carbon-based adsorbents, offer promising alternatives to current high-pressure compression techniques for on-board hydrogen storage. These materials exhibit high storage capacities, with metal–organic frameworks reaching up to 10 wt.% and covalent organic frameworks achieving around 6 wt.%. Additionally, high-entropy alloys and advanced composites demonstrate improved stability and hydrogen uptake. One significant breakthrough identified in this review is the transformative power of machine learning techniques in predicting and designing efficient storage materials. Machine learning has played a crucial role in overcoming data challenges and has facilitated the development of innovative materials with enhanced storage performance. This integration of machine learning with hydrogen storage research opens up new possibilities for accelerated material discovery and optimization. The findings of this review emphasize the potential of innovative materials and techniques in overcoming the limitations of current hydrogen storage methods. By leveraging solid-state materials and machine learning, it becomes possible to achieve safer, more efficient, and economically viable hydrogen storage solutions. These advancements have far-reaching implications for the global transition towards a low-carbon future and the development of a reliable and accessible hydrogen supply chain. Overall, this review provides valuable insights into the development of efficient and sustainable hydrogen storage systems. The identified advancements in sorbent materials, machine learning techniques, and the understanding of storage efficiency contribute significantly to overcoming the technical challenges of hydrogen storage. By embracing these findings, we can accelerate the realization of a cleaner and more sustainable energy landscape, where hydrogen plays a vital role in mitigating climate change and ensuring a greener future for generations to come.
References
Abe JO, Popoola API, Ajenifuja E, Popoola OM (2019) Hydrogen energy, economy and storage: review and recommendation. Int J Hydrog Energy 44:15072–15086. https://doi.org/10.1016/j.ijhydene.2019.04.068
Aboutalebi SH, Aminorroaya-Yamini S, Nevirkovets I et al (2012) Enhanced hydrogen storage in graphene oxide-MWCNTs composite at room temperature. Adv Energy Mater 2:1439–1446. https://doi.org/10.1002/aenm.201200154
Agarwal RK, Noh JS, Schwarz JA, Davini P (1987) Effect of surface acidity of activated carbon on hydrogen storage. Carbon 25:219–226
Ahmed A, Siegel DJ (2021) Predicting hydrogen storage in MOFs via machine learning. Patterns 2(7):100291
Ahmed A, Seth S, Purewal J et al (2019) Exceptional hydrogen storage achieved by screening nearly half a million metal–organic frameworks. Nat Commun 10:1568. https://doi.org/10.1038/s41467-019-09365-w
Allendorf MD, Hulvey Z, Gennett T et al (2018) An assessment of strategies for the development of solid-state adsorbents for vehicular hydrogen storage. Energy Environ Sci 11:2784–2812. https://doi.org/10.1039/C8EE01085D
Alazawi AM, Mohammed MY, Alheety MA et al (2023) Effect of Zn precursors on hydrogen storage in MWCNTs-ZnO nanocomposites. Results Chem 5:100948. https://doi.org/10.1016/j.rechem.2023.100948
Alcántar V, Aceves SM, Ledesma E et al (2017) Optimization of Type 4 composite pressure vessels using genetic algorithms and simulated annealing. Int J Hydrog Energy 42:15770–15781. https://doi.org/10.1016/j.ijhydene.2017.03.032
Ali A, Khan MA, Abbas N, Choi H (2022) Prediction of hydrogen storage in dibenzyltoluene empowered with machine learning. J Energy Storage 55:105844. https://doi.org/10.1016/j.est.2022.105844
Anwar S, Khan F, Zhang Y, Djire A (2021) Recent development in electrocatalysts for hydrogen production through water electrolysis. Int J Hydrog Energy 46:32284–32317. https://doi.org/10.1016/j.ijhydene.2021.06.191
Ao ZM, Tan TT, Li S, Jiang Q (2009) Molecular hydrogen storage in Al-doped bulk graphite with wider layer distances. Solid State Commun 149:1363–1367. https://doi.org/10.1016/j.ssc.2009.05.022
Ariharan A, Viswanathan B, Nandhakumar V (2017) Nitrogen doped graphene as potential material for hydrogen storage. Graphene 6:41–60
Ariharan A, Ramesh K, Vinayagamoorthi R et al (2021) Biomass derived phosphorous containing porous carbon material for hydrogen storage and high-performance supercapacitor applications. J Energy Storage 35:102185
Arshad SHM, Ngadi N, Aziz AA et al (2016) Preparation of activated carbon from empty fruit bunch for hydrogen storage. J Energy Storage 8:257–261
Assfour B, Seifert G (2010a) Hydrogen adsorption sites and energies in 2D and 3D covalent organic frameworks. Chem Phys Lett 489:86–91. https://doi.org/10.1016/j.cplett.2010.02.046
Assfour B, Seifert G (2010b) Adsorption of hydrogen in covalent organic frameworks: comparison of simulations and experiments. Microporous Mesoporous Mater 133:59–65
Bader N, Zacharia R, Abdelmottaleb O, Cossement D (2018) How the activation process modifies the hydrogen storage behavior of biomass-derived activated carbons. J Porous Mater 25:221–234
Balat M (2008) Potential importance of hydrogen as a future solution to environmental and transportation problems. Int J Hydrog Energy 33:4013–4029. https://doi.org/10.1016/j.ijhydene.2008.05.047
Bambalaza SE, Langmi HW, Mokaya R et al (2018) Compaction of a zirconium metal–organic framework (UiO-66) for high density hydrogen storage applications. J Mater Chem A 6:23569–23577. https://doi.org/10.1039/C8TA09227C
Barthélémy H, Weber M, Barbier F (2017) Hydrogen storage: recent improvements and industrial perspectives. Int J Hydrog Energy 42:7254–7262
Batalović K, Radaković J, Kuzmanović B et al (2023) Machine learning-based high-throughput screening of Mg-containing alloys for hydrogen storage and energy conversion applications. J Energy Storage 68:107720. https://doi.org/10.1016/j.est.2023.107720
Bicil Z, Dogan M (2021) Characterization of activated carbons prepared from almond shells and their hydrogen storage properties. Energy Fuels 35:10227–10240
Borja NK, Fabros CJE, Doma BT Jr (2024) Prediction of hydrogen adsorption and moduli of metal–organic frameworks (MOFs) using machine learning strategies. Energies 17:927
Bosu S, Rajamohan N (2023) Recent advancements in hydrogen storage: comparative review on methods, operating conditions and challenges. Int J Hydrog Energy. https://doi.org/10.1016/j.ijhydene.2023.01.344
Bouša D, Luxa J, Sedmidubskỳ D et al (2016) Nanosized graphane (C 1 H 1.14) n by hydrogenation of carbon nanofibers by Birch reduction method. RSC Adv 6:6475–6485
Broom DP, Webb CJ, Hurst KE et al (2016) Outlook and challenges for hydrogen storage in nanoporous materials. Appl Phys A 122:151. https://doi.org/10.1007/s00339-016-9651-4
Butova VV, Soldatov MA, Guda AA et al (2016) Metal-organic frameworks: structure, properties, methods of synthesis and characterization. Russ Chem Rev 85:280. https://doi.org/10.1070/RCR4554
