
Abstract
Comparisons of MHC gene content and diversity among closely related species can provide insights into the evolutionary mechanisms shaping immune system variation. After chimpanzees and bonobos, gorillas are humans’ closest living relatives; but in contrast, relatively little is known about the structure and variation of gorilla MHC class I genes (Gogo). Here, we combined long-range amplifications and long-read sequencing technology to analyze full-length MHC class I genes in 35 gorillas. We obtained 50 full-length genomic sequences corresponding to 15 Gogo-A alleles, 4 Gogo-Oko alleles, 21 Gogo-B alleles, and 10 Gogo-C alleles including 19 novel coding region sequences. We identified two previously undetected MHC class I genes related to Gogo-A and Gogo-B, respectively, thereby illustrating the potential of this approach for efficient and highly accurate MHC genotyping. Consistent with their phylogenetic position within the hominid family, individual gorilla MHC haplotypes share characteristics with humans and chimpanzees as well as orangutans suggesting a complex history of the MHC class I genes in humans and the great apes. However, the overall MHC class I diversity appears to be low further supporting the hypothesis that gorillas might have experienced a reduction of their MHC repertoire.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Molecules encoded by the MHC class I genes play an important role in the defense against intracellular pathogens (Klein and Figueroa 1986). Through the recognition and presentation of antigens, MHC class I molecules function as stimulatory ligands for lymphocytes of the adaptive and innate immune system. Cytotoxic CD8+ T cells recognize antigens that are presented by MHC class I molecules on the cell surface while some MHC class I allotypes, in addition, interact with killer cell immunoglobulin-like receptors (KIR receptors) that are primarily expressed on natural killer cells (NK cells) and regulate the inhibition and activation of NK cell responses (Parham and Ohta 1996; Lanier 2005). Due to host-pathogen interactions, the genes encoding MHC class I molecules are subject to rapid evolution (Hedrick 1994; Hughes and Hughes 1995). High-sequence polymorphism of MHC class I genes provides diversity in antigen-binding repertoires while recombination between MHC class I genes facilitates the generation of novel specificities (Ohta 1983; Jakobsen et al. 1998). In humans and other nonhuman primates, the MHC class I multigene family has diversified through recurrent duplications and deletions leading to species-specific patterns of intra- and interlocus variability (Adams and Parham 2001; Kulski et al. 2002; Kelley et al. 2005). Thus, interspecies comparisons can provide important insights into the evolution of MHC class I genes and possibly the resistance and susceptibility to infectious diseases (Sommer 2005; Shiina et al. 2006).
Sharing a common ancestor approximately 10 million years ago, gorillas are humans’ closest living relatives after chimpanzees and bonobos (Ruvolo et al. 1991; Scally et al. 2012). However, in contrast to humans and chimpanzees, surprisingly little is known about the structure and variation of MHC genes in gorillas (Hans et al. 2015). In general, it is presumed that gorillas have a MHC class I region similar to humans as concluded from the presence of gorilla orthologues (Gogo-A, -B, and -C) of all classical human MHC class I genes (HLA-A, -B, -C) (Venditti et al. 1996; Lawlor et al. 1991; Watkins et al. 1991; Adams and Parham 2001). The previous sampling of Gogo class I alleles derived from only four individuals showed that Gogo-A alleles segregate only into one of the two ancient lineages of HLA-A, the A2 lineage (Lawlor et al. 1991; Adams and Parham 2001). However, among the five identified complementary DNA (cDNA) sequences related to HLA-A, two appeared particularly divergent (Lawlor et al. 1991; Adams and Parham 2001). The allele Gogo-A*05:01 shares remarkable sequence similarity with the human pseudogene HLA-Y (also designated as HLA-BEL, -COQ, and -DEL) and was speculated to represent an ancestral A locus allele predating the divergence of the gorilla and human MHC (Lawlor et al. 1991; Williams et al. 1999; Coquillard et al. 2004; Marsh et al. 2010). The Gogo-A3 or -Oko allele, however, shares sequence homology with the human pseudogene H and other classical A locus alleles but until recently, it was unclear whether Gogo-Oko is a divergent A locus allele or a separate gene (Lawlor et al. 1991; Watkins et al. 1991; Adams and Parham 2001). Comparative analysis of a single gorilla MHC haplotype showed that Gogo-Oko is syntenic to the chimpanzee A-related locus Patr-AL (Gleimer et al. 2011). Patr-AL is present on only half of the surveyed chimpanzee MHC haplotypes and is in strong linkage disequilibrium with Patr-A, the chimpanzee orthologue of HLA-A (Adams et al. 2001; Geller et al. 2002). In contrast, the gorilla MHC haplotype with Gogo-Oko lacks the genomic block containing the A locus (Gleimer et al. 2011). Thus, gorillas possess an additional and presumably functional A-related locus which is characterized by an unusual haplotype structure but little is known about its frequency and variation (Lawlor et al. 1991; Watkins et al. 1991). Nevertheless, that certain Gogo-A alleles are orthologous to HLA-A suggests that some gorilla MHC haplotypes have retained the genomic block with the A locus (Gleimer et al. 2011). In sum, there are indications for a complex history of the A and A-related genes in gorillas and their closest relatives.
