
Abstract
Background
Inclusions of TAR DNA-binding protein 43 kDa (TDP-43) has been designated limbic-predominant, age-related TDP-43 encephalopathy (LATE), with or without co-occurrence of Alzheimer’s disease (AD). Approximately, 30–70% AD cases present TDP-43 proteinopathy (AD-TDP), and a greater disease severity compared to AD patients without TDP-43 pathology. However, it remains unclear to what extent TDP-43 dysfunction is involved in AD pathogenesis.
Methods
To investigate whether TDP-43 dysfunction is a prominent feature in AD-TDP cases, we evaluated whether non-conserved cryptic exons, which serve as a marker of TDP-43 dysfunction in amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD-TDP), accumulate in AD-TDP brains. We assessed a cohort of 192 post-mortem brains from three different brain regions: amygdala, hippocampus, and frontal cortex. Following RNA and protein extraction, qRT-PCR and immunoassays were performed to quantify the accumulation of cryptic RNA targets and phosphorylated TDP-43 pathology, respectively.
Results
We detected the accumulation of misspliced cryptic or skiptic RNAs of STMN2, KCNQ2, UNC13A, CAMK2B, and SYT7 in the amygdala and hippocampus of AD-TDP cases. The topographic distribution of cryptic RNA accumulation mimicked that of phosphorylated TDP-43, regardless of TDP-43 subtype classification. Further, cryptic RNAs efficiently discriminated AD-TDP cases from controls.
Conclusions
Overall, our results indicate that cryptic RNAs may represent an intriguing new therapeutic and diagnostic target in AD, and that methods aimed at detecting and measuring these species in patient biofluids could be used as a reliable tool to assess TDP-43 pathology in AD. Our work also raises the possibility that TDP-43 dysfunction and related changes in cryptic splicing could represent a common molecular mechanism shared between AD-TDP and FTLD-TDP.
Similar content being viewed by others


Background
Alzheimer’s disease (AD) is the most common cause of dementia worldwide, displaying an insidious onset with a gradual progression of memory loss and cognitive decline. The neuropathological mechanisms of AD are related to abnormalities in the proteins β-amyloid and tau, which aggregate in neuritic plaques and neurofibrillary tangles (NFTs), respectively [1]. Approximately, 30–70% of AD patients are also affected by TAR DNA-binding protein 43 kDa (TDP-43) proteinopathy [2,3,4,5,6,7,8]. In fact, TDP-43 pathology can be found in aging brains, which has been termed limbic-predominant, age-related TDP-43 encephalopathy (LATE), and may be concomitant with AD neuropathological changes [9,10,11]. TDP-43 is an RNA-binding protein strongly linked to the majority of cases of amyotrophic lateral sclerosis (ALS) and about 50% of cases of frontotemporal lobar degeneration (FTLD-TDP) [12, 13]. AD cases affected by TDP-43 proteinopathy (AD-TDP) present with a greater disease severity characterized by worse memory and greater hippocampal atrophy compared to AD patients without TDP-43 pathology [14,15,16]. Currently, there is a lack of understanding of the mechanism/s underlying TDP-43-associated neurodegeneration in AD pathogenesis.
TDP-43 is a nuclear protein with roles in maintaining RNA homeostasis [17,18,19]. In TDP-43 proteinopathies, TDP-43 becomes insoluble and either aggregates in the nucleus or mislocalizes to the cytoplasm and forms inclusions there – ultimately leading to a loss of its nuclear function [12, 13, 20]. The molecular pattern of TDP-43 deposition has been used to establish a staging scheme for AD-TDP [21,22,23]: the amygdala becomes affected in the first stages followed by progression into hippocampal regions. In the most severe and advanced cases, TDP-43 inclusions can also be detected in the frontal cortex. In contrast, in FTLD-TDP cases, TDP-43 positive inclusions are initially detected in the amygdala, but progress through the medial frontal cortex, hippocampus and, in latter stages, spread toward motor cortex, spinal cord, and occipital lobe [24]. Further, TDP-43 histopathological pattern deposition is heterogeneous, and has been used to classify TDP-43 cases into different subtypes. In approximately 54% of AD-TDP cases, TDP-43 inclusions resemble the aggregates described in FTLD-TDP type A cases, which are characterized by neuronal cytoplasmic and intranuclear inclusions, dystrophic neurites, and are more widely distributed (AD-TDP type α) [25, 26]. In the remaining 46% of AD-TDP cases, TDP-43 inclusions are accompanied by TDP-43 associated with NFTs, which has been referred to as AD-TDP type β [25, 26]. Nevertheless, although AD-TDP and FTLD-TDP are classified by distinct neuropathological and clinical presentations, TDP-43 dysfunction in both disorders suggests they have a common molecular mechanism.
The role of TDP-43 as a splicing repressor has recently received increased attention in light of the discovery that loss of TDP-43 function leads to inclusion of non-conserved cryptic exons in ALS/FTLD-TDP and AD cases [19, 27,28,29,30,31,32,33,34]. The two most studied cryptic RNAs are the cryptic transcript variants of stathmin-2 (STMN2) and unc-13 homolog A (UNC13A). STMN2 is a microtubule-associated phosphoprotein involved in signal transduction and neuronal growth [31, 32, 35], and with roles in maintaining neuromuscular junctions [36,37,38,39]. Whereas UNC13A, a gene harboring ALS and FTD-linked risk variants, encodes a protein involved in neurotransmitter release [29, 30]. Both cryptic species accumulate in the frontal cortex of FTLD-TDP cases, and associate with the burden of phosphorylated TDP-43 (pTDP-43). Further, they are also associated with an earlier age of disease onset and a shorter survival [29, 35]. The fate of different cryptic RNAs varies: some species, including the cryptic forms of STMN2 and UNC13A, incorporate premature stop codons resulting in their rapid degradation without undergoing translation. In contrast, other cryptic RNAs are effectively translated, and novel peptides have been detected in neurons and cerebrospinal fluid (CSF) of ALS with or without frontotemporal dementia (FTD) patients [34, 40], potentially serving as markers of TDP-43 dysfunction. Regardless whether the cryptic RNA may lead to a truncated variant or not, the accumulation of cryptic RNAs results in downregulation of the corresponding full-length RNA and protein variants, many of which have important roles in maintaining neuronal functions [29,30,31,32, 35, 41]. Rescuing these missplicing events may present a novel approach to mitigate TDP-43-related neurotoxicity [32, 42]. Thus, cryptic RNA accumulation due to TDP-43 dysfunction may have potential not only as a biomarker to stratify patients with and without TDP-43 pathology, but may also have important therapeutic implications for TDP-43 proteinopathies.
Although aberrant cryptic transcripts have been systematically reported in ALS/FTLD-TDP cases, the degree to which these RNAs accumulate in other TDP-43 proteinopathies such as AD, was unknown. Therefore, we aimed to investigate if cryptic RNAs detected in FTLD-TDP also accumulate in AD-TDP in three brain regions differentially affected by TDP-43 deposition (amygdala, hippocampus, and frontal cortex) [21,22,23]. Compared to cognitively normal controls and AD cases without TDP-43 pathology, AD-TDP cases accumulated cryptic RNAs in regions most affected by TDP-43 deposition. Similar to what has been observed for FTLD-TDP, some cryptic RNAs in AD-TDP best associated with the levels of pTDP-43 and discriminated from TDP-43 negative controls. These results suggest that different TDP-43 proteinopathies may share common pathological mechanisms, and TDP-43-targeted interventions and associated diagnostic tools may be of benefit to multiple conditions, including AD-TDP as well as FTLD-TDP.