Cao L, Yu IKM, Xiong X et al (2020) Biorenewable hydrogen production through biomass gasification: a review and future prospects. Environ Res 186:109547. https://doi.org/10.1016/j.envres.2020.109547
Chakraborty S, Kumar NM, Jayakumar A et al (2021) Selected aspects of sustainable mobility reveals implementable approaches and conceivable actions. Sustainability 13:12918
Chang C, Gao P, Bao D et al (2014) Ball-milling preparation of one-dimensional Co–carbon nanotube and Co–carbon nanofiber core/shell nanocomposites with high electrochemical hydrogen storage ability. J Power Sources 255:318–324. https://doi.org/10.1016/j.jpowsour.2014.01.034
Chen P, Zhu M (2008) Recent progress in hydrogen storage. Mater Today 11:36–43
Cheng H-M, Yang Q-H, Liu C (2001) Hydrogen storage in carbon nanotubes. Carbon 39:1447–1454
Chilev Ch, Darkrim Lamari F, Ljutzkanov L et al (2012) Hydrogen storage systems using modified sorbents for application in automobile manufacturing. Int J Hydrog Energy 37:10172–10181. https://doi.org/10.1016/j.ijhydene.2012.03.016
Choi YJ, Lee JW, Choi JH, Kang JK (2008) Ideal metal-decorated three dimensional covalent organic frameworks for reversible hydrogen storage. Appl Phys Lett 92:173102
Chung K-H (2010) High-pressure hydrogen storage on microporous zeolites with varying pore properties. Energy 35:2235–2241. https://doi.org/10.1016/j.energy.2010.02.010
Colson JW, Dichtel WR (2013) Rationally synthesized two-dimensional polymers. Nat Chem 5:453–465. https://doi.org/10.1038/nchem.1628
Côté AP, El-Kaderi HM, Furukawa H et al (2007) Reticular synthesis of microporous and mesoporous 2D covalent organic frameworks. J Am Chem Soc 129:12914–12915. https://doi.org/10.1021/ja0751781
Dash SK, Chakraborty S, Elangovan D (2023) A brief review of hydrogen production methods and their challenges. Energies 16:1141. https://doi.org/10.3390/en16031141
Davoodi S, Vo Thanh H, Wood DA et al (2023) Machine-learning models to predict hydrogen uptake of porous carbon materials from influential variables. Sep Purif Technol 316:123807. https://doi.org/10.1016/j.seppur.2023.123807
Deng W-Q, Xu X, Goddard WA (2004) New alkali doped pillared carbon materials designed to achieve practical reversible hydrogen storage for transportation. Phys Rev Lett 92:166103. https://doi.org/10.1103/PhysRevLett.92.166103
Dillon AC, Jones KM, Bekkedahl TA et al (1997) Storage of hydrogen in single-walled carbon nanotubes. Nature 386:377–379. https://doi.org/10.1038/386377a0
Dogu MN, McCarthy E, McCann R et al (2022) Digitisation of metal AM for part microstructure and property control. Int J Mater Form 15:30. https://doi.org/10.1007/s12289-022-01686-4
Dong S, Wang Y, Li J et al (2023) Exploration and design of Mg alloys for hydrogen storage with supervised machine learning. Int J Hydrog Energy 48:38412–38424
Dreher A, Bexten T, Sieker T et al (2022) AI agents envisioning the future: forecast-based operation of renewable energy storage systems using hydrogen with deep reinforcement learning. Energy Convers Manag 258:115401. https://doi.org/10.1016/j.enconman.2022.115401
Du X, Wu E (2006) Physisorption of hydrogen in A, X and ZSM-5 types of zeolites at moderately high pressures. Chin J Chem Phys 19:457. https://doi.org/10.1360/cjcp2006.19(5).457.6
Duan X-Q, Li G-X, Zhang W-H et al (2023) Ti3AlCN MAX for tailoring MgH2 hydrogen storage material: from performance to mechanism. Rare Met 42:1923–1934. https://doi.org/10.1007/s12598-022-02231-7
Ekpete OA, Orie KJ (2023) Fullerenes: synthesis and application. Fac Nat Appl Sci J Sci Innov 4:221–236
El-Kaderi HM, Hunt JR, Mendoza-Cortés JL et al (2007) Designed synthesis of 3D covalent organic frameworks. Science 316:268–272. https://doi.org/10.1126/science.1139915
Eng AYS, Sofer Z, Bouša D et al (2017) Near-stoichiometric bulk graphane from halogenated graphenes (X = Cl/Br/I) by the Birch reduction for high density energy storage. Adv Funct Mater 27:1605797. https://doi.org/10.1002/adfm.201605797
Ensafi AA, Jafari-Asl M, Nabiyan A et al (2016) Hydrogen storage in hybrid of layered double hydroxides/reduced graphene oxide using spillover mechanism. Energy 99:103–114. https://doi.org/10.1016/j.energy.2016.01.042
Fomkin A, Pribylov A, Men’shchikov I et al (2021) Adsorption-based hydrogen storage in activated carbons and model carbon structures. Reactions 2:209–226
François-Lavet V, Taralla D, Ernst D, Fonteneau R (2016) Deep reinforcement learning solutions for energy microgrids management. In European Workshop on Reinforcement Learning (EWRL 2016)
Furukawa H, Yaghi OM (2009) Storage of hydrogen, methane, and carbon dioxide in highly porous covalent organic frameworks for clean energy applications. In: ACS Publ. https://doi.org/10.1021/ja9015765. Accessed 8 Sep 2023
Ghayoor H, Rouhi M, Hoa SV, Hojjati M (2017) Use of curvilinear fibers for improved bending-induced buckling capacity of elliptical composite cylinders. Int J Solids Struct 109:112–122. https://doi.org/10.1016/j.ijsolstr.2017.01.012
Giappa RM, Tylianakis E, Di Gennaro M et al (2021) A combination of multi-scale calculations with machine learning for investigating hydrogen storage in metal organic frameworks. Int J Hydrog Energy 46:27612–27621. https://doi.org/10.1016/j.ijhydene.2021.06.021
Gogotsi Y, Portet C, Osswald S et al (2009) Importance of pore size in high-pressure hydrogen storage by porous carbons. Int J Hydrog Energy 34:6314–6319
González-Navarro MF, Giraldo L, Moreno-Piraján JC (2014) Preparation and characterization of activated carbon for hydrogen storage from waste African oil-palm by microwave-induced LiOH basic activation. J Anal Appl Pyrol 107:82–86
Grigorova E, Nihtianova D, Tsyntsarski B, Stoycheva I (2020) Investigation of hydrogen storage characteristics of MgH2 based materials with addition of Ni and activated carbon. Inorganics 8:12. https://doi.org/10.3390/inorganics8020012
Gu C, Gao G-H, Yu Y-X, Mao Z-Q (2001) Simulation study of hydrogen storage in single walled carbon nanotubes. Int J Hydrog Energy 26:691–696
Gu J, Zhang X, Fu L, Pang A (2019) Study on the hydrogen storage properties of the dual active metals Ni and Al doped graphene composites. Int J Hydrog Energy 44:6036–6044. https://doi.org/10.1016/j.ijhydene.2019.01.057
Guo J-H, Zhang H, Liu Z-P, Cheng X-L (2012) Multiscale study of hydrogen adsorption, diffusion, and desorption on Li-doped phthalocyanine covalent organic frameworks. J Phys Chem C 116:15908–15917. https://doi.org/10.1021/jp305949q
Guo J-H, Li S-J, Su Y, Chen G (2020) Theoretical study of hydrogen storage by spillover on porous carbon materials. Int J Hydrog Energy 45:25900–25911. https://doi.org/10.1016/j.ijhydene.2019.12.146
Gutiérrez-Martín F, García-De María JM, Baïri A, Laraqi N (2009) Management strategies for surplus electricity loads using electrolytic hydrogen. Int J Hydrog Energy 34:8468–8475. https://doi.org/10.1016/j.ijhydene.2009.08.018
Han Y-J, Park S-J (2017) Influence of nickel nanoparticles on hydrogen storage behaviors of MWCNTs. Appl Surf Sci 415:85–89. https://doi.org/10.1016/j.apsusc.2016.12.108