In contrast, it is presumed that, similar to humans and chimpanzees, gorillas possess a single MHC-B and MHC-C gene, respectively, whereas orangutan haplotypes have multiple copies of MHC-B and some lack the MHC-C (Chen et al. 1992; Adams et al. 1999; Adams and Parham 2001; de Groot et al. 2016). For Gogo-B, the gorilla orthologue of HLA-B, 11 alleles have been characterized among 18 individuals which implies that Gogo-B is, like its human and chimpanzee equivalent, the most polymorphic of the gorilla MHC class I genes (Lawlor et al. 1991; Urvater et al. 2001; Martínez-Laso et al. 2006; Abi-Rached et al. 2011). Therefore, analysis of additional gorillas will likely increase the number of alleles identified at this locus. As with MHC-B, it appears that the MHC-C gene is present on all MHC haplotypes in humans and the African great apes and accordingly, a total of seven Gogo-C alleles have been identified among seven individuals (Lawlor et al. 1991; Urvater et al. 2001). However, to gain a more comprehensive understanding of the gorilla MHC class I diversity and the evolution of these loci in great apes and humans, characterization of more individuals is needed.
Despite their importance, MHC class I genes are rarely characterized comprehensively which at least in part is due to the technical difficulty of unambiguously determining the sequences of both alleles using conventional sequencing approaches. Indeed, the majority of Gogo class I alleles have been described from cDNA sequences. Emerging long-read sequencing technologies have the ability to reliably characterize full-length MHC class I genes, as has recently been shown in humans (Mayor et al. 2015). Similarly, we here applied long-range PCR (LR-PCR) to amplify full-length gorilla MHC class I genes from high-quality genomic DNA comprising complete coding region sequences and intervening introns. This approach has several advantages. First, sequencing of polymorphism within non-coding regions increases the level of detail at which an allele is characterized. Thus, the higher resolution enables the recognition of heterozygous genotypes if individual alleles are identical in coding region sequences. Second, amplification from genomic DNA allows the characterization of low- or non-expressed alleles which are undetectable at the cDNA level. Third, LR-PCR primers can be placed in conserved regions outside the genes thereby allowing the characterization of the entire allelic variation. Here, we use this approach to characterize 35 gorillas including a large number of wild-born individuals to achieve a more comprehensive description of MHC class I variation in gorillas with comparison to data from human and chimpanzees.
Material and methods
Sample collection and DNA extraction
Taxonomically, gorillas are classified into two species: the western gorilla (Gorilla gorilla) and the eastern gorilla (Gorilla beringei), each of which have been further subdivided into two subspecies. The western species is comprised of western lowland gorillas (Gorilla gorilla gorilla) and Cross River gorillas (Gorilla gorilla diehli) while mountain gorillas (Gorilla beringei beringei) and eastern lowland gorillas (Gorilla beringei graueri) comprise the eastern species (Groves 2001). Our study consisted of samples from 35 captive individuals including one eastern lowland gorilla, one Cross River gorilla, and 33 western lowland gorillas. As our aim was to assess MHC class I gene variation among natural populations, our sampling focused on wild-born individuals (n = 25) as well as captive-born individuals of the first and second generation with at least one wild-born parent (n = 9) (Online Resources 1 and 2). Based on reported capture locations, we could broadly infer geographical origins of most wild-born gorillas (Online Resource 1). Genomic DNA was extracted from 17 whole blood, 1 buffy coat, and 5 tissue samples using either the Gentra Puregene Blood Kit (QIAGEN, Hilden, Germany) or the DNeasy Blood & Tissue Kit (QIAGEN) according to the manufacturer’s instructions. In addition, 11 pre-extracted whole blood samples collected as part of a previous study were purified using the QIAamp DNA Mini Kit (QIAGEN) (see for details Thalmann et al. 2007). Furthermore, commercially available genomic DNA from the gorilla cell line “EB (JC)” was purchased from the European Collection of Cell Cultures (ECACC) (Public Health England, Porton Down, Salisbury, UK) (Online Resource 1). Quantities of DNA templates were estimated through spectrophotometric measurements using a NanoDrop ND-1000 (Thermo Scientific, Waltham, MA, USA) and concentrations were adjusted to approximately 25 ng/μL for the LR-PCR amplifications. Collection of samples was conducted under the supervision of ethical committees and, if required, CITES permissions (Convention on International Trade in Endangered Species of Wild Fauna and Flora) were obtained.