Methods
Study approval and sample selection
Human postmortem brain tissues from amygdala, hippocampus, and frontal cortex were provided by the Mayo Clinic Florida Brain Bank. Diagnosis was independently ascertained by trained neurologists and neuropathologists upon neurological and pathological examinations, respectively. All participants, or the next of kin, provided written informed consent, and all protocols were reviewed and approved by the Mayo Clinic Institutional Review Board and Ethics Committee. Samples were selected based on neuropathological diagnosis of FTLD-TDP, AD with and without TDP-43 pathology, and cognitively normal controls. Of note, the presence/absence of Lewy bodies was not a criterion for sample selection. A total of 15 AD-TDP cases (21%) had Lewy bodies in the amygdala, but none had Lewy bodies in the brainstem or cortex. Size of the study cohort was determined by sample availability for all three brain regions, TDP-43 subtype characterization available, and both males and females were included in the study. A summary of the study cohort is described in Table 1.
Neuropathological assessments
Immunohistochemistry was performed on 5-µm-thick sections of formalin-fixed, paraffin-embedded tissue mounted on glass slides. Sections were deparaffinized and processed for phospho-TDP-43 (pS409/410, mouse monoclonal, 1:5,000; Cosmo Bio, Tokyo, Japan) [25], and Thioflavin S (Sigma-Aldrich, St. Louis, MO) for Braak staging [43] and Thal phasing [44], according to previously published methods.
Tissue sampling for biochemical assessments
In order to determine pTDP-43 protein and cryptic RNA levels, 50-60 mg of human tissue for protein and 40 mg tissue for RNA extractions were dissected from the amygdala, hippocampus and frontal cortex. Specifically, the amygdala was cut from a coronal section at the level of the uncus, the hippocampus was cut from a coronal section at the level of the lateral geniculate, and the middle frontal gyrus samples were cut from Brodmann area 9.
Protein extraction from human brain tissues
To measure pTDP-43 from postmortem tissues, we performed protein fractionation as previously described [45]. Briefly, tissues were homogenized in 5 volumes (w/v) of cold RIPA buffer (25 mM Tris-HCl [pH 7.6], 150 mM NaCl, 1% sodium deoxycholate, 1% Nonidet P-40, 0.1% sodium dodecyl sulfate, and protease and phosphatase inhibitor cocktail), sonicated on ice, and centrifuged at 100,000 x g for 30 min at 4 °C. The supernatant was then collected as RIPA-soluble fraction. The pellet was resuspended in RIPA buffer, sonicated and centrifuged again. The supernatant was discarded, and the remaining pellet was dissolved in urea buffer (30 mM Tris-HCl [pH 8.5], 7 M urea, 2 M thiourea, and 4% CHAPS [(3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate]) for 1 h at room temperature with continuous agitation. Following incubation, samples were sonicated and centrifuged at 100,000 x g for 30 min at 22 °C. The resulting supernatant, referred as urea-soluble or RIPA-insoluble fraction, was then collected. Protein concentrations of urea-soluble fractions were determined by Bradford assay (ThermoFisher).
Phosphorylated TDP-43 immunoassay
Assessment of pTDP-43 was performed in the urea-soluble fraction from the amygdala, hippocampus, and frontal cortex of our study cohort using a sandwich Meso Scale Discovery (MSD) immunoassay [45]. A rabbit polyclonal antibody specific for TDP-43 phosphorylated at serines 409/410 (3 µg/mL, 22309-1-AP, Proteintech) was used for capture, and a sulfo-tagged rabbit polyclonal C-terminal TDP-43 antibody (3 µg/mL, 12892-1-AP, Proteintech) for detection. All samples were tested in duplicate wells, and controls were included in every plate to account for any interplate variability. MSD QUICKPLEX SQ120 technology was used to acquire the response values corresponding to the intensity of emitted light upon electrochemical stimulation.
RNA extraction from human brain samples
RNA was extracted using the RNAeasy Plus Mini Kit (Qiagen) per manufacturer’s instructions, and as previously described [35, 45]. RNA from up to three cuts was extracted, and only extractions with high quality RNA were kept for downstream analyses. RNA concentration was determined using Nanodrop technologies (Thermo Fisher), and an Agilent 2100 bioanalyzer (Agilent Technologies) was used to evaluate the RNA integrity number (RIN). Median RIN values for all samples were > 9 for all brain regions (Table S1).
Evaluation of cryptic RNA accumulation by qRT-PCR
Following RNA extraction, 500 ng of total RNA were transcribed into complementary DNA (cDNA) using the High-Capacity cDNA Transcription Kit (Applied Biosystems) according to manufacturer’s directions. Then, quantitative real-time PCR (qRT-PCR) was performed, in triplicates, using SYBR GreenER qPCR SuperMix (Invitrogen) on a QuantStudio™ 7 Flex Real-Time PCR System (Applied Biosystems). All samples from amygdala, hippocampus and frontal cortex were assessed at the same time, and control samples were included in every plate to account for any interplate variability. Relative quantification of cryptic RNAs was determined using the ΔΔCt method and normalized to two endogenous controls, GAPDH and RPLP0. The following primers were used: cryptic STMN2 forward: 5′-GGACTCGGCAGAAGACCTTC-3′, cryptic STMN2 reverse: 5′-GCAGGCTGTCTGTCTCTCTC-3′; skiptic KCNQ2 forward: 5′-TATGCCCACAGCAAGATCAC-3′, skiptic KCNQ2 reverse: 5′-AGACACCGATGAGGGTGAAG; cryptic UNC13A forward: 5′-TGGATGGAGAGATGGAACCT, cryptic UNC13A reverse: 5′-GGGCTGTCTCATCGTAGTAAAC; cryptic CAMK2B forward: 5′-CTGCTCCGTGGTCTTAATGAT, cryptic CAMK2B reverse: 5′-GAGTGCAGAGACTTCCCCC; cryptic SYT7 forward: 5′-GCAGTGAGAAGAAGGCTATCAA; cryptic SYT7 reverse 5′-CGGCAGACTGGAGCCT; GAPDH forward: 5′-GTTCGACAGTCAGCCGCATC, GAPDH reverse: 5′-GGAATTTGCCATGGGTGGA; and RPLP0 forward: 5′-TCTACAACCCTGAAGTGCTTGAT; RPLP0 reverse: 5′-CAATCTGCAGACAGACACTGG.
Statistics
Statistical analyses were performed in GraphPad Prism 9 (GraphPad Software). To evaluate differences in pTDP-43 protein or cryptic RNA levels between disease cases and controls, for each region, we performed One-way ANOVA followed by Dunn’s multiple comparison tests as indicated in the figure legends. When comparing cryptic RNA levels between AD-TDP to AD no TDP cases, the Mann-Whitney test was used. Further, we also performed single-variable (unadjusted) and multivariable linear regression models (adjusted) to evaluate differences between AD-TDP and other study groups (Tables). Multivariable models were adjusted for age at death, sex and RIN for cryptic RNAs, and for age at death and sex for pTDP-43 protein. Due to the skewed distribution of the data, cryptic RNA and pTDP-43 were analyzed on the base 10 logarithmic scale. The regression coefficients (β) and 95% confidence intervals (CIs) were estimated and interpreted as the difference in the means, bases on the 10 logarithmic scale, between all AD-TDP cases (reference group) and the other study groups. For pTDP-43, P values less than 0.0167 were considered statistically significant after adjusting for the three separate AD-TDP vs. CN, AD-TDP vs. AD no TDP, and AD-TDP vs. FTLD-TDP analyses. For cryptic RNA, P values less than 0.025 were considered statistically significant after adjusting for the three separate AD-TDP vs. controls (CN + AD no TDP), and AD-TDP vs. FTLD-TDP analyses.