Han SS, Furukawa H, Yaghi OM, Goddard WAI (2008) Covalent organic frameworks as exceptional hydrogen storage materials. J Am Chem Soc 130:11580–11581. https://doi.org/10.1021/ja803247y
Han SS, Mendoza-Cortés JL, Iii WAG (2009) Recent advances on simulation and theory of hydrogen storage in metal–organic frameworks and covalent organic frameworks. Chem Soc Rev 38:1460–1476. https://doi.org/10.1039/B802430H
Hassan IA, Ramadan HS, Saleh MA, Hissel D (2021) Hydrogen storage technologies for stationary and mobile applications: Review, analysis and perspectives. Renew Sustain Energy Rev 149:111311. https://doi.org/10.1016/j.rser.2021.111311
Hassan NS, Jalil AA, Rajendran S et al (2024a) Recent review and evaluation of green hydrogen production via water electrolysis for a sustainable and clean energy society. Int J Hydrog Energy 52:420–441. https://doi.org/10.1016/j.ijhydene.2023.09.068
Hassan Q, Tabar VS, Sameen AZ et al (2024b) A review of green hydrogen production based on solar energy; techniques and methods. Energy Harvest Syst. https://doi.org/10.1515/ehs-2022-0134
Heo Y-J, Park S-J (2015) Synthesis of activated carbon derived from rice husks for improving hydrogen storage capacity. J Ind Eng Chem 31:330–334
Hermosilla-Lara G, Momen G, Marty PH et al (2007) Hydrogen storage by adsorption on activated carbon: investigation of the thermal effects during the charging process. Int J Hydrog Energy 32:1542–1553. https://doi.org/10.1016/j.ijhydene.2006.10.048
Heydariyan Z, Monsef R, Dawi EA, Salavati-Niasari M (2023) EuMnO3/EuMn2O5/MWCNT nanocomposites: insights into synthesis and application as potential materials for development of hydrogen storage capacity. Fuel 351:128885. https://doi.org/10.1016/j.fuel.2023.128885
Hosseini A, Ghoreyshi AA, Pirzadeh K, Mohammadi M (2017) Enhancement of hydrogen storage on multi-walled carbon nanotube through KOH activation and nickel nanoparticle deposition. Sci Iran 24:1230–1240. https://doi.org/10.24200/sci.2017.4107
Hou P-X, Xu S-T, Ying Z et al (2003) Hydrogen adsorption/desorption behavior of multi-walled carbon nanotubes with different diameters. Carbon 41:2471–2476
Hu W, Li Y, Zheng M et al (2021) Degradation of biomass components to prepare porous carbon for exceptional hydrogen storage capacity. Int J Hydrog Energy 46:5418–5426
Huang C-C, Chen H-M, Chen C-H (2010) Hydrogen adsorption on modified activated carbon. Int J Hydrog Energy 35:2777–2780
Hunt JR, Doonan CJ, LeVangie JD et al (2008) Reticular synthesis of covalent organic borosilicate frameworks. J Am Chem Soc 130:11872–11873. https://doi.org/10.1021/ja805064f
Islam M, Sohaib M, Kim J, Kim J-M (2018) Crack classification of a pressure vessel using feature selection and deep learning methods. Sensors 18:4379. https://doi.org/10.3390/s18124379
Jackson KT, Reich TE, El-Kaderi HM (2012) Targeted synthesis of a porous borazine-linked covalent organic framework. Chem Commun 48:8823–8825. https://doi.org/10.1039/C2CC33583B
Jaiswal A, Chakraborty B, Sahu S (2022) High capacity reversible hydrogen storage in Si substituted and Li decorated C20 fullerene: acumen from density functional theory simulations. Int J Energy Res 46:19521–19537. https://doi.org/10.1002/er.8524
Jordá-Beneyto M, Suárez-García F, Lozano-Castelló D et al (2007) Hydrogen storage on chemically activated carbons and carbon nanomaterials at high pressures. Carbon 45:293–303. https://doi.org/10.1016/j.carbon.2006.09.022
Jung H, Park KT, Gueye MN et al (2016) Bio-inspired graphene foam decorated with Pt nanoparticles for hydrogen storage at room temperature. Int J Hydrog Energy 41:5019–5027. https://doi.org/10.1016/j.ijhydene.2015.12.016
Kalidindi SB, Fischer RA (2013) Covalent organic frameworks and their metal nanoparticle composites: prospects for hydrogen storage. Phys Status Solidi B 250:1119–1127. https://doi.org/10.1002/pssb.201248477
Kang P-C, Ou Y-S, Li G-L et al (2021) Room-temperature hydrogen adsorption via spillover in Pt nanoparticle-decorated UiO-66 nanoparticles: implications for hydrogen storage. ACS Appl Nano Mater 4:11269–11280. https://doi.org/10.1021/acsanm.1c02862
Kaveh A, DadrasEslamlou A, Javadi SM, Geran Malek N (2021) Machine learning regression approaches for predicting the ultimate buckling load of variable-stiffness composite cylinders. Acta Mech 232:921–931. https://doi.org/10.1007/s00707-020-02878-2
Kaye SS, Dailly A, Yaghi OM, Long JR (2007) Impact of preparation and handling on the hydrogen storage properties of Zn4O(1,4-benzenedicarboxylate)3 (MOF-5). J Am Chem Soc 129:14176–14177. https://doi.org/10.1021/ja076877g
Kazansky VB, Borovkov VY, Serich A, Karge HG (1998) Low temperature hydrogen adsorption on sodium forms of faujasites: barometric measurements and drift spectra1. Microporous Mesoporous Mater 22:251–259. https://doi.org/10.1016/S1387-1811(98)00084-5
Ke Z, Cheng Y, Yang S et al (2017) Modification of COF-108 via impregnation/functionalization and Li-doping for hydrogen storage at ambient temperature. Int J Hydrog Energy 42:11461–11468. https://doi.org/10.1016/j.ijhydene.2017.01.143
Kim C-U, Hong C-S, Kim C-G, Kim J-Y (2005a) Optimal design of filament wound type 3 tanks under internal pressure using a modified genetic algorithm. Compos Struct 71:16–25. https://doi.org/10.1016/j.compstruct.2004.09.006
Kim C-U, Kang J-H, Hong C-S, Kim C-G (2005b) Optimal design of filament wound structures under internal pressure based on the semi-geodesic path algorithm. Compos Struct 67:443–452. https://doi.org/10.1016/j.compstruct.2004.02.003
Kim TH, Bae J, Lee TH et al (2016) Room-temperature hydrogen storage via two-dimensional potential well in mesoporous graphene oxide. Nano Energy 27:402–411. https://doi.org/10.1016/j.nanoen.2016.07.027
Klontzas E, Tylianakis E, Froudakis GE (2008) Hydrogen storage in 3D covalent organic frameworks. a multiscale theoretical investigation. J Phys Chem C 112:9095–9098. https://doi.org/10.1021/jp711326g
Klontzas E, Tylianakis E, Froudakis GE (2010) Designing 3D COFs with enhanced hydrogen storage capacity. Nano Lett 10:452–454. https://doi.org/10.1021/nl903068a
Kostoglou N, Koczwara C, Stock S et al (2022) Nanoporous polymer-derived activated carbon for hydrogen adsorption and electrochemical energy storage. Chem Eng J 427:131730
Kothari R, Buddhi D, Sawhney RL (2008) Comparison of environmental and economic aspects of various hydrogen production methods. Renew Sustain Energy Rev 12:553–563. https://doi.org/10.1016/j.rser.2006.07.012
Kuhn P, Antonietti M, Thomas A (2008) Porous, covalent triazine-based frameworks prepared by ionothermal synthesis. Angew Chem Int Ed 47:3450–3453. https://doi.org/10.1002/anie.200705710
Lan J, Cao D, Wang W (2009) Li12Si60H60 fullerene composite: a promising hydrogen storage medium. ACS Nano 3:3294–3300. https://doi.org/10.1021/nn900842j
Lan L, You L, Zhang Z et al (2020) Generative adversarial networks and its applications in biomedical informatics. Front Public Health 8:164