Primer design, LR-PCR, and sequencing of MHC class I genes
To design gene-specific primers, we analyzed all available MHC class I gene sequences from the gorilla and its closest relatives, human, chimpanzee, and bonobo. Following the construction of multiple alignments using ClustalW as implemented in BioEdit version 7.2.0 (Hall 1999), primers were manually placed in interspecies conserved regions and where necessary, degenerate bases were included to ensure the amplification of the complete allelic variation of each gene. Following the equation given in SantaLucia (1998), primers were chosen to have melting temperatures of at least 64 °C to enhance reaction specificity. Finally, primers were checked in silico for potential self-complementary and hairpin formation using the program OligoCalc (Kibbe 2007). Primers were designed to encompass entire coding sequences with the forward primer located in the promoter-enhancer region and the reverse primer located in the 3′-untranslated region. Due to high sequence similarity outside of the coding regions, a universal primer pair was designed to nonspecifically amplify both the gorilla MHC-A and -Oko genes. Sequences of primers used in the present study are given in Online Resource 3.
Individual LR-PCRs (gorilla MHC-A/Oko, -B, and -C) were performed in a final volume of 50 μL consisting of 1× Crimson LongAmp Taq Reaction Buffer, 100 μM dNTPs, 0.2 μM of each primer, 0.1 unit Crimson LongAmp Taq Polymerase (New England Biolabs, Frankfurt am Main, Germany), and 2 μL of DNA template. PCR amplifications consisted of a two-step PCR program with an initial denaturation at 94 °C for 2 min, followed by 40 cycles of 94 °C for 20 s and 68 °C for 9 min and a final extension at 68 °C for 15 min. After confirmation of successful amplification by gel electrophoresis, amplicons were individually purified using 1.0× ratio of Agencourt AMPure XP beads (Beckman Coulter, Krefeld, Germany) according to the manufacturer’s instructions, and concentrations were measured with a NanoDrop ND-1000 spectrophotometer (Thermo Scientific). The three amplicons of each individual were pooled in equimolar ratios and used to construct single-molecule real-time (SMRT) cell libraries using PacBio barcoded adapters for multiplex sequencing following the manufacturer’s protocol (Pacific Biosciences, Menlo Park, CA, USA). After blunt-end ligation of a symmetric 16-bp barcoded adapter, up to 10 uniquely barcoded amplicon pools were multiplexed prior to two DNA damage repair steps and exonuclease treatment. The quantity and quality of each library was checked using an Agilent DNA 7500 chip and the 2100 Bionanalyzer (Agilent Technologies, Santa Clara, CA, USA). The DNA/Polymerase Binding kit P6 v2 (Pacific Biosciences) was used to bind polymerases to templates followed by using the MagBead kit (Pacific Biosciences) for uniform loading of SMRT cells. Libraries with a concentration of 40 pM were sequenced on the PacBio RS II instrument (Pacific Biosciences) using the C4 chemistry, stage start, and a movie collection time of 240 min. A total of nine SMRT cells were sequenced yielding 654,289 polymerase reads (mean per SMRT cell 79,464 ± 14,220). The long amplicon analysis (LAA) as implemented in the SMRT Analysis (version 2.3.0) was used to determine phased consensus sequences followed by manual trimming of primer sequences. To resolve individual allele ambiguities between replicates, we applied the minor variant analysis implemented in the same software package.
Genotype estimation and validation
As detailed below, we validated individual MHC genotypes through (i) the comparison of consensus sequences obtained from two independent amplifications; (ii) the analysis of four individuals with completely or partially known genotypes from previous studies; and (iii) the analysis of allelic inheritance patterns among related individuals, i.e., one parent-offspring trio and one father-offspring dyad. Full-length genomic coding sequences were submitted to the MHC-IPD NHP database and given official allele designations by the nonhuman primate MHC nomenclature committee (Maccari et al. 2017). In this context, the MHC of a species is abbreviated by a four-letter code which corresponds to its scientific name. Specifically, the MHC of the western gorilla (G. gorilla) is defined by Gogo, whereas Gobe refers to the MHC of the eastern gorilla (G. beringei) (Klein et al. 1990; de Groot et al. 2012). As listed in Table 1, the majority of alleles identified in the eastern lowland gorilla were also found in western gorillas. However, please note that allele designations differ between the two gorilla species.
As mentioned above, LR-PCR amplifications and sequencing were performed twice for each gorilla sample to validate generated sequences. However, after consensus clustering of barcoded subreads, 5 of 35 samples showed low coverage for gorilla Gogo-A/Oko amplicons in both replicates, probably due to unequal pooling of LR-PCR products and thus were repeated. The total number of subreads per sample and replicate ranged from 1016 to 3172, with an average of 2165 ± 518. More specifically, total numbers of subreads per sample and replicate ranged from 264 to 1414 (mean 801 ± 273) for the gorilla Gogo-A/Oko amplicons, from 206 to 1464 (751 ± 278) for the Gogo-B amplicons, and from 500 to 1000 (613 ± 198) for the Gogo-C amplicons. Subread coverage of consensus sequences, which by default cannot exceed 500, was on average 270 ± 158 (range 25–500), 370 ± 138 (51–500), and 447 ± 97 (225–500) for Gogo-A/Oko, -B, and -C, respectively (ConsensusTools v2.3.0 Documentation 2014). The mean expected accuracy of consensus sequences based on the consensus-calling algorithm Quiver was 99.958% (±0.102%, range 98.132–99.994%).