Associations between cryptic RNA and pTDP-43 protein levels were evaluated in AD-TDP and FTLD-TDP cases using single-variable and multivariable linear regression models. Both cryptic RNA and pTDP-43 protein levels were analyzed using the base 10 logarithmic scale. The multivariable model was adjusted for age, sex, RIN, and TDP-43 subtype, and P values < 0.01 were considered as statistically significant. For assessing differences in cryptic RNA levels within TDP-43 subtypes, in AD-TDP and FTLD-TDP, single-variable and multivariable linear regression models were also performed as described, and where TDP-43 type A/α was set as the reference group. The multivariable model was adjusted for age, sex, RIN, and pTDP-43 levels, and P values < 0.05 were considered as statistically significant.
To evaluate the ability of cryptic RNAs to discriminate AD-TDP or FTLD-TDP cases from controls, we estimated the area under the receiver operating characteristic curve (AUC) along with a 95% confidence interval (CI) for each of the cryptic RNAs in each region. Of note, an AUC value of 0.5 corresponds to predictive ability equal to that of chance, and an AUC of 1.0 represents perfect predictive ability.
Results
Study cohort characteristics
Our study cohort included a total of 192 postmortem cases classified into four main groups: 27 cognitively normal (CN) cases (median Braak NFT stage II, Thal phase 0), 27 AD cases without TDP-43 pathology (AD no TDP), 71 AD cases with confirmed TDP-43 pathology (AD-TDP), and 67 FTLD-TDP cases (Table 1). All AD cases had a median Braak NFT stage of VI and Thal phase of 5, while FTLD-TDP cases presented lower Braak NFT (median I-II, range: 0 to IV-V) and Thal phase (median 1, range: 0–5) (Table 1). The median age of death for the AD-TDP group was 84 years, the FTLD-TDP median age of death was lower at 71 years, and the median age of death of CN and AD no TDP-43 were 79 and 78 years, respectively (Table 1). Our study included similar numbers of males and females, when possible, based on tissue availability. The AD-TDP and CN groups showed the highest and lowest proportion of females, respectively (Table 1). Disease duration in AD cases, irrespective of the presence of TDP-43 pathology, was 9–10 years and with a median age of disease onset was 70–73 years (Table 1). As expected, disease duration in FTLD-TDP cases was shorter (median 5 years, range: 1–25 years) and disease onset was earlier (median 64 years, range: 42–90 years) (Table 1). Both, AD-TDP and FTLD-TDP groups included similar number of cases from two TDP-43 subtypes: type α (N = 36) and β (N = 35) in AD-TDP, and type A (N = 32) and B (N = 35) in FTLD-TDP (Table 1).
TDP-43 pathology accumulates in amygdala and hippocampus of AD-TDP
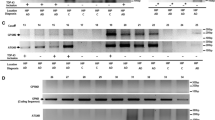
To assess TDP-43 dysfunction in AD-TDP, we first evaluated post-mortem brain tissues from three different brain regions: amygdala, hippocampus, and frontal cortex. The criteria for selecting these specific areas were based on TDP-43 deposition scheme in AD that describes an initial TDP-43 accumulation in the amygdala (stage 1) followed by pathology in hippocampus and occipitotemporal gyrus (stages 2–3), basal forebrain and ventral striatum (stages 4–5) and last in the frontal cortex (stage 6) (Fig. 1A) [21,22,23]. Importantly, these same regions are known to accumulate TDP-43 pathology in FTLD-TDP [24, 46], allowing us to evaluate whether changes resulting from TDP-43 dysfunction in FTLD-TDP are also observed in AD-TDP. Given that pTDP-43 is a sensitive and specific marker of TDP-43 pathology [29, 35, 45], we quantified pTDP-43 protein levels across all three brain regions using a Meso Scale Discovery (MSD) based immunoassay. We found a significant accumulation of pTDP-43 protein in the urea soluble fraction of all three regions (amygdala, hippocampus, and frontal cortex) of FTLD-TDP cases (Fig. 1B). In AD-TDP, pTDP-43 protein levels were markedly elevated in the amygdala, the primarily region affected by TDP-43 pathology; levels were also significantly elevated in the hippocampus, albeit to a lesser extent (Fig. 1B). No significant pTDP-43 accumulation was found in the frontal cortex of AD-TDP cases (Fig. 1B), consistent with AD-TDP pathophysiology where a very small proportion of cases present TDP-43 pathology in this region [21,22,23]. As expected, all control cases, regardless of their classification as cognitively normal (CN) or AD without TDP-43 (AD no TDP), showed no pTDP-43 accumulation in any region (Fig. 1B). Further, the accumulation of pTDP-43 in the amygdala and hippocampus of AD-TDP cases was significantly increased compared to controls (CN and AD no TDP), and significantly lower than FTLD-TDP cases after adjusting for sex, and age at death (Table S2).

TDP-43 pathology accumulates in amygdala and hippocampus of AD-TDP. (A) Graphical overview of AD-TDP staging based on TDP-43 deposition pattern from amygdala (stage 1), followed by the hippocampus and occipitotemporal gyrus (stages 2–3), basal forebrain and ventral striatum (stages 4–5) and last in the frontal cortex (stage 6). Created with BioRender.com. (B) Quantification of pTDP-43 protein levels in cognitively normal controls (CN), AD and FTLD cohorts across three brain regions: amygdala, hippocampus, and frontal cortex (see Table 1), using an immunoassay (see Methods). Data are presented as mean ± SEM. Number of cases is included in the figures. Statistical analyses were performed by One-way ANOVA following Dunn’s multiple comparison tests: *P < 0.05, **P < 0.005, *** P < 0.0005, ****P < 0.0001, ns: not significant
Aberrant cryptic RNAs accumulate in the amygdala and hippocampus of AD-TDP
Having validated the presence of TDP-43 pathology in different regions of AD-TDP and FTLD-TDP brains, we next sought to evaluate whether these same regions demonstrated evidence of TDP-43 dysfunction. The aberrant accumulation of cryptic RNA targets in ALS and FTLD tissues with TDP-43 pathology (e.g., frontal cortex, motor cortex, spinal cord, etc.) has been the focus of recent studies [29,30,31,32, 34, 35]. These studies found that, in the absence of TDP-43, cryptic RNAs are expressed from genes with key functions in neuronal networks. In particular, cryptic RNAs encoded from STMN2 (neuronal growth, axonal regeneration, signal transduction), KCNQ2 (neuronal excitability), UNC13A (neurotransmitter release at the synapse), CAMK2B (neuronal plasticity and synapse formation), and SYT7 (exocytosis of secretory and synaptic vesicles) have been validated in frontal cortex of ALS and FTLD-TDP [29, 34, 35]. Moreover, some are detectable as peptides in ALS/FTD CSF [34]. In fact, we validated the accumulation of five cryptic RNAs in the frontal cortex of FTLD-TDP in this study cohort (Fig. S1, Table S3). Of note, we did not detect significant accumulation in the frontal cortex of AD-TDP cases, most likely due to the paucity of TDP-43 pathology in this region (see Fig. 1). In contrast, we found significant accumulations of cryptic RNAs in both amygdala (Fig. 2A) and hippocampus (Fig. 2B) of AD-TDP compared to controls (Ctrl: CN + AD no TDP), even after adjusting for potential confounding variables [age at death, sex and RNA integrity number (RIN), Table S4]. Of note, we combined CN and AD no TDP groups, referred as controls from herein, since both groups are negative for pTDP-43 accumulation. Nonetheless, similar results were found when comparing AD-TDP to AD no TDP (Fig. S2). Moreover, while the accumulation of cryptic RNAs appeared greater in FTLD-TDP, it was not significantly higher than in AD-TDP after adjusting for age at death, sex, and RIN (Table S4). Taken together, our results indicate that AD with TDP-43 pathology accumulate cryptic RNAs in brain regions affected by TDP-43 pathology.