Langmi HW, Walton A, Al-Mamouri MM et al (2003) Hydrogen adsorption in zeolites A, X, Y and RHO. J Alloys Compd 356–357:710–715. https://doi.org/10.1016/S0925-8388(03)00368-2
Langmi HW, Book D, Walton A et al (2005) Hydrogen storage in ion-exchanged zeolites. J Alloys Compd 404–406:637–642. https://doi.org/10.1016/j.jallcom.2004.12.193
Lee S-Y, Park S-J (2011) Effect of platinum doping of activated carbon on hydrogen storage behaviors of metal-organic frameworks-5. Int J Hydrog Energy 36:8381–8387. https://doi.org/10.1016/j.ijhydene.2011.03.038
Lee SM, An KH, Lee YH et al (2001) A hydrogen storage mechanism in single-walled carbon nanotubes. J Am Chem Soc 123:5059–5063. https://doi.org/10.1021/ja003751+
Leh D, Magneville B, Saffré P et al (2015) Optimisation of 700 bar type IV hydrogen pressure vessel considering composite damage and dome multi-sequencing. Int J Hydrog Energy 40:13215–13230. https://doi.org/10.1016/j.ijhydene.2015.06.156
Leng H, Miao N, Li Q (2020) Improved hydrogen storage properties of MgH2 by the addition of KOH and graphene. Int J Hydrog Energy 45:28183–28189. https://doi.org/10.1016/j.ijhydene.2020.03.070
Li Y, Yang RT (2006a) Hydrogen storage in metal−organic frameworks by bridged hydrogen spillover. J Am Chem Soc 128:8136–8137. https://doi.org/10.1021/ja061681m
Li Y, Yang RT (2006b) Significantly enhanced hydrogen storage in metal−organic frameworks via spillover. J Am Chem Soc 128:726–727. https://doi.org/10.1021/ja056831s
Li Y, Yang RT (2007) Hydrogen storage on platinum nanoparticles doped on superactivated carbon. J Phys Chem C 111:11086–11094. https://doi.org/10.1021/jp072867q
Li F, Zhao J, Johansson B, Sun L (2010) Improving hydrogen storage properties of covalent organic frameworks by substitutional doping. Int J Hydrog Energy 35:266–271. https://doi.org/10.1016/j.ijhydene.2009.10.061
Li G, Kobayashi H, Taylor JM et al (2014) Hydrogen storage in Pd nanocrystals covered with a metal–organic framework. Nat Mater 13:802–806. https://doi.org/10.1038/nmat4030
Li S, Zhai Y, Wei X et al (2021) Catalytic performance of MIL-88B(V) and MIL-101(V) MOFs for the selective catalytic reduction of NO with NH3. ChemCatChem 13:940–951. https://doi.org/10.1002/cctc.202001622
Li F, Chen X, Xu P et al (2023a) Optimal design of thin-layered composites for type IV vessels: finite element analysis enhanced by ANN. Thin-Walled Struct 187:110752. https://doi.org/10.1016/j.tws.2023.110752
Li J, Jing Z, Bai H et al (2023b) Optimizing hydrogen production by alkaline water decomposition with transition metal-based electrocatalysts. Environ Chem Lett 21:2583–2617. https://doi.org/10.1007/s10311-023-01616-z
Li Y, Guo Q, Ding Z et al (2024) MOFs-based materials for solid-state hydrogen storage: strategies and perspectives. Chem Eng J 485:149665. https://doi.org/10.1016/j.cej.2024.149665
Lim D-W, Yoon JW, Ryu KY, Suh MP (2012) Magnesium nanocrystals embedded in a metal–organic framework: hybrid hydrogen storage with synergistic effect on physi- and chemisorption. Angew Chem Int Ed 51:9814–9817. https://doi.org/10.1002/anie.201206055
Liu X (2020) Metal-organic framework UiO-66 membranes. Front Chem Sci Eng 14:216–232. https://doi.org/10.1007/s11705-019-1857-5
Liu C, Shi Y (2020) Design optimization for filament wound cylindrical composite internal pressure vessels considering process-induced residual stresses. Compos Struct 235:111755. https://doi.org/10.1016/j.compstruct.2019.111755
Liu C, Fan YY, Liu M et al (1999) Hydrogen storage in single-walled carbon nanotubes at room temperature. Science 286:1127–1129
Liu C, Chen Y, Wu C-Z et al (2010) Hydrogen storage in carbon nanotubes revisited. Carbon 48:452–455. https://doi.org/10.1016/j.carbon.2009.09.060
Liu Y, Du H, Zhang X et al (2015) Superior catalytic activity derived from a two-dimensional Ti3C2 precursor towards the hydrogen storage reaction of magnesium hydride. Chem Commun 52:705–708. https://doi.org/10.1039/C5CC08801A
Liu Y, Gao H, Zhu Y et al (2019) Excellent catalytic activity of a two-dimensional Nb4C3Tx (MXene) on hydrogen storage of MgH2. Appl Surf Sci 493:431–440. https://doi.org/10.1016/j.apsusc.2019.07.037
Liu L, Cui W, Lu C et al (2020) Analyzing the adsorptive behavior of Amoxicillin on four Zr-MOFs nanoparticles: functional groups dependence of adsorption performance and mechanisms. J Environ Manage 268:110630. https://doi.org/10.1016/j.jenvman.2020.110630
Liu H, Lu C, Wang X et al (2021) Combinations of V2C and Ti3C2 MXenes for boosting the hydrogen storage performances of MgH2. ACS Appl Mater Interfaces 13:13235–13247. https://doi.org/10.1021/acsami.0c23150
Liu P, Jia Y, Ning Y et al (2024) Enhanced room-temperature hydrogen storage by CuNi nanoparticles decorated in metal–organic framework UiO-66(Zr). Int J Hydrog Energy 56:315–322. https://doi.org/10.1016/j.ijhydene.2023.12.178
Lotfi R, Saboohi Y (2014) A comparative study on hydrogen interaction with defective graphene structures doped by transition metals. Phys E Low-Dimens Syst Nanostruct 60:104–111. https://doi.org/10.1016/j.physe.2014.02.004
Lu Z-Y, Yu H-J, Lu X et al (2021) Two-dimensional vanadium nanosheets as a remarkably effective catalyst for hydrogen storage in MgH2. Rare Met 40:3195–3204. https://doi.org/10.1007/s12598-021-01764-7
Lu C, Liu H, Xu L et al (2022a) Two-dimensional vanadium carbide for simultaneously tailoring the hydrogen sorption thermodynamics and kinetics of magnesium hydride. J Magnes Alloys 10:1051–1065. https://doi.org/10.1016/j.jma.2021.03.030
Lu Z, Wang J, Wu Y et al (2022b) Predicting hydrogen storage capacity of V-Ti–Cr–Fe alloy via ensemble machine learning. Int J Hydrog Energy 47:34583–34593. https://doi.org/10.1016/j.ijhydene.2022.08.050
Luo L, Zhang B, Zhang G et al (2021) Rapid prediction and inverse design of distortion behaviors of composite materials using artificial neural networks. Polym Adv Technol 32:1049–1060. https://doi.org/10.1002/pat.5152
Ma Y, Xia Y, Zhao M, Ying M (2002) Hydrogen storage capacity in single-walled carbon nanotubes. Phys Rev B 65:155430. https://doi.org/10.1103/PhysRevB.65.155430
Ma Z, Zou J, Hu C et al (2019) Effects of trimesic acid-Ni based metal organic framework on the hydrogen sorption performances of MgH2. Int J Hydrog Energy 44:29235–29248. https://doi.org/10.1016/j.ijhydene.2019.01.288
Ma Z, Zhang Q, Panda S et al (2020) In situ catalyzed and nanoconfined magnesium hydride nanocrystals in a Ni-MOF scaffold for hydrogen storage. Sustain Energy Fuels 4:4694–4703. https://doi.org/10.1039/D0SE00818D
Ma Z, Panda S, Zhang Q et al (2021) Improving hydrogen sorption performances of MgH2 through nanoconfinement in a mesoporous CoS nano-boxes scaffold. Chem Eng J 406:126790. https://doi.org/10.1016/j.cej.2020.126790
Macili A, Vlamidis Y, Pfusterschmied G et al (2023) Study of hydrogen absorption in a novel three-dimensional graphene structure: Towards hydrogen storage applications. Appl Surf Sci 615:156375. https://doi.org/10.1016/j.apsusc.2023.156375