Overall, 424 of 446 high-quality consensus sequences were completely identical between the two LR-PCR reactions. The remaining 22 sequences differed in homopolymer stretches or had highly similar alleles but the variants could be resolved. In detail, 16 Gogo-B and -Oko consensus sequences obtained among the replicates of seven samples were nearly identical except for inconsistencies in homopolymer regions, a common issue of most sequencing technologies (Mayor et al. 2015). Although it is unclear whether these inaccuracies reflect PCR-mediated errors due to “polymerase slippage” or are a result of the SMRT DNA sequencing technology, the affected bases were located in the 3′ untranslated region and hence had no impact on further analyses (but see Ross et al. 2013). The remaining six inconsistent sequences were observed among the replicates of two samples and consisted of three highly similar Gogo-B consensus sequences, each of which differed from one another at a single base-pair position. Regarding the first sample (“Carlos”), a deletion was observed in the consensus sequence of the first replicate whereas consensus sequences of the second replicate showed two different bases at that specific position. Translation of the consensus sequence with the deletion resulted in a frameshift and premature termination. In contrast, the consensus sequences with different bases translated into two different amino acid sequences and identical coding sequences containing each of the two base variants were found in other samples, including its offspring “Kwan”, thus indicating two alleles. Similarly, consensus sequences of the second sample (“Binti Jua”) differed at one position in an intronic region, with a deletion observed in the consensus sequence of the first replicate and consensus sequences of the second replicate containing two base variants. Only sequences containing each of the two bases were observed in other samples, thus indicating again two different alleles. These inaccuracies probably occurred as a result of the high homogeneity shared between the two sequences (personal communication, Philip Lobb from Pacific Biosciences). If there is an insufficient number of reads, consensus and variant calling performed by the LAA cannot differentiate between the two alleles to segregate reads into multiple haplotypes (ConsensusTools v2.3.0 Documentation 2014). However, to further resolve discordant alleles between replicates of these two samples, we used a reference-based analysis method which enables the detection and quantification of minor variants in a heterogeneous sample. The minor variant analysis verified the presence of two alleles differing by a single base-pair in each of the replicates of the two samples, respectively. Moreover, we did not detect pronounced deviations between replicates from both the frequency of each variant (allele frequencies in first and second replicate of “Carlos”: 0.439/0.561 and 0.419/0.581; and “Binti Jua”: 0.319/0.681 and 0.310/0.690) and its respective depth of coverage (allele coverage in first and second replicate of “Carlos”: 395/504 and 392/544; and “Binti Jua”: 696/1480 and 689/1538) which demonstrates that these inaccuracies are not due to unequal amplification efficiencies and/or allelic dropout but are a result of the LAA. However, we did not detect any differences between replicates of the other samples. Overall, we therefore conclude that obtained consensus sequences are highly reproducible and therefore accurately reflect individual MHC class I sequences.
To further validate our approach, we included three gorilla individuals with previously described MHC class I genotypes: “Banga”, Gogo-A*04:01, -B*01:01, -B*01:03, -C*01:01, and -C*02:01 (Lawlor et al. 1991); “Oko”, Gogo-Oko, -B*02:01, and -C*02:02 (Lawlor et al. 1991); “Beta”, Gogo-A*04:01, -A*05:01, -B*01:01, -B*03:01, -C*01:01, and -C*01:03 (Urvater et al. 2001); as well as individuals with the following full-length genomic Gogo-B alleles: “Beta”, Gogo-B*07:01; “Oko”, Gogo-B*02:01; and “Machi”, Gogo-B*04:01 (Abi-Rached et al. 2011). Comparison of our generated sequences to previously described cDNAs or full-length genomic sequences revealed complete identity, as summarized in Table 2. However, for the gorilla “Banga” we identified the allele Gogo-C*02:03 which was previously mischaracterized as Gogo-C*02:01 (see below).
As a third validation step, we confirmed sharing of identical alleles within one parent-offspring trio and one father-offspring dyad (Table 2). Due to the analysis of one parent-offspring trio which included Banga’s offspring “Rok” and its sire “Ozoum”, we were able to confirm the Gogo-C*02:03 allele for “Banga” (Table 2). Taken together, we are confident that our LR-PCR approach in combination with the SMRT sequencing technology enables the reliable estimation of the entire MHC class I gene variation in gorillas.