Aberrant cryptic RNAs accumulate in the amygdala and hippocampus of AD-TDP. Cryptic RNA (STMN2, KCNQ2, UNC13A, CAMK2B, and SYT7) levels were measured by qRT-PCR in amygdala (A) and hippocampus (B) of controls (Ctrl: CN, represented by white circles + AD no TDP, represented by orange circles), AD-TDP, and FTLD-TDP cases. Number of cases is included in the figures. Data are presented as mean ± SEM. Statistical analyses were performed by One-way ANOVA following Dunn’s multiple comparison tests: *P < 0.05, **P < 0.005, *** P < 0.0005, ****P < 0.0001, ns: not significant
Cryptic RNA accumulation associates with pTDP-43 burden but does not generally differ between TDP-43 subtypes
In previous studies, we and others demonstrated that pTDP-43 burden is strongly associated with the levels of cryptic RNAs in the frontal cortex of ALS and FTLD-TDP. We also observed a correlation between pTDP-43 levels and the accumulation of all five cryptic RNAs in FTLD-TDP frontal cortex (Table S5). Interestingly, the correlation of pTDP-43 protein and cryptic RNAs was most significant in frontal cortex and amygdala, but less in the hippocampus (Table S5). In AD-TDP, we found significant associations between cryptic RNAs and pTDP-43 in both amygdala and hippocampus, even after adjusting for age at death, sex, RIN, and TDP-43 subtype (Table 2). Among all cryptic RNAs evaluated, KCNQ2 cryptic RNA consistently associated with higher pTDP-43 burden in both brain regions (Table 2).
Next, we sought to evaluate whether the type and distribution of TDP-43 inclusions associated with the levels of cryptic RNAs. In FTLD-TDP, TDP-43 type A and B are the most frequent subtypes. TDP-43 type A has the most widely distributed TDP-43 pathology and presents a wider range of inclusion types, while TDP-43 type B is predominantly characterized by neuronal cytoplasmic inclusions and is sometimes associated with motor neuron disease [47]. In our study cohort, we observed that FTLD-TDP type B tended to have lower levels of pTDP-43 burden in the frontal cortex, although it did not reach statistical significance (Fig. S3A). Of all five cryptic RNAs, the levels of KCNQ2 cryptic RNA in the frontal cortex and CAMK2B cryptic RNA in the amygdala were significantly lower in TDP-43 type B FTLD-TDP, after adjusting for age at death, sex, RIN and pTDP-43 levels (Table S6). No subtype-specific differences were observed for the other three cryptic RNAs (Table S6). In AD-TDP a similar TDP-43 staging scheme has also been described, with TDP-43 type α resembling TDP-43 type A in FTLD-TDP, and TDP-43 type β often associated with neurons that also have tau aggregates in the form of NFTs [25, 26]. Similar to what was observed in FTLD-TDP type B, AD-TDP cases with TDP-43 type β showed a lower accumulation of pTDP-43, but this was not significant (Fig. S3B). Only STMN2 cryptic RNA levels in the amygdala were significantly lower in AD-TDP type β after adjusting for age at death, sex, RIN and pTDP-43 levels (Table S7). Once again, there was no change in the levels of the other cryptic RNAs across TDP-43 subtypes (Table S7). Overall, our results confirm strong associations of cryptic RNA accumulation with pTDP-43 burden in FTLD-TDP and AD-TDP. Moreover, our data demonstrated that cryptic RNA accumulation was largely independent of TDP-43 subtype.
Cryptic RNAs can discriminate TDP-43 cases from controls
To evaluate the ability of each of the cryptic RNAs to distinguish TDP-43 positive from TDP-43 negative cases, we performed receiver operating characteristic (ROC) analyses and calculated the area under the curve (AUC) for each cryptic RNA in FTLD-TDP and AD-TDP compared to controls. As expected, the discriminatory ability of all five cryptic RNAs was highly significant (P < 0.0001) in the frontal cortex and amygdala in FTLD-TDP, with AUC ranging 0.79–0.98 in amygdala and 0.81–0.94 in frontal cortex (Fig. S4). The discriminatory ability of the cryptic RNAs in the hippocampus of FTLD-TDP cases was more variable (AUC: 0.67–0.95), but still statistically significant (P < 0.005, Fig. S4). In AD-TDP, four (STMN2, KCNQ2, UNC13A, SYT7) out of the five cryptic RNA were able to distinguish TDP-43 cases from controls, in both amygdala and hippocampus, although the significance and AUC values were lower in the latter region (Fig. 3A). Interestingly, KCNQ2 and UNC13A cryptic RNA levels had the most significant discriminatory ability in AD-TDP. Of note, the pattern of discriminatory ability in AD-TDP was similar to that of FTLD-TDP, with KCNQ2 and UNC13A cryptic RNAs showing the highest significance and AUC values (Fig. 3B). Overall, our data demonstrate that cryptic RNA levels display a reliable ability to discriminate TDP-43 positive cases from controls in both FTLD-TDP and AD-TDP.

Cryptic RNA can discriminate AD-TDP cases from controls. (A) Representative images of the discriminatory ability of cryptic RNAs to distinguish AD-TDP cases from controls, evaluated by receiver operating characteristic (ROC) analyses, in amygdala (left; AD-TDP, N = 69; controls, N = 49) and hippocampus (right; AD-TDP, N = 71; controls, N = 54). The area under the curve (AUC) values, 95% confidence intervals (CI), and P values for each cryptic RNA are included in the bottom table. (B) Representative image of the comparable discriminatory ability pattern of cryptic RNAs for AD-TDP and FTLD-TDP cases in the amygdala
Discussion
TDP-43 dysfunction results in a lack of splicing repression leading to the aberrant accumulation of cryptic RNAs in ALS and FTLD-TDP brain regions with marked TDP-43 pathology [19, 27,28,29,30,31,32,33,34,35]. Here, we aimed to evaluate to what extent TDP-43 dysfunction is also present in AD-TDP by assessing accumulation of cryptic RNAs characteristic of FTLD-TDP in the amygdala, hippocampus and frontal cortex. Consistent with the topographic distribution of TDP-43 inclusions, pTDP-43 significantly accumulated in all three regions in FTLD-TDP, with amygdala and frontal cortex showing the highest burden. In contrast, AD-TDP had greater accumulation in the amygdala, followed by the hippocampus, and no detectable accumulation in frontal cortex. The latter is not surprising since only a small proportion of AD-TDP cases has TDP-43 pathology in the frontal cortex [21,22,23]. Interestingly, the levels of pTDP-43 in amygdala and hippocampus of AD-TDP were lower than those in FTLD-TDP, even after correcting for confounding variables. Given that the time course of FTLD-TDP is relatively shorter than that of AD-TDP, it is tempting to speculate that enhanced pTDP-43 deposition may correlate with the more aggressive disease course in FTLD-TDP. On the other hand, accumulation of pTDP-43 protein may represent only a fraction of reduced TDP-43 nuclear function. In fact, loss of nuclear TDP-43 without overt TDP-43 pathology has been associated with neurodegeneration [48, 49], and is sufficient to accumulate TDP-43 cryptic RNA targets [28, 41]. Thus, evaluating splicing changes resulting from aggregation or reduced levels of nuclear TDP-43 may provide additional tools to assess the severity of TDP-43 dysfunction.