Mahamiya V, Shukla A, Chakraborty B (2022) Scandium decorated C24 fullerene as high capacity reversible hydrogen storage material: insights from density functional theory simulations. Appl Surf Sci 573:151389
Maulana Kusdhany MI, Lyth SM (2021) New insights into hydrogen uptake on porous carbon materials via explainable machine learning. Carbon 179:190–201. https://doi.org/10.1016/j.carbon.2021.04.036
Mazloomi K, Sulaiman NB, Moayedi H (2012) Electrical efficiency of electrolytic hydrogen production. Int J Electrochem Sci 7:3314–3326. https://doi.org/10.1016/S1452-3981(23)13957-5
Meduri S, Nandanavanam J (2023) Materials for hydrogen storage at room temperature—an overview. Mater Today Proc 72:1–8
Mehrabi M, Parvin P, Reyhani A, Mortazavi SZ (2017) Hydrogen storage in multi-walled carbon nanotubes decorated with palladium nanoparticles using laser ablation/chemical reduction methods. Mater Res Express 4:095030. https://doi.org/10.1088/2053-1591/aa87f6
Mehrabi M, Reyhani A, Parvin P, Mortazavi SZ (2019) Surface structural alteration of multi-walled carbon nanotubes decorated by nickel nanoparticles based on laser ablation/chemical reduction methods to enhance hydrogen storage properties. Int J Hydrog Energy 44:3812–3823. https://doi.org/10.1016/j.ijhydene.2018.12.122
Miracle DB, Senkov ON (2017) A critical review of high entropy alloys and related concepts. Acta Mater 122:448–511. https://doi.org/10.1016/j.actamat.2016.08.081
Modak P, Chakraborty B, Banerjee S (2012) Study on the electronic structure and hydrogen adsorption by transition metal decorated single wall carbon nanotubes. J Phys Condens Matter 24:185505. https://doi.org/10.1088/0953-8984/24/18/185505
Mohan M, Sharma VK, Kumar EA, Gayathri V (2019) Hydrogen storage in carbon materials—a review. Energy Storage 1:e35. https://doi.org/10.1002/est2.35
Moradi R, Groth KM (2019) Hydrogen storage and delivery: review of the state of the art technologies and risk and reliability analysis. Int J Hydrog Energy 44:12254–12269. https://doi.org/10.1016/j.ijhydene.2019.03.041
Morse JR, Zugell DA, Matis BR et al (2020) Macroscale evaluation and testing of chemically hydrogenated graphene for hydrogen storage applications. Int J Hydrog Energy 45:2135–2144
Morse JR, Zugell DA, Patterson E et al (2021) Hydrogenated graphene: important material properties regarding its application for hydrogen storage. J Power Sources 494:229734. https://doi.org/10.1016/j.jpowsour.2021.229734
Musyoka NM, Mutuma BK, Manyala N (2020a) Onion-derived activated carbons with enhanced surface area for improved hydrogen storage and electrochemical energy application. RSC Adv 10:26928–26936
Musyoka NM, Wdowin M, Rambau KM et al (2020b) Synthesis of activated carbon from high-carbon coal fly ash and its hydrogen storage application. Renew Energy 155:1264–1271
Nachtane M, Tarfaoui M, Abichou MA et al (2023) An overview of the recent advances in composite materials and artificial intelligence for hydrogen storage vessels design. J Compos Sci 7:119. https://doi.org/10.3390/jcs7030119
Nagar R, Srivastava S, Hudson SL et al (2023) Recent developments in state-of-the-art hydrogen energy technologies: review of hydrogen storage materials. Sol Compass 5:100033. https://doi.org/10.1016/j.solcom.2023.100033
Nair AAS, Sundara R, Anitha N (2015) Hydrogen storage performance of palladium nanoparticles decorated graphitic carbon nitride. Int J Hydrog Energy 40:3259–3267. https://doi.org/10.1016/j.ijhydene.2014.12.065
Navaid HB, Emadi H, Watson M (2023) A comprehensive literature review on the challenges associated with underground hydrogen storage. Int J Hydrog Energy 48:10603–10635. https://doi.org/10.1016/j.ijhydene.2022.11.225
Nguyen TA, Gupta RK (eds) (2022) Covalent organic frameworks: chemistry, properties, and energy applications for a sustainable future. CRC Press, Boca Raton
Nguyen VG, Nguyen-Thi TX, Phong Nguyen PQ et al (2024) Recent advances in hydrogen production from biomass waste with a focus on pyrolysis and gasification. Int J Hydrog Energy 54:127–160. https://doi.org/10.1016/j.ijhydene.2023.05.049
Niaz S, Manzoor T, Pandith AH (2015) Hydrogen storage: materials, methods and perspectives. Renew Sustain Energy Rev 50:457–469. https://doi.org/10.1016/j.rser.2015.05.011
Nijkamp MG, Raaymakers JEMJ, van Dillen AJ, de Jong KP (2001) Hydrogen storage using physisorption—materials demands. Appl Phys A 72:619–623. https://doi.org/10.1007/s003390100847
Noh JS, Agarwal RK, Schwarz JA (1987) Hydrogen storage systems using activated carbon. Int J Hydrog Energy 12:693–700. https://doi.org/10.1016/0360-3199(87)90132-7
Oni AO, Anaya K, Giwa T et al (2022) Comparative assessment of blue hydrogen from steam methane reforming, autothermal reforming, and natural gas decomposition technologies for natural gas-producing regions. Energy Convers Manag 254:115245. https://doi.org/10.1016/j.enconman.2022.115245
Oriňáková R, Oriňák A (2011) Recent applications of carbon nanotubes in hydrogen production and storage. Fuel 90:3123–3140. https://doi.org/10.1016/j.fuel.2011.06.051
Osman AI, Mehta N, Elgarahy AM et al (2022) Hydrogen production, storage, utilisation and environmental impacts: a review. Environ Chem Lett 20:153–188. https://doi.org/10.1007/s10311-021-01322-8
Osman AI, Elgarahy AM, Eltaweil AS et al (2023a) Biofuel production, hydrogen production and water remediation by photocatalysis, biocatalysis and electrocatalysis. Environ Chem Lett 21:1315–1379. https://doi.org/10.1007/s10311-023-01581-7
Osman AI, Lai ZY, Farghali M et al (2023b) Optimizing biomass pathways to bioenergy and biochar application in electricity generation, biodiesel production, and biohydrogen production. Environ Chem Lett 21:2639–2705. https://doi.org/10.1007/s10311-023-01613-2
Osman AI, Nasr M, Farghali M et al (2024a) Machine learning for membrane design in energy production, gas separation, and water treatment: a review. Environ Chem Lett 22:505–560. https://doi.org/10.1007/s10311-023-01695-y
Osman AI, Nasr M, Farghali M et al (2024b) Optimizing biodiesel production from waste with computational chemistry, machine learning and policy insights: a review. Environ Chem Lett. https://doi.org/10.1007/s10311-024-01700-y
Osman AI, Redpath D, Lichtfouse E, Rooney DW (2024c) Synergy between vertical farming and the hydrogen economy. Environ Chem Lett 22:1–6. https://doi.org/10.1007/s10311-023-01648-5
Pal DB, Singh A, Bhatnagar A (2022) A review on biomass based hydrogen production technologies. Int J Hydrog Energy 47:1461–1480. https://doi.org/10.1016/j.ijhydene.2021.10.124
Panella B, Hirscher M, Roth S (2005) Hydrogen adsorption in different carbon nanostructures. Carbon 43:2209–2214. https://doi.org/10.1016/j.carbon.2005.03.037
Parambhath VB, Nagar R, Sethupathi K, Ramaprabhu S (2011) Investigation of spillover mechanism in palladium decorated hydrogen exfoliated functionalized graphene. J Phys Chem C 115:15679–15685. https://doi.org/10.1021/jp202797q