Phylogenetic analyses
Phylogenetic analyses were performed using full-length coding region sequences of human, chimpanzee, bonobo, gorilla, and orangutan selected to represent the range of MHC class I variation in each species. Sequences were aligned using MAFFT (Katoh et al. 2002) and phylogenies were reconstructed using the neighbor-joining (NJ) method with the Tamura-Nei model and 500 bootstrap replicates as implemented in MEGA version 6 (Tamura et al. 2013). However, inference of orthologous relationships between MHC class I genes can be confounded by inter- and intralocus recombination. Thus, we combined domain-by-domain phylogenetic analyses with recombination detection methods to examine relationships among MHC class I genes of humans and great apes (Gleimer et al. 2011). Available genomic full-length sequences were aligned using MAFFT and manually edited. Sequence alignments were investigated for patterns of recombination with the program RDP4 which utilizes a variety of methods to identify and detect recombination events (Martin et al. 2015). Based on the results obtained from the RDP4 analysis, MHC class I sequence alignments were divided into various segments, each of which was validated through NJ analyses using the parameters described above.
Interspecies sequence diversity analyses
To assess variation of MHC class I genes in gorillas, we compared sequence diversity measures of the 33 unrelated gorillas to a similar-sized sample of chimpanzees and humans, respectively. Individual sequence data of 24 chimpanzees were obtained from the study of Adams et al. (2000) which included 19 western chimpanzees (Pan troglodytes verus), 3 central chimpanzees (Pan troglodytes troglodytes) and two eastern chimpanzees (Pan troglodytes schweinfurthii). Genotypes of 30 unrelated Yoruba (YRI) individuals from Ibadan, Nigeria, characterized at the classical HLA genes as part of the HapMap project, were taken from the study by de Bakker et al. (2006). Corresponding HLA allele sequences were downloaded from the IPD-IMGT/HLA database (Robinson et al. 2015). For each locus separately, pairwise allelic differences of full-length coding region sequences were calculated using MEGA. For the calculation of nucleotide diversity (π), number of variable sites, mean nonsynonymous (Ka) and synonymous (Ks) substitution rates within antigen-binding sites (ABS), we used the program DnaSP version 5.10 (Librado and Rozas 2009). Fisher’s exact test was applied to test the null hypothesis of equal rates of synonymous and nonsynonymous changes.
Results
We used high-quality genomic DNAs from 35 gorillas as templates for individual LR-PCR amplifications of MHC class I genes with primers designed to encompass complete coding region sequences including intervening introns. LR-PCR products were multiplexed and sequenced using PacBio long-read sequencing technology. A total of 50 full-length MHC class I alleles corresponding to 15 Gogo-A alleles, 4 Gogo-Oko, 21 Gogo-B alleles, and 10 Gogo-C alleles were identified among the 35 gorillas analyzed. Comparison with previously described Gogo class I alleles showed that 19 coding region sequences were novel (Table 1 and Fig. 1). Identification of up to four Gogo-A alleles and three Gogo-B alleles in certain gorillas indicates the presence of two Gogo-A and -B genes, respectively (Table 2). Evolutionary relationships of MHC class I genes from gorillas and the other great apes, including humans, were phylogenetically investigated using complete coding region sequences selected to represent the species-specific range of allelic structure at each locus (Figs. 2 and 3). To further examine the evolutionary relationship of the newly identified Gogo class I genes, we extended phylogenetic analyses using available full-length genomic MHC class I sequences of human, chimpanzee, and orangutan. Interspecies comparison of sequence diversity measures allowed us to assess the variation at MHC class I genes in gorillas (Fig. 4 and Table 3). Results for each locus will be discussed in turn.