Several cryptic RNAs were found to accumulate in both AD-TDP and FTLD-TDP brains, in regions most affected by TDP-43 proteinopathy, but were absent from cognitively normal controls and AD no TDP control brains. In particular, we evaluated cryptic RNAs in STMN2 and UNC13A, both of which we previously validated in FTLD-TDP cases [29, 35]. STMN2 is a neuronal phosphoprotein highly expressed during neuronal development, and in the adult brain [50]. It has been implicated in microtubule dynamics, has a role in axonal regeneration [51,52,53], and is important for maintenance of the neuromuscular junction [36,37,38,39]. While STMN2 misprocessing due to TDP-43 loss of function has not been previously reported in AD, other stathmin protein family members have been implicated with AD pathogenesis. For example, STMN1 protein and RNA levels were decreased and upregulated, respectively, in AD brains [54]. Interestingly, the levels of STMN1 protein and number of NFT, but not plaques, were inversely correlated [54]. The accumulation of cryptic STMN2 RNA in ALS/FTLD-TDP is accompanied by a decrease in full-length STMN2 RNA and protein [31, 32, 35]. Of interest, full-length STMN2 was found to be unaltered at both RNA and protein levels in AD; however, the TDP-43 status of these cases was not reported [55]. Thus, it is possible that cryptic STMN2, or the consequent decrease in full-length STMN2 may play a pathogenic role in AD-TDP.
Genetic variants at the UNC13A locus were previously reported to confer an increased risk for ALS/FTD [56, 57], and recent studies demonstrate that the risk haplotype not only locates within the cryptic exon itself, but also leads to decreased ability of TDP-43 and other hnRNPs to bind and repress cryptic exon inclusion in UNC13A [29, 30, 58]. Of interest, reduced levels of Unc13a have been associated with impaired processing of amyloid precursor protein [59, 60], suggesting that changes affecting UNC13A splicing downstream of TDP-43 dysfunction may be detrimental in AD.
We also observed the accumulation of CAMK2B and SYT7 cryptic RNAs in AD-TDP. These two genes encode proteins involved in calcium-dependent regulation of membrane trafficking and synaptic transmission. While less is known about whether these genes can influence AD, both have been associated with synaptic plasticity changes in mouse models of AD [61, 62]. Thus, our data further highlights the importance of changes in these genes to AD.
Dysfunction of TDP-43 in FTLD-TDP and AD-TDP also causes missplicing in the potassium channel regulator KCNQ2. In contrast to the other cryptic splicing events, loss of TDP-43 leads to the exclusion of an in-frame canonical exon in KCNQ2, rather than the inclusion of a novel exon. This “skiptic” event leads to an RNA encoding a protein lacking a sequence of the pore domain, which is required for potassium conduction. KCNQ2 forms part of M channels, and mutations in KCNQ2 are associated with decreased neuronal excitability and neonatal epilepsy [63]. In fact, KCNQ2 has been associated with cognitive decline during normal aging [64], and M channel blockers have been proposed as potential therapeutic targets to treat cognitive decline in AD [63, 65]. Moreover, it has been suggested that amyloid beta deposition can affect KCNQ2 levels [66, 67], further supporting its potential contribution to AD pathophysiology.
The accumulation of cryptic RNAs paralleled the anatomic pattern of pTDP-43 accumulation, with high levels in both the amygdala and hippocampus of AD-TDP and FTLD-TDP. While the burden of cryptic RNAs initially seemed lower in AD-TDP cases compared to FTLD-TDP in both amygdala and hippocampus, these differences disappeared after correcting for age and sex. The accumulation of cryptic RNAs was also highly significant in the frontal cortex of FTLD-TDP, where the burden was similar to that in the amygdala, but AD-TDP frontal cortex showed no pTDP-43 deposition and no cryptic RNA accumulation. Given that the FTLD-TDP cases analyzed had a shorter disease duration (median 5 years, range: 1–25 years) than AD-TDP cases (median 10 years, range: 3–23 years), our results suggest that the accumulation of cryptic RNAs may be faster in FTLD-TDP. At the same time, deposition of TDP-43 pathology and cryptic RNAs in the frontal cortex in FTLD-TDP may explain a more aggressive disease phenotype in FTLD-TDP compared to AD-TDP. Why TDP-43 dysfunction in the frontal cortex is an early event in FTLD-TDP, but late in AD-TDP remains to be further studied.
We also evaluated whether different TDP-43 subtypes could influence the accumulation of TDP-43-regulated cryptic RNA targets. While some cryptic RNAs were present to a lower degree in FTLD-TDP type B and AD-TDP type β, there were not consistent expression patterns across targets or brain regions. This is intriguing for AD-TDP, since co-deposition of TDP-43 in NFT is common in AD-TDP type β, but it did not seem to affect TDP-43 dysfunction compared to AD-TDP type α. Overall, our studies suggest that cryptic RNA accumulation in both AD-TDP and FTLD-TDP results from loss of TDP-43 nuclear function, independently of TDP-43 inclusion type, and TDP-43 dysfunction in both AD and FTLD may result into shared disease mechanisms.
The specific contribution of cryptic RNAs to disease pathogenesis remains unclear. Inclusion of cryptic exons in some of these genes leads to premature stop codons (STMN2, UNC13A, SYT7, CAMK2B), which may lead to rapid degradation of the RNA or their respective truncated encoded proteins. Others result in the incorporation of in-frame exons without introducing stop codons (KCNQ2), and lead to the synthesis of stable proteins with potential deleterious functions. Indeed, some these predicted cryptic peptides (KCNQ2, CAMK2B, SYT7) have been identified in neurons depleted of TDP-43 and in the CSF of patients with ALS/FTD [34]. These studies not only underscore the importance of understanding the role of these de-novo proteins in disease pathogenesis, but also suggest that some may serve as markers of TDP-43 dysfunction. The ability to detect cryptic RNAs may differ due to transcript abundance, KCNQ2 and UNC13A best associated with pTDP-43 burden in both AD-TDP and FTLD-TDP, and consistently differentiated TDP-43-positive cases from controls. Our observation that some of the same targets identified in AD-TDP as in ALS and FTLD-TDP suggests that biomarkers of TDP-43 dysfunction developed in ALS and FTD may serve to identify AD patients with TDP-43 pathology. Since TDP-43 pathology is associated with worse outcomes in AD [14,15,16], further understanding the role of missplicing events in AD is paramount.