Pareek A, Dom R, Gupta J et al (2020) Insights into renewable hydrogen energy: recent advances and prospects. Mater Sci Energy Technol 3:319–327. https://doi.org/10.1016/j.mset.2019.12.002
Paul D, Mane P, Sarkar U, Chakraborty B (2023) Yttrium decorated fullerene C30 as potential hydrogen storage material: perspectives from DFT simulations. Theor Chem Acc 142:94
Pei P, Whitwick MB, Sun WL et al (2017) Enhanced hydrogen adsorption on graphene by manganese and manganese vanadium alloy decoration. Nanoscale 9:4143–4153. https://doi.org/10.1039/C6NR09545C
Peng Z, Xu Y, Luo W et al (2020) Conversion of biomass wastes into activated carbons by chemical activation for hydrogen storage. ChemistrySelect 5:11221–11228
Pinjari S, Bera T, Kjeang E (2023) Room temperature hydrogen storage enhancement in copper-doped zeolitic imidazolate frameworks with trioctylamine. Sustain Energy Fuels 7:3142–3151. https://doi.org/10.1039/D3SE00277B
Prins R (2012) Hydrogen spillover. Facts and fiction. Chem Rev 112:2714–2738. https://doi.org/10.1021/cr200346z
Pumera M (2011) Graphene-based nanomaterials for energy storage. Energy Environ Sci 4:668–674. https://doi.org/10.1039/C0EE00295J
Pupysheva OV, Farajian AA, Yakobson BI (2008) Fullerene nanocage capacity for hydrogen storage. Nano Lett 8:767–774. https://doi.org/10.1021/nl071436g
Qian C, Sun K, Bao W (2022) Recent advance on machine learning of MXenes for energy storage and conversion. Int J Energy Res 46:21511–21522. https://doi.org/10.1002/er.7833
Rahimi M, Abbaspour-Fard MH, Rohani A (2021) Machine learning approaches to rediscovery and optimization of hydrogen storage on porous bio-derived carbon. J Clean Prod 329:129714. https://doi.org/10.1016/j.jclepro.2021.129714
Rajaura RS, Srivastava S, Sharma V et al (2016) Role of interlayer spacing and functional group on the hydrogen storage properties of graphene oxide and reduced graphene oxide. Int J Hydrog Energy 41:9454–9461. https://doi.org/10.1016/j.ijhydene.2016.04.115
Rambau KM, Musyoka NM, Manyala N et al (2018) Mechanochemical approach in the synthesis of activated carbons from waste tyres and its hydrogen storage applications. Mater Today Proc 5:10505–10513
Ramesh T, Rajalakshmi N, Dhathathreyan KS (2017) Synthesis and characterization of activated carbon from jute fibers for hydrogen storage. Renew Energy Environ Sustain 2:4. https://doi.org/10.1051/rees/2017001
Rather S, Nahm KS (2014) Hydrogen uptake of high-energy ball milled nickel-multiwalled carbon nanotube composites. Mater Res Bull 49:525–530. https://doi.org/10.1016/j.materresbull.2013.09.022
Rather S, Zacharia R, Hwang SW et al (2007) Hydrogen uptake of palladium-embedded MWCNTs produced by impregnation and condensed phase reduction method. Chem Phys Lett 441:261–267. https://doi.org/10.1016/j.cplett.2007.05.006
Rather S, Zacharia R, Naik M et al (2008) Surface adsorption and micropore filling of the hydrogen in activated MWCNTs. Int J Hydrog Energy 33:6710–6718. https://doi.org/10.1016/j.ijhydene.2008.08.040
Ratnakar RR, Gupta N, Zhang K et al (2021) Hydrogen supply chain and challenges in large-scale LH2 storage and transportation. Int J Hydrog Energy 46:24149–24168. https://doi.org/10.1016/j.ijhydene.2021.05.025
Raymundo-Piñero E, Azaïs P, Cacciaguerra T et al (2005) KOH and NaOH activation mechanisms of multiwalled carbon nanotubes with different structural organisation. Carbon 43:786–795. https://doi.org/10.1016/j.carbon.2004.11.005
Reiter G, Lindorfer J (2015) Global warming potential of hydrogen and methane production from renewable electricity via power-to-gas technology. Int J Life Cycle Assess 20:477–489. https://doi.org/10.1007/s11367-015-0848-0
Ren L, Zhu W, Li Y et al (2022a) Oxygen vacancy-rich 2D TiO2 nanosheets: a bridge toward high stability and rapid hydrogen storage kinetics of nano-confined MgH2. Nano-Micro Lett 14:144. https://doi.org/10.1007/s40820-022-00891-9
Ren L, Zhu W, Zhang Q et al (2022b) MgH2 confinement in MOF-derived N-doped porous carbon nanofibers for enhanced hydrogen storage. Chem Eng J 434:134701. https://doi.org/10.1016/j.cej.2022.134701
Rikards R, Abramovich H, Auzins J et al (2004) Surrogate models for optimum design of stiffened composite shells. Compos Struct 63:243–251. https://doi.org/10.1016/S0263-8223(03)00171-5
Rimza T, Saha S, Dhand C et al (2022) Carbon-based sorbents for hydrogen storage: challenges and sustainability at operating conditions for renewable energy. Chemsuschem 15:e202200281. https://doi.org/10.1002/cssc.202200281
Romanos J, Beckner M, Prosniewski M et al (2019) Boron-neutron capture on activated carbon for hydrogen storage. Sci Rep 9:2971. https://doi.org/10.1038/s41598-019-39417-6
Rosi NL, Eckert J, Eddaoudi M et al (2003) Hydrogen storage in microporous metal–organic frameworks. Science 300:1127–1129
Rouhi M, Ghayoor H, Hoa SV, Hojjati M (2014) Effect of structural parameters on design of variable-stiffness composite cylinders made by fiber steering. Compos Struct 118:472–481. https://doi.org/10.1016/j.compstruct.2014.08.021
Rouhi M, Ghayoor H, Hoa SV, Hojjati M (2015) Multi-objective design optimization of variable stiffness composite cylinders. Compos Part B Eng 69:249–255. https://doi.org/10.1016/j.compositesb.2014.10.011
Rouhi M, Ghayoor H, Hoa SV et al (2016) Stiffness tailoring of elliptical composite cylinders for axial buckling performance. Compos Struct 150:115–123. https://doi.org/10.1016/j.compstruct.2016.05.007
Rouhi M, Ghayoor H, Hoa SV, Hojjati M (2017) Computational efficiency and accuracy of multi-step design optimization method for variable stiffness composite structures. Thin-Walled Struct 113:136–143. https://doi.org/10.1016/j.tws.2017.01.019
Rowlandson JL, Edler KJ, Tian M, Ting VP (2020) Toward process-resilient lignin-derived activated carbons for hydrogen storage applications. ACS Sustain Chem Eng 8:2186–2195
Sahar T, Rauf M, Murtaza A et al (2023) Anomaly detection in laser powder bed fusion using machine learning: a review. Results Eng 17:100803. https://doi.org/10.1016/j.rineng.2022.100803
Sahlberg M, Karlsson D, Zlotea C, Jansson U (2016) Superior hydrogen storage in high entropy alloys. Sci Rep 6:36770. https://doi.org/10.1038/srep36770
Sahoo RK, Chakraborty B, Sahu S (2021) Reversible hydrogen storage on alkali metal (Li and Na) decorated C20 fullerene: a density functional study. Int J Hydrog Energy 46:40251–40261
Samantaray SS, Mangisetti SR, Ramaprabhu S (2019) Investigation of room temperature hydrogen storage in biomass derived activated carbon. J Alloys Compd 789:800–804. https://doi.org/10.1016/j.jallcom.2019.03.110
Sazali N (2020) Emerging technologies by hydrogen: a review. Int J Hydrog Energy 45:18753–18771. https://doi.org/10.1016/j.ijhydene.2020.05.021
Schäfer RA, Englert JM, Wehrfritz P et al (2013) On the way to graphane-pronounced fluorescence of polyhydrogenated graphene. Angew Chem Int Ed Engl 52:754–757. https://doi.org/10.1002/anie.201206799
Schäfer RA, Dasler D, Mundloch U et al (2016) Basic insights into tunable graphene hydrogenation. J Am Chem Soc 138:1647–1652. https://doi.org/10.1021/jacs.5b11994