Alignment of amino acid sequences of the alpha-1 and -2 domains of gorilla class I sequences. Identity to consensus sequence is denoted by a dash. For differences from the consensus sequence, the amino acid is shown. Highlighted in bold are previously unreported alleles. Gogo, Gorilla gorilla, Gobe, Gorilla beringei

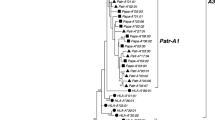
Phylogenetic tree constructed from full-length coding region sequences of MHC-A and -A-related genes. Relevant bootstrap values (≥70%) are shown. Previously described gorilla alleles identified in the present study are highlighted in bold. Newly identified alleles are underlined. (A2) and (A3) indicate the two phylogenetic A lineages, respectively. HLA human, Patr Pan troglodytes, Papa Pan paniscus, Gogo Gorilla gorilla, Popy Pongo pygmaeus

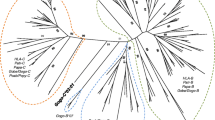
Phylogenetic tree constructed from full-length coding region sequences of MHC-B and -C genes. Relevant bootstrap values (≥70%) are shown. Previously described gorilla alleles identified in the present study are highlighted in bold. Newly identified alleles are underlined. (B) and (C) indicate the phylogenetic clusters of the orthologous MHC-B and -C genes, respectively. HLA human, Patr Pan troglodytes, Papa Pan paniscus, Gogo Gorilla gorilla, Gobe Gorilla beringei, Popy Pongo pygmaeus

Distribution of pairwise nucleotide differences between complete coding region sequences of MHC class I A, B, and C alleles in gorillas, chimpanzees, and humans. Gogo, Patr, and HLA distributions are shown in black, gray, and white, respectively. Gogo-Oko and Gogo-B*07 distributions are shown in dashed black, respectively. In the upper left corner, the number of alleles used for the calculation of the mean pairwise difference is shown
A and A-related genes in gorillas
Among the 35 gorillas analyzed, we identified 19 full-length alleles corresponding to 15 Gogo-A alleles and 4 Gogo-Oko alleles (Table 1). Comparison with known Gogo-A and -Oko alleles showed that 8 of the 11 coding region sequences have not been previously described (Table 1). The novel allele Gogo-A*01:01:02 varied from Gogo-A*01:01 by one synonymous substitution, whereas Gogo-A*07:01 differed at 11 nucleotide positions from Gogo-A*01:01 and -A*04:01, respectively (Lawlor et al. 1991). Interestingly, many gorilla individuals were shown to be heterozygous for Gogo-A*04:01 with differences between alleles observed only in non-coding region sequences. In addition, we identified six individuals being homozygous for either Gogo-A*01:01, -A*04:01, or -A*07:01 alleles (Table 1). Furthermore, three coding region sequences were most similar to the previously described allele Gogo-A*05:01 but differed at 4–6 nucleotide positions (Lawlor et al. 1991). For Gogo-Oko, we identified, besides the sole previously described allele, three novel alleles differing at 1–31 nucleotide positions (Lawlor et al. 1991; Watkins et al. 1991). Among the 35 gorillas analyzed, we identified three individuals homozygous for Gogo-Oko alleles (Table 2). Some Gogo-A and -Oko alleles are shared between gorilla species and subspecies as expected given the trans-species polymorphism of MHC genes (reviewed in Klein et al. 1998) (Table 1). In this context, it has to be noted that the allele Gobe-Oko*01:01 identified in the eastern lowland gorilla is identical to the Gogo-Oko*01:02 allele found in western lowland gorillas. Overall, translation of the 11 coding region sequences resulted in 10 unique amino acid sequences. However, comparison of the three polypeptide chains obtained from the four alleles Gogo-A*01:01:01, -A*01:01:02, -A*04:01, and -A*07:01 revealed very similar sequences suggesting low functional variation (Fig. 1). In contrast to the previously described Gogo-A*05:01 allele, we identified in each of the three novel Gogo-A*05 alleles a nonsynonymous substitution resulting in a stop codon at position 257 within the exon 4, presumably causing a loss of function. Indeed, identical nonsense mutations were identified in the null alleles HLA-A*02:15N, -A*24:183N, and -A*02:356N (Ishikawa et al. 1996; Shimizu et al. 2013). These findings strongly suggest that molecules of Gogo-A*05 alleles from the present study are nonfunctional and therefore, were denoted by the suffix N. Similar to the HLA null alleles, it is unlikely that identified Gogo-A*05 alleles are expressed due to their lack of the transmembrane and intracellular regions encoded by the exons downstream of the stop codon. That in a previous study, cDNA was obtained for a Gogo-A*05 null allele in the gorilla “Beta” but not in the gorilla “Banga” argues for varying levels of messenger RNA (mRNA) transcription which, however, does not contradict our conclusions (Lawlor et al. 1991; Urvater et al. 2001) (Table 2). For Gogo-Oko, however, translation of the four alleles resulted in four unique amino acid sequences indicating allelic polymorphism.
In the phylogenetic tree (Fig. 2), coding region sequences group alleles of Gogo-A (gorilla), Patr-A (chimpanzee), Papa-A (bonobo), and HLA-A (human) together indicating that this clade represents the A orthologue of gorilla, chimpanzee, bonobo, and human. Within this clade, alleles are divided into two groups, the ancient lineages A2 and A3 (Lawlor et al. 1990). Although HLA-A alleles segregate in both lineages, the two novel and three previously described alleles Gogo-A*01:01, -A*01:01:02, -A*02:01, -A*04:01, and -A*07:01 segregate only into the A2 lineage together with alleles of the HLA-A19 family. In contrast, Patr-A and Papa-A alleles group only into the A3 lineage with alleles of the HLA-A1/A3/A11 family, as has been shown (Lawlor et al. 1995; McAdam et al. 1995; Adams et al. 2000; de Groot et al. 2000). The previously described Gogo-A*05:01 allele and the novel Gogo-A*05 alleles, however, fall outside of both the A2 and A3 lineages instead forming a cluster with the human pseudogene HLA-Y, previously known as HLA-BEL, -COQ, and -DEL (Williams et al. 1999; Coquillard et al. 2004; Marsh et al. 2010). Contrary to previous suggestions, Gogo-A*05 alleles cannot represent a divergent A lineage because we identified individual gorillas which have Gogo-A*05 alleles in addition to two Gogo-A alleles showing that these are definitely separate genes (Table 2) (Lawlor et al. 1991; Coquillard et al. 2004). Thus, our findings strongly suggest that Gogo-A*05 represents the gorilla orthologue of HLA-Y. Similarly, alleles of the Gogo-Oko locus are phylogenetically distinct, segregating with H locus alleles of human, chimpanzee, and gorilla consistent with previous findings showing that Gogo-Oko, through recombination, shares partial sequence homology with the pseudogene H (Lawlor et al. 1991; Watkins et al. 1991; Adams and Parham 2001; Gleimer et al. 2011). Thus, our results indicate that gorillas possess, besides Gogo-A and Gogo-Oko, an additional A-related locus consisting of Gogo-A*05 alleles which likely represents a pseudogene in the gorillas analyzed.