Our study capitalized on a large cohort of clinically and pathologically well-characterized postmortem tissues, with analyses of three different brain regions relevant to TDP-43 deposition. In addition to inclusion of cognitively normal controls, cases of AD without TDP-43 pathology clearly aids in confirming that cryptic RNA accumulation in AD is dependent on TDP-43 pathology. Additional strengths of the study include use of high-quality tissues (high RIN) and our ability to quantify pTDP-43 and cryptic RNA burden. Our study also has some limitations. While we evaluated a few cryptic RNAs, additional cryptic RNAs have been described in ALS/FTLD-TDP which may also be misregulated in AD-TDP. One limitation of this study is that we do not know the specific contribution of these targets to disease. Thus, defining the functional impact of cryptic RNA accumulation to the pathobiology if AD is an essential future step. Another limitation is that only one method was used to determine the presence of cryptic RNAs. Additional methods such as analyses of RNA sequencing data or RNA-based imaging studies, while have their limitations, would be of added benefit as we have previously shown [29, 35]. Finally, while our data shows that UNC13A and KCNQ2 were best able to discriminate TDP-43 cases from controls and they correlated better with pTDP-43 burden in both AD-TDP and FTLD-TDP, we cannot conclude which RNA or combination of RNAs may be the best candidate for biomarker development. In fact, measuring the levels of cryptic proteins may be more relevant to biomarker discovery efforts.
Conclusions
Overall, we found that cryptic RNAs regulated by TDP-43 accumulate in tissues and diseases of different etiologies but with TDP-43 pathology. The fact that cryptic RNAs in ALS/FTLD-TDP also accumulate in AD-TDP cases suggest a common mechanism involving changes in RNA metabolism in both diseases. Our findings have significant implications for broadening our understanding of AD-TDP pathomechanisms and aiding in the development of biomarkers to identify AD cases with TDP-43 pathology. Future efforts to restore missplicing events resulting from TDP-43 dysfunction may be a promissing therapeutic approach for AD-TDP and ALS/FTLD-TDP. For example, restoring STMN2 splicing is sufficient to restore axonal regeneration deficits resulting from TDP-43 dysfunction [32]. Further, the use of antisense oligonucleotides may serve as an effective tool to restore STMN2 splicing defects in vivo [42], and it is the goal of ongoing clinical trials (QRL-201 trial: NCT05633459). In all, TDP-43-regulated cryptic RNAs are expected to facilitate the generation of tools not only to assess TDP-43 dysfunction in ante-mortem biofluids, but also represent novel targets for therapeutic intervention, which would benefit multiple TDP-43 proteinopathies, including AD.
Data availability
All data generated or analyzed during this study are included in this published article [and its supplementary information files], or available from the corresponding author on reasonable request.
Abbreviations
- ALS:
-
Amyotrophic lateral sclerosis
- AD:
-
Alzheimer’s disease
- AD-TDP:
-
AD with TDP-43 pathology
- AUC:
-
Area under the curve
- CAMK2B:
-
Calcium/calmodulin dependent protein kinase II beta
- CI:
-
Confidence interval
- CN:
-
Cognitively normal
- CSF:
-
Cerebrospinal fluid
- FTLD:
-
Frontotemporal lobar degeneration
- FTLD-TDP:
-
FTLD with TDP-43 pathology
- KCNQ2:
-
Potassium voltage-gated channel subfamily member 2
- LATE:
-
Limbic-predominant, age-related TDP-43 encephalopathy
- NFT:
-
Neurofibrillary tangle
- RIN:
-
RNA integrity number
- ROC:
-
Receiver operating characteristic
- SEM:
-
Standard error of the mean
- STMN2:
-
Stathmin 2
- SYT7:
-
Synaptotagmin 7
- pTDP-43:
-
Phosphorylated TDP-43
- TDP-43:
-
TAR DNA-binding protein 43 kDa
- UNC13A:
-
Unc-12 homolog A
References
DeTure MA, Dickson DW. The neuropathological diagnosis of Alzheimer’s disease. Mol neurodegeneration. 2019;14(1):1–18.
Amador-Ortiz C, Lin WL, Ahmed Z, Personett D, Davies P, Duara R, et al. TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer’s disease. Ann Neurol. 2007;61(5):435–45.
Arai T, Mackenzie IR, Hasegawa M, Nonoka T, Niizato K, Tsuchiya K, et al. Phosphorylated TDP-43 in Alzheimer’s disease and dementia with Lewy bodies. Acta Neuropathol. 2009;117(2):125–36.
Higashi S, Iseki E, Yamamoto R, Minegishi M, Hino H, Fujisawa K, et al. Concurrence of TDP-43, tau and alpha-synuclein pathology in brains of Alzheimer’s disease and dementia with Lewy bodies. Brain Res. 2007;1184:284–94.
Hu WT, Josephs KA, Knopman DS, Boeve BF, Dickson DW, Petersen RC, et al. Temporal lobar predominance of TDP-43 neuronal cytoplasmic inclusions in Alzheimer disease. Acta Neuropathol. 2008;116(2):215–20.
Kadokura A, Yamazaki T, Lemere CA, Takatama M, Okamoto K. Regional distribution of TDP-43 inclusions in Alzheimer disease (AD) brains: their relation to AD common pathology. Neuropathology: official journal of the Japanese Society of Neuropathology. 2009;29(5):566–73.
Uryu K, Nakashima-Yasuda H, Forman MS, Kwong LK, Clark CM, Grossman M, et al. Concomitant TAR-DNA-binding protein 43 pathology is present in Alzheimer disease and corticobasal degeneration but not in other tauopathies. J Neuropathol Exp Neurol. 2008;67(6):555–64.
Josephs KA, Dickson DW, Tosakulwong N, Weigand SD, Murray ME, Petrucelli L, et al. Rates of hippocampal atrophy and presence of post-mortem TDP-43 in patients with Alzheimer’s disease: a longitudinal retrospective study. Lancet Neurol. 2017;16(11):917–24.
Robinson JL, Porta S, Garrett FG, Zhang P, Xie SX, Suh E, et al. Limbic-predominant age-related TDP-43 encephalopathy differs from frontotemporal lobar degeneration. Brain. 2020;143(9):2844–57.
Besser LM, Teylan MA, Nelson PT. Limbic predominant age-related TDP-43 Encephalopathy (LATE): Clinical and Neuropathological Associations. J Neuropathol Exp Neurol. 2020;79(3):305–13.
Nelson PT, Dickson DW, Trojanowski JQ, Jack CR, Boyle PA, Arfanakis K, et al. Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report. Brain. 2019;142(6):1503–27.
Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351(3):602–11.
Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130–3.
Bejanin A, Murray ME, Martin P, Botha H, Tosakulwong N, Schwarz CG, et al. Antemortem volume loss mirrors TDP-43 staging in older adults with non-frontotemporal lobar degeneration. Brain. 2019;142(11):3621–35.
Wennberg AM, Whitwell JL, Tosakulwong N, Weigand SD, Murray ME, Machulda MM, et al. The influence of tau, amyloid, alpha-synuclein, TDP-43, and vascular pathology in clinically normal elderly individuals. Neurobiol Aging. 2019;77:26–36.
Buciuc M, Botha H, Murray ME, Schwarz CG, Senjem ML, Jones DT et al. Utility of FDG-PET in diagnosis of Alzheimer-related TDP-43-proteinopathy. Neurology. 2020:In press.
Ling SC, Polymenidou M, Cleveland DW. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron. 2013;79(3):416–38.
Polymenidou M, Lagier-Tourenne C, Hutt KR, Huelga SC, Moran J, Liang TY, et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat Neurosci. 2011;14(4):459–68.
Ling JP, Pletnikova O, Troncoso JC, Wong PC. TDP-43 repression of nonconserved cryptic exons is compromised in ALS-FTD. Science. 2015;349(6248):650–5.