Schimmel HG, Kearley GJ, Nijkamp MG et al (2003) Hydrogen adsorption in carbon nanostructures: comparison of nanotubes, fibers, and coals. Chem Eur J 9:4764–4770. https://doi.org/10.1002/chem.200304845
Sdanghi G, Canevesi RL, Celzard A et al (2020) Characterization of carbon materials for hydrogen storage and compression. C 6:46
Serrano L, Moussa M, Yao J-Y et al (2023) Development of Ti-V-Nb-Cr-Mn high entropy alloys for hydrogen storage. J Alloys Compd 945:169289. https://doi.org/10.1016/j.jallcom.2023.169289
Sethia G, Sayari A (2016) Activated carbon with optimum pore size distribution for hydrogen storage. Carbon 99:289–294
Shahi RR, Gupta AK, Kumari P (2023) Perspectives of high entropy alloys as hydrogen storage materials. Int J Hydrog Energy 48:21412–21428. https://doi.org/10.1016/j.ijhydene.2022.02.113
Shekhar S, Chowdhury C (2024a) Prediction of hydrogen storage in metal-organic frameworks: a neural network based approach. Results Surf Interfaces 14:100166. https://doi.org/10.1016/j.rsurfi.2023.100166
Shekhar S, Chowdhury C (2024b) Topological data analysis enhanced prediction of hydrogen storage in metal–organic frameworks (MOFs). Mater Adv 5:820–830. https://doi.org/10.1039/D3MA00591G
Shen Z, Wang Z, Zhang M et al (2018) A novel solid-solution MXene (Ti0.5V0.5)3C2 with high catalytic activity for hydrogen storage in MgH2. Materialia 1:114–120. https://doi.org/10.1016/j.mtla.2018.04.007
Shet SP, Shanmuga Priya S, Sudhakar K, Tahir M (2021) A review on current trends in potential use of metal–organic framework for hydrogen storage. Int J Hydrog Energy 46:11782–11803. https://doi.org/10.1016/j.ijhydene.2021.01.020
Singh G, Ramadass K, DasiReddy VD et al (2023) Material-based generation, storage, and utilisation of hydrogen. Prog Mater Sci 135:101104. https://doi.org/10.1016/j.pmatsci.2023.101104
Smith Milton R, Bittner EW, Shi W et al (2003) Chemical activation of single-walled carbon nanotubes for hydrogen adsorption. J Phys Chem B 107:3752–3760. https://doi.org/10.1021/jp027631v
Song Y, Dai JH (2013) Mechanisms of dopants influence on hydrogen uptake in COF-108: a first principles study. Int J Hydrog Energy 38:14668–14674. https://doi.org/10.1016/j.ijhydene.2013.09.025
Subrahmanyam KS, Kumar P, Maitra U et al (2011) Chemical storage of hydrogen in few-layer graphene. Proc Natl Acad Sci 108:2674–2677. https://doi.org/10.1073/pnas.1019542108
Suksaengrat P, Amornkitbamrung V, Srepusharawoot P, Ahuja R (2016) Density functional theory study of hydrogen adsorption in a Ti-decorated Mg-based metal–organic framework-74. ChemPhysChem 17:879–884. https://doi.org/10.1002/cphc.201500981
Sumida K, Hill MR, Horike S et al (2009) Synthesis and hydrogen storage properties of Be12(OH)12(1,3,5-benzenetribenzoate)4. J Am Chem Soc 131:15120–15121. https://doi.org/10.1021/ja9072707
Sunny MR, Kabir MA, Naheen IT, Ahad MT (2020) Residential energy management: a machine learning perspective. In: 2020 IEEE green technologies conference (GreenTech), pp 229–234
Suwarno S, Dicky G, Suyuthi A et al (2022) Machine learning analysis of alloying element effects on hydrogen storage properties of AB2 metal hydrides. Int J Hydrog Energy 47:11938–11947. https://doi.org/10.1016/j.ijhydene.2022.01.210
Taherkhani K, Eischer C, Toyserkani E (2022) An unsupervised machine learning algorithm for in-situ defect-detection in laser powder-bed fusion. J Manuf Process 81:476–489. https://doi.org/10.1016/j.jmapro.2022.06.074
Thanh HV, EbrahimniaTaremsari S, Ranjbar B et al (2023) Hydrogen storage on porous carbon adsorbents: rediscovery by nature-derived algorithms in random forest machine learning model. Energies 16:2348. https://doi.org/10.3390/en16052348
Thaweelap N, Plerdsranoy P, Poo-arporn Y et al (2021) Ni-doped activated carbon nanofibers for storing hydrogen at ambient temperature: experiments and computations. Fuel 288:119608. https://doi.org/10.1016/j.fuel.2020.119608
Tiba AA, Tivanski AV, MacGillivray LR (2019) Size-dependent mechanical properties of a metal-organic framework: increase in flexibility of ZIF-8 by crystal downsizing. Nano Lett 19:6140–6143. https://doi.org/10.1021/acs.nanolett.9b02125
Tozzini V, Pellegrini V (2013) Prospects for hydrogen storage in graphene. Phys Chem Chem Phys 15:80–89. https://doi.org/10.1039/C2CP42538F
ullah Rather S (2020) Preparation, characterization and hydrogen storage studies of carbon nanotubes and their composites: a review. Int J Hydrog Energy 45:4653–4672
ullah Rather S, Hwang SW (2016) Comparative hydrogen uptake study on titanium–MWCNTs composite prepared by two different methods. Int J Hydrog Energy 41:18114–18120. https://doi.org/10.1016/j.ijhydene.2016.07.194
Üner O (2019) Hydrogen storage capacity and methylene blue adsorption performance of activated carbon produced from Arundo donax. Mater Chem Phys 237:121858
Usman MR (2022) Hydrogen storage methods: review and current status. Renew Sustain Energy Rev 167:112743. https://doi.org/10.1016/j.rser.2022.112743
Vaidyanathan A, Kandasamy M, Ramaniah LM et al (2024) Hydrogen storage in Sc-decorated Ψ-graphene via density functional theory simulations. Int J Hydrog Energy 52:376–389. https://doi.org/10.1016/j.ijhydene.2023.08.356
Vellingiri L, Annamalai K, Kandasamy R, Kombiah I (2018) Characterization and hydrogen storage properties of SnO2 functionalized MWCNT nanocomposites. Int J Hydrog Energy 43:10396–10409. https://doi.org/10.1016/j.ijhydene.2018.04.120
Verdinelli V, Juan A, German E (2019) Ruthenium decorated single walled carbon nanotube for molecular hydrogen storage: a first-principle study. Int J Hydrog Energy 44:8376–8383. https://doi.org/10.1016/j.ijhydene.2019.02.004
Verma R, Jaggi N (2024) A DFT investigation of Osmium decorated single walled carbon nanotubes for hydrogen storage. Int J Hydrog Energy 54:1507–1520
Vinayan BP, Nagar R, Ramaprabhu S (2013) Solar light assisted green synthesis of palladium nanoparticle decorated nitrogen doped graphene for hydrogen storage application. J Mater Chem A 1:11192–11199. https://doi.org/10.1039/C3TA12016C
Wang H, Gao Q, Hu J (2009) High hydrogen storage capacity of porous carbons prepared by using activated carbon. J Am Chem Soc 131:7016–7022. https://doi.org/10.1021/ja8083225
Wang Z, Sun L, Xu F et al (2016) Nitrogen-doped porous carbons with high performance for hydrogen storage. Int J Hydrog Energy 41:8489–8497. https://doi.org/10.1016/j.ijhydene.2016.03.023
Wang L, Chen X, Du H et al (2018) First-principles investigation on hydrogen storage performance of Li, Na and K decorated borophene. Appl Surf Sci 427:1030–1037. https://doi.org/10.1016/j.apsusc.2017.08.126
Wang Y, Lan Z, Huang X et al (2019a) Study on catalytic effect and mechanism of MOF (MOF = ZIF-8, ZIF-67, MOF-74) on hydrogen storage properties of magnesium. Int J Hydrog Energy 44:28863–28873. https://doi.org/10.1016/j.ijhydene.2019.09.110