To further examine the evolutionary relationship of the novel Gogo-A*05 with Gogo-A, -Oko, and related MHC class I genes of the other hominid species, we performed domain-by-domain phylogenetic analyses using available genomic full-length sequences (Online Resource 4). As expected, Gogo-A*05 is most closely related to the human pseudogene HLA-Y. Except for exon 3 and its flanking introns which show higher sequence similarity to other Gogo-A locus alleles, Gogo-A*05 is orthologous to HLA-Y throughout the gene. Thus, our results clearly show that Gogo-A*05 is the gorilla orthologue of HLA-Y. In this context, HLA-Y has been suggested to be the human equivalent of the chimpanzee A-related locus Patr-AL (Gleimer et al. 2011). Consistent with previous findings, domain-by-domain phylogenetic analyses demonstrate that both HLA-Y and Gogo-A*05 have segments orthologous to Patr-AL (Gleimer et al. 2011). In the 5′ part which includes the 5′ flanking region, exon 1, intron 1, and partial exon 2, both HLA-Y and Gogo-A*05 have sequences in common with Patr-AL. Otherwise, HLA-Y and Gogo-A*05 share sequence similarity with A locus alleles of the A2 lineage. In contrast, Patr-AL is most closely related to the orangutan Popy-A which likewise shares sequence similarity with HLA-Y and Gogo-A*05 in the 5′ flanking region. Furthermore, Gogo-Oko appears to be orthologous to Popy-A/Gogo-A*05/Patr-AL/HLA-Y in the 5′ flanking region, and indeed, analysis of a gorilla MHC haplotype revealed that Gogo-Oko is located in the same genomic position as Patr-AL (Gleimer et al. 2011). Otherwise, Gogo-Oko shares sequence similarity with the nonfunctional H and other A locus alleles consistent with previous findings (Lawlor et al. 1991; Watkins et al. 1991; Adams and Parham 2001; Gleimer et al. 2011). Taken together, our results, in combination with previous findings, demonstrate the orthologous relationship of the A-related genes in human (HLA-Y), chimpanzee (Patr-AL), gorilla (Gogo-A*05 and Gogo-Oko), and orangutan (Popy-A).
To assess the gorilla MHC class I diversity, we compared allelic and sequence variability to a similar-sized sample of chimpanzees and humans. Based on the number of MHC-A alleles, gorillas are much less diverse than chimpanzees and humans. Among the 33 unrelated gorillas analyzed, we identified only 4 different coding region sequences for Gogo-A whereas a similar sized study of chimpanzees (n = 24) identified 19 different Patr-A alleles. Among 30 unrelated Yoruba individuals, a total of 21 HLA-A alleles have been described. Further evidence showing that the MHC-A diversity in gorillas is reduced compared to chimpanzees and humans, comes from the distribution of pairwise allelic differences (Fig. 4). The mean pairwise difference of Gogo-A alleles (mean = 12.7) is much lower than the means of Patr-A alleles (mean = 21.2) and HLA-A alleles (mean = 34.2), respectively. The reduced diversity in gorillas is not only influenced by the absence of A3 lineage alleles but is also due to the homogeneity of Gogo-A alleles, as implied by Figs. 1 and 4. In contrast, chimpanzees appear to have accumulated variation at Patr-A despite their lack of alleles belonging to the A2 lineage. The distribution of pairwise differences between HLA-A alleles, however, reflects the presence of A2 and A3 lineage alleles in the Yoruba population. As expected from the comparison of pairwise allelic differences, nucleotide diversity was lowest for Gogo-A (π = 0.01154) compared to Patr-A (π = 0.01975) and HLA-A (π = 0.04088), respectively (Table 3). The overall numbers of variable sites of coding region sequences were 22, 67, and 70 for gorillas, chimpanzees and humans, respectively. Within the antigen-binding sites (ABS), numbers of variable sites were 13 for Gogo-A alleles, 31 for Patr-A alleles, and 35 for HLA-A alleles (Table 3). Although Fisher’s exact test showed no significant differences between substitution rates at Gogo-A, the nonsynonymous substitution rate (Ka) is substantially lower in gorillas compared to chimpanzees and humans. The synonymous substitution rate (Ks), however, appears to be highest in gorillas which might suggest that selection acts against amino acid altering mutations at Gogo-A (Table 3). In contrast, the Ka/Ks ratio in humans is strikingly higher compared to gorillas and chimpanzees which can be attributed to the significantly lower synonymous substitution rate at HLA-A (Table 3).