Hatanpaa KJ, Bigio EH, Cairns NJ, Womack KB, Weintraub S, Morris JC, et al. TAR DNA-binding protein 43 immunohistochemistry reveals extensive neuritic pathology in FTLD-U: a midwest-southwest consortium for FTLD study. J Neuropathol Exp Neurol. 2008;67(4):271–9.
Josephs KA, Murray ME, Whitwell JL, Parisi JE, Petrucelli L, Jack CR, et al. Staging TDP-43 pathology in Alzheimer’s disease. Acta Neuropathol. 2014;127(3):441–50.
Tan RH, Kril JJ, Fatima M, McGeachie A, McCann H, Shepherd C, et al. TDP-43 proteinopathies: pathological identification of brain regions differentiating clinical phenotypes. Brain. 2015;138(Pt 10):3110–22.
Josephs KA, Murray ME, Whitwell JL, Tosakulwong N, Weigand SD, Petrucelli L, et al. Updated TDP-43 in Alzheimer’s disease staging scheme. Acta Neuropathol. 2016;131(4):571–85.
Brettschneider J, Del Tredici K, Irwin DJ, Grossman M, Robinson JL, Toledo JB, et al. Sequential distribution of pTDP-43 pathology in behavioral variant frontotemporal dementia (bvFTD). Acta Neuropathol. 2014;127(3):423–39.
Josephs KA, Murray ME, Tosakulwong N, Weigand SD, Serie AM, Perkerson RB, et al. Pathological, imaging and genetic characteristics support the existence of distinct TDP-43 types in non-FTLD brains. Acta Neuropathol. 2019;137(2):227–38.
Tomé SO, Vandenberghe R, Ospitalieri S, Van Schoor E, Tousseyn T, Otto M, et al. Distinct molecular patterns of TDP-43 pathology in Alzheimer’s disease: relationship with clinical phenotypes. Acta Neuropathol Commun. 2020;8(1):61.
Jeong YH, Ling JP, Lin SZ, Donde AN, Braunstein KE, Majounie E, et al. Tdp-43 cryptic exons are highly variable between cell types. Mol Neurodegener. 2017;12(1):13.
Sun M, Bell W, LaClair KD, Ling JP, Han H, Kageyama Y, et al. Cryptic exon incorporation occurs in Alzheimer’s brain lacking TDP-43 inclusion but exhibiting nuclear clearance of TDP-43. Acta Neuropathol. 2017;133(6):923–31.
Ma XR, Prudencio M, Koike Y, Vatsavayai SC, Kim G, Harbinski F, et al. TDP-43 represses cryptic exon inclusion in the FTD-ALS gene UNC13A. Nature. 2022;603(7899):124–30.
Brown AL, Wilkins OG, Keuss MJ, Hill SE, Zanovello M, Lee WC, et al. TDP-43 loss and ALS-risk SNPs drive mis-splicing and depletion of UNC13A. Nature. 2022;603(7899):131–7.
Melamed Z, Lopez-Erauskin J, Baughn MW, Zhang O, Drenner K, Sun Y, et al. Premature polyadenylation-mediated loss of stathmin-2 is a hallmark of TDP-43-dependent neurodegeneration. Nat Neurosci. 2019;22(2):180–90.
Klim JR, Williams LA, Limone F, Guerra San Juan I, Davis-Dusenbery BN, Mordes DA, et al. ALS-implicated protein TDP-43 sustains levels of STMN2, a mediator of motor neuron growth and repair. Nat Neurosci. 2019;22(2):167–79.
Humphrey J, Emmett W, Fratta P, Isaacs AM, Plagnol V. Quantitative analysis of cryptic splicing associated with TDP-43 depletion. BMC Med Genomics. 2017;10(1):38.
Seddighi S, Qi YA, Brown AL, Wilkins OG, Bereda C, Belair C et al. Mis-spliced transcripts generate de novo proteins in TDP-43-related ALS/FTD. bioRxiv. 2023.
Prudencio M, Humphrey J, Pickles S, Brown AL, Hill SE, Kachergus JM et al. Truncated stathmin-2 is a marker of TDP-43 pathology in frontotemporal dementia. J Clin Invest. 2020.
Krus KL, Strickland A, Yamada Y, Devault L, Schmidt RE, Bloom AJ, et al. Loss of Stathmin-2, a hallmark of TDP-43-associated ALS, causes motor neuropathy. Cell Rep. 2022;39(13):111001.
Graf ER, Heerssen HM, Wright CM, Davis GW, DiAntonio A. Stathmin is required for stability of the Drosophila neuromuscular junction. J Neurosci. 2011;31(42):15026–34.
Guerra San Juan I, Nash LA, Smith KS, Leyton-Jaimes MF, Qian M, Klim JR, et al. Loss of mouse Stmn2 function causes motor neuropathy. Neuron. 2022;110(10):1671–88. e6.
Duncan JE, Lytle NK, Zuniga A, Goldstein LS. The Microtubule Regulatory protein stathmin is required to maintain the Integrity of Axonal Microtubules in Drosophila. PLoS ONE. 2013;8(6):e68324.
Irwin KE, Jasin P, Braunstein KE, Sinha I, Bowden KD, Moghekar A et al. A fluid biomarker reveals loss of TDP-43 splicing repression in pre-symptomatic ALS. bioRxiv. 2023.
Pickles S, Gendron TF, Koike Y, Yue M, Song Y, Kachergus JM, et al. Evidence of cerebellar TDP-43 loss of function in FTLD-TDP. Acta Neuropathol Commun. 2022;10(1):107.
Baughn MW, Melamed Z, Lopez-Erauskin J, Beccari MS, Ling K, Zuberi A, et al. Mechanism of STMN2 cryptic splice-polyadenylation and its correction for TDP-43 proteinopathies. Science. 2023;379(6637):1140–9.
Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82(4):239–59.
Murray ME, Graff-Radford NR, Ross OA, Petersen RC, Duara R, Dickson DW. Neuropathologically defined subtypes of Alzheimer’s disease with distinct clinical characteristics: a retrospective study. Lancet Neurol. 2011;10(9):785–96.
Prudencio M, Gonzales PK, Cook CN, Gendron TF, Daughrity LM, Song Y, et al. Repetitive element transcripts are elevated in the brain of C9orf72 ALS/FTLD patients. Hum Mol Genet. 2017;26(17):3421–31.
Brettschneider J, Del Tredici K, Toledo JB, Robinson JL, Irwin DJ, Grossman M, et al. Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann Neurol. 2013;74(1):20–38.
Lee EB, Porta S, Michael Baer G, Xu Y, Suh E, Kwong LK, et al. Expansion of the classification of FTLD-TDP: distinct pathology associated with rapidly progressive frontotemporal degeneration. Acta Neuropathol. 2017;134(1):65–78.
Nana AL, Sidhu M, Gaus SE, Hwang JL, Li L, Park Y, et al. Neurons selectively targeted in frontotemporal dementia reveal early stage TDP-43 pathobiology. Acta Neuropathol. 2019;137(1):27–46.
LaClair KD, Donde A, Ling JP, Jeong YH, Chhabra R, Martin LJ, et al. Depletion of TDP-43 decreases fibril and plaque beta-amyloid and exacerbates neurodegeneration in an Alzheimer’s mouse model. Acta Neuropathol. 2016;132(6):859–73.