Wang Z, Zhang X, Ren Z et al (2019b) In situ formed ultrafine NbTi nanocrystals from a NbTiC solid-solution MXene for hydrogen storage in MgH2. J Mater Chem A 7:14244–14252. https://doi.org/10.1039/C9TA03665B
Wang P, Yang Y, Moghaddam NS (2022) Process modeling in laser powder bed fusion towards defect detection and quality control via machine learning: the state-of-the-art and research challenges. J Manuf Process 73:961–984. https://doi.org/10.1016/j.jmapro.2021.11.037
Weitkamp J, Fritz M, Ernst S (1993) Zeolites as media for hydrogen storage. In: von Ballmoos R, Higgins JB, Treacy MMJ (eds) Proceedings from the ninth international zeolite conference. Butterworth-Heinemann, pp 11–19
Wenelska K, Michalkiewicz B, Chen X, Mijowska E (2014) Pd nanoparticles with tunable diameter deposited on carbon nanotubes with enhanced hydrogen storage capacity. Energy 75:549–554. https://doi.org/10.1016/j.energy.2014.08.016
Whitener KE (2018) Hydrogenated graphene: a user’s guide. J Vac Sci Technol A 36:05G401
Xia L, Wang F (2016) Prediction of hydrogen storage properties of Zr-based MOFs. Inorganica Chim Acta 444:186–192. https://doi.org/10.1016/j.ica.2016.01.039
Xiao Y, Dong H, Long C et al (2014) Melaleuca bark based porous carbons for hydrogen storage. Int J Hydrog Energy 39:11661–11667
Xu W-C, Takahashi K, Matsuo Y et al (2007) Investigation of hydrogen storage capacity of various carbon materials. Int J Hydrog Energy 32:2504–2512. https://doi.org/10.1016/j.ijhydene.2006.11.012
Yadav S, Zhu Z, Singh CV (2014) Defect engineering of graphene for effective hydrogen storage. Int J Hydrog Energy 39:4981–4995
Yadav MK, Panwar N, Singh S, Kumar P (2020) Preheated self-aligned graphene oxide for enhanced room temperature hydrogen storage. Int J Hydrog Energy 45:19561–19566. https://doi.org/10.1016/j.ijhydene.2020.05.083
Yang SJ, Jung H, Kim T et al (2012) Effects of structural modifications on the hydrogen storage capacity of MOF-5. Int J Hydrog Energy 37:5777–5783. https://doi.org/10.1016/j.ijhydene.2011.12.163
Yang Y, Li Y, Huang Z, Huang X (2016) (C1. 04H) n: a nearly perfect pure graphane. Carbon 107:154–161
Yang L, Yu LL, Wei HW et al (2019) Hydrogen storage of dual-Ti-doped single-walled carbon nanotubes. Int J Hydrog Energy 44:2960–2975. https://doi.org/10.1016/j.ijhydene.2018.12.028
Ye Y, Ahn CC, Witham C et al (1999) Hydrogen adsorption and cohesive energy of single-walled carbon nanotubes. Appl Phys Lett 74:2307–2309. https://doi.org/10.1063/1.123833
Yildirim T, Íñiguez J, Ciraci S (2005) Molecular and dissociative adsorption of multiple hydrogen molecules on transition metal decorated C 60. Phys Rev B 72:153403. https://doi.org/10.1103/PhysRevB.72.153403
Yoon M, Yang S, Wang E, Zhang Z (2007) Charged fullerenes as high-capacity hydrogen storage media. Nano Lett 7:2578–2583. https://doi.org/10.1021/nl070809a
Younas M, Shafique S, Hafeez A et al (2022) An overview of hydrogen production: current status, potential, and challenges. Fuel 316:123317
Yu X, Tang Z, Sun D et al (2017) Recent advances and remaining challenges of nanostructured materials for hydrogen storage applications. Prog Mater Sci 88:1–48. https://doi.org/10.1016/j.pmatsci.2017.03.001
Yuan B, Guss GM, Wilson AC et al (2018) Machine-learning-based monitoring of laser powder bed fusion. Adv Mater Technol 3:1800136. https://doi.org/10.1002/admt.201800136
Zacharia R, Kim KY, Fazle Kibria AKM, Nahm KS (2005) Enhancement of hydrogen storage capacity of carbon nanotubes via spill-over from vanadium and palladium nanoparticles. Chem Phys Lett 412:369–375. https://doi.org/10.1016/j.cplett.2005.07.020
Zacharia R, Rather S, Hwang SW, Nahm KS (2007) Spillover of physisorbed hydrogen from sputter-deposited arrays of platinum nanoparticles to multi-walled carbon nanotubes. Chem Phys Lett 434:286–291. https://doi.org/10.1016/j.cplett.2006.12.022
Zhan G, Zeng HC (2018) Hydrogen spillover through Matryoshka-type (ZIFs@)n−1ZIFs nanocubes. Nat Commun 9:3778. https://doi.org/10.1038/s41467-018-06269-z
Zhang X (2023) Hydrogen Storage Property of Carbon Nitride Nanotubes. Thesis, UNSW Sydney
Zhang Y, Cheng X (2018) Hydrogen storage property of alkali and alkaline-earth metal atoms decorated C24 fullerene: a DFT study. Chem Phys 505:26–33. https://doi.org/10.1016/j.chemphys.2018.03.010
Zhang L, Hu YH (2011) Structure distortion of Zn4O13C24H12 framework (MOF-5). Mater Sci Eng B 176:573–578. https://doi.org/10.1016/j.mseb.2011.01.014
Zhang L, Nyahuma FM, Zhang H et al (2023) Metal organic framework supported niobium pentoxide nanoparticles with exceptional catalytic effect on hydrogen storage behavior of MgH2. Green Energy Environ 8:589–600. https://doi.org/10.1016/j.gee.2021.09.004
Zhao Y, Kim Y-H, Dillon AC et al (2005) Hydrogen storage in novel organometallic buckyballs. Phys Rev Lett 94:155504. https://doi.org/10.1103/PhysRevLett.94.155504
Zhao W, Fierro V, Zlotea C et al (2012) Activated carbons doped with Pd nanoparticles for hydrogen storage. Int J Hydrog Energy 37:5072–5080. https://doi.org/10.1016/j.ijhydene.2011.12.058
Zhao W, Luo L, Wang H, Fan M (2017) Synthesis of bamboo-based activated carbons with super-high specific surface area for hydrogen storage. BioResources 12:1246–1262
Zhou C, Szpunar JA, Cui X (2016) Synthesis of Ni/graphene nanocomposite for hydrogen storage. ACS Appl Mater Interfaces 8:15232–15241. https://doi.org/10.1021/acsami.6b02607
Zhou L, Zhou Y, Sun Y (2006) Studies on the mechanism and capacity of hydrogen uptake by physisorption-based materials. Int J Hydrog Energy 31:259–264. https://doi.org/10.1016/j.ijhydene.2005.04.048
Zhu ZW, Zheng QR (2023) Investigation of cryo-adsorption hydrogen storage capacity of rapidly synthesized MOF-5 by mechanochemical method. Int J Hydrog Energy 48:5166–5174
Züttel A, Nützenadel C, Sudan P et al (2002) Hydrogen sorption by carbon nanotubes and other carbon nanostructures. J Alloys Compd 330–332:676–682. https://doi.org/10.1016/S0925-8388(01)01659-0
Acknowledgements
This work was supported by the Finnish Cultural Foundation (grant number: 00240256). Additionally, the authors would like to express their gratitude to Prince Sultan University for their support. The authors wish to acknowledge the support of The Bryden Centre project (Project ID VA5048), which was awarded by The European Union’s INTERREG VA Programme, managed by the Special EU Programmes Body (SEUPB), with match funding provided by the Department for the Economy in Northern Ireland and the Department of Business, Enterprise, and Innovation in the Republic of Ireland. Dr. Ahmed I. Osman and Dr. Mahmoud Nasr to dedicate this work to the spirit of the distinguished Egyptian professor Dr. Samih A. Halawy, who passed away on the 2nd of September 2022.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Osman, A.I., Abd-Elaziem, W., Nasr, M. et al. Enhanced hydrogen storage efficiency with sorbents and machine learning: a review. Environ Chem Lett (2024). https://doi.org/10.1007/s10311-024-01741-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10311-024-01741-3