Bieche I, Maucuer A, Laurendeau I, Lachkar S, Spano AJ, Frankfurter A, et al. Expression of stathmin family genes in human tissues: non-neural-restricted expression for SCLIP. Genomics. 2003;81(4):400–10.
Shin JE, Geisler S, DiAntonio A. Dynamic regulation of SCG10 in regenerating axons after injury. Exp Neurol. 2014;252:1–11.
Shin JE, Miller BR, Babetto E, Cho Y, Sasaki Y, Qayum S, et al. SCG10 is a JNK target in the axonal degeneration pathway. Proc Natl Acad Sci U S A. 2012;109(52):E3696–705.
Morii H, Shiraishi-Yamaguchi Y, Mori N. SCG10, a microtubule destabilizing factor, stimulates the neurite outgrowth by modulating microtubule dynamics in rat hippocampal primary cultured neurons. J Neurobiol. 2006;66(10):1101–14.
Jin LW, Masliah E, Iimoto D, Deteresa R, Mallory M, Sundsmo M, et al. Neurofibrillary tangle-associated alteration of stathmin in Alzheimer’s disease. Neurobiol Aging. 1996;17(3):331–41.
Okazaki T, Wang H, Masliah E, Cao M, Johnson SA, Sundsmo M, et al. SCG10, a neuron-specific growth-associated protein in Alzheimer’s disease. Neurobiol Aging. 1995;16(6):883–94.
van Es MA, Veldink JH, Saris CG, Blauw HM, van Vught PW, Birve A, et al. Genome-wide association study identifies 19p13.3 (UNC13A) and 9p21.2 as susceptibility loci for sporadic amyotrophic lateral sclerosis. Nat Genet. 2009;41(10):1083–7.
Diekstra FP, van Vught PW, van Rheenen W, Koppers M, Pasterkamp RJ, van Es MA, et al. UNC13A is a modifier of survival in amyotrophic lateral sclerosis. Neurobiol Aging. 2012;33(3):630e3–8.
Koike Y, Pickles S, Estades Ayuso V, Jansen-West K, Qi YA, Li Z, et al. TDP-43 and other hnRNPs regulate cryptic exon inclusion of a key ALS/FTD risk gene, UNC13A. PLoS Biol. 2023;21(3):e3002028.
Hartlage-Rubsamen M, Waniek A, Rossner S. Munc13 genotype regulates secretory amyloid precursor protein processing via postsynaptic glutamate receptors. Int J Dev Neurosci. 2013;31(1):36–45.
Rossner S, Fuchsbrunner K, Lange-Dohna C, Hartlage-Rubsamen M, Bigl V, Betz A, et al. Munc13-1-mediated vesicle priming contributes to secretory amyloid precursor protein processing. J Biol Chem. 2004;279(27):27841–4.
Barthet G, Jorda-Siquier T, Rumi-Masante J, Bernadou F, Muller U, Mulle C. Presenilin-mediated cleavage of APP regulates synaptotagmin-7 and presynaptic plasticity. Nat Commun. 2018;9(1):4780.
Habif M, Do Carmo S, Baez MV, Colettis NC, Cercato MC, Salas DA, et al. Early long-term memory impairment and changes in the expression of synaptic Plasticity-Associated genes, in the McGill-R-Thy1-APP rat model of Alzheimer’s-Like Brain Amyloidosis. Front Aging Neurosci. 2020;12:585873.
Surti TS, Jan LY. A potassium channel, the M-channel, as a therapeutic target. Curr Opin Investig Drugs. 2005;6(7):704–11.
Bonham LW, Evans DS, Liu Y, Cummings SR, Yaffe K, Yokoyama JS. Neurotransmitter pathway genes in cognitive decline during aging: evidence for GNG4 and KCNQ2 genes. Am J Alzheimers Dis Other Demen. 2018;33(3):153–65.
Song MK, Cui YY, Zhang WW, Zhu L, Lu Y, Chen HZ. The facilitating effect of systemic administration of Kv7/M channel blocker XE991 on LTP induction in the hippocampal CA1 area independent of muscarinic activation. Neurosci Lett. 2009;461(1):25–9.
Duran-Gonzalez J, Michi ED, Elorza B, Perez-Cordova MG, Pacheco-Otalora LF, Touhami A, et al. Amyloid beta peptides modify the expression of antioxidant repair enzymes and a potassium channel in the septohippocampal system. Neurobiol Aging. 2013;34(8):2071–6.
Mayordomo-Cava J, Yajeya J, Navarro-Lopez JD, Jimenez-Diaz L. Amyloid-beta(25–35) modulates the expression of GirK and KCNQ Channel genes in the Hippocampus. PLoS ONE. 2015;10(7):e0134385.
Acknowledgements
We thank Wanhao Chi and Evangelos Kiskinis for designing and sharing the KCNQ2 qRT-PCR primers. We also thank all the patients and their families for their contribution to this study.
Funding
This work was supported by the National Institutes of Health (U54NS123743 to LP & MP; RF1NS120992 to MP and KAJ; R35NS097273 to LP; P01NS084974 to LP; R01AG37491 to K.J.; P30AG062677 to RCP and LP; U19AG063911 to BFB and LP), Target ALS (to MP), the Robert Packard Center for ALS Research at Johns Hopkins University (to LP), a BrightFocus ADR Grant (A2020279F to SP).
Author information
Authors and Affiliations
Contributions
VEA, KAJ, DWD, LP and MP conceptualized and designed the study. VEA and MP carried out the investigation. VEA, SP, MY, KJ-W, YS, JGB and MP developed the laboratory work. BR, MD, NRG, BFB, DSK, RCP, DWD, KAJ and LP provided resources. VEA and MP performed the data curation, formal analysis, and wrote the original manuscript draft, which was reviewed by all co-authors and further edited by VEA, SP, TT, DSK, DWD, KAJ, and MP. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Human postmortem brain tissues from amygdala, hippocampus, and frontal cortex were provided by the Mayo Clinic Florida Brain Bank. Diagnosis was independently ascertained by trained neurologists and neuropathologists upon neurological and pathological examinations, respectively. All participants, or the next of kin, provided written informed consent, and all protocols were reviewed and approved by the Mayo Clinic Institutional Review Board and Ethics Committee.
Consent for publication
All authors have reviewed the final manuscript and consent to publication.
Competing interests
BFB receives institutional research grant support from Alector, Biogen, Transposon, Cognition Therapeutics, and GE Healthcare. BFB receives honorarium for SAB activities for the Tau Consortium. LP is a consultant for Expansion Therapeutics. DSK serves on a Data Safety Monitoring Board for the Dominantly Inherited Alzheimer Network Treatment Unit study. DSK served on a Data Safety monitoring Board for a tau therapeutic for Biogen (until 2021) but received no personal compensation. DSK is an investigator in clinical trials sponsored by Biogen, Lilly Pharmaceuticals, and the University of Southern California. DSK has served as a consultant for Roche, Samus Therapeutics, Magellan Health, Biovie and Alzeca Biosciences but receives no personal compensation. DSK attended an Eisai advisory board meeting for lecanemab on December 2, 2022, but received no compensation. DSK receives funding from the NIH. All other authors declare no disclosures or conflicts of interest related to the content of the article.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Estades Ayuso, V., Pickles, S., Todd, T. et al. TDP-43-regulated cryptic RNAs accumulate in Alzheimer’s disease brains. Mol Neurodegeneration 18, 57 (2023). https://doi.org/10.1186/s13024-023-00646-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13024-023-00646-z