B genes in gorillas
For Gogo-B, we identified a total of 21 genomic full-length alleles corresponding to 18 different coding region sequences (Table 1). Comparison with previously described Gogo-B alleles revealed that 8 of the 18 coding region sequences were novel. The allele Gogo-B*01:04 differed from Gogo-B*01:03 by one nonsynonymous substitution, whereas coding region sequences of Gogo-B*03:02 and -B*03:03 differed from Gogo-B*03:01 at five and seven nucleotide positions, respectively (Lawlor et al. 1991; Urvater et al. 2001). The novel allele Gogo-B*07:02 varied from Gogo-B*07:01 by one nonsynonymous substitution (Abi-Rached et al. 2011). On the contrary, the alleles Gogo-B*07:03 and -B*07:04 were most similar to Gogo-B*07:01 but shared sequence similarity in exons 5–7 with other Gogo-B alleles, a likely result of interallelic gene conversion. The novel allele Gogo-B*12:01 was most similar to Gogo-B*04:01 differing at 20 nucleotide positions, whereas the allele Gobe-B*13:01, which was only observed in the eastern lowland gorillas, differed from the previously described allele Gogo-B*06:01 at 35 nucleotide positions (Abi-Rached et al. 2011). Unlike Gogo-A and -Oko alleles, we identified only one Gogo-B allele shared between the two western gorilla subspecies, suggesting the rapid diversification of B locus alleles (Table 1). The 18 different coding region sequences translated into 18 unique amino acid sequences. Comparison of polypeptide chains revealed that many amino acid motifs are shared between alleles indicating that recombination plays an important role in the generation of allelic variation (Fig. 1).
The phylogenetic tree including full-length MHC-B coding region sequences shows little substructure with alleles predominantly forming species-specific clusters (Fig. 3). The absence of clearly defined B lineages has been attributed to frequent recombination and interlocus gene conversion at this MHC locus, as reflected by low bootstrap support values (Lawlor et al. 1990; Adams and Parham 2001). Consistent with previous findings, alleles of human, chimpanzee, gorilla, and orangutan mostly group by species with the exception of certain gorilla and chimpanzee B alleles segregating with HLA-B*73:01 to form a clade which has been suggested to represent an ancient B lineage (Parham et al. 1994; Hoffmann et al. 1995; Abi-Rached et al. 2011; Yasukochi and Ohashi 2016). Distinguishing the ancient B lineage, all these alleles share a 3-bp insertion in exon 5 which adds another amino acid to the transmembrane region (Adams and Parham 2001). Included in this clade are the gorilla B alleles Gogo-B*02:01, -B*03:01, -B*03:02, -B*03:03, -B*04:01, -B*06:01, -B*12:01, and Gobe-B*13:01 whereas the alleles Gogo-B*01:01, -B*01:02, -B*01:03, -B*01:04, -B*05:01, and -B*05:02 form a separate cluster within the clade that consists of the other human and chimpanzee B alleles. Despite the relatively few MHC-B alleles found in gorillas, our findings indicate that gorillas have two distinct B loci because we identified seven individuals each of which possessing three Gogo-B alleles (Table 2). Divergent from the other Gogo-B alleles, the alleles Gogo-B*07:01, -B*07:02, -B*07:03, and -B*07:04 clearly segregate apart and appear to be most closely related to Popy-B alleles of orangutans. Strong linkage disequilibrium between alleles of Gogo-B*03 and -B*07 demonstrates that these are located on the same haplotype and in accordance, we identified one individual which was homozygous for Gogo-B*03 and -B*07, respectively (Table 2). Our data thus suggest that, in addition to Gogo-B, gorillas possess another B locus which is present only on some gorilla B haplotypes.
To further examine the evolutionary relationships of the novel Gogo-B gene, we extended our phylogenetic analyses by using available genomic full-length DNA sequences of human, chimpanzee, and orangutan (Online Resource 5). Domain-by-domain analyses of full-length sequences show that, as expected, alleles of Gogo-B*03 co-segregating with Gogo-B*07 are most similar to other gorilla B alleles. In the 5′ region, Gogo-B*03 alleles clearly segregate with Gogo-B*04:01 whereas in the remaining part of the gene Gogo-B*03 alleles form a cluster with Gogo-B*02:01, -B*04:01 and -B*12:01 indicating that Gogo-B*03 alleles belong to the gorilla B orthologue of HLA-B and Patr-B. The novel Gogo-B*07 locus, however, has a complex recombinant structure. In the 5′ region and the intron 3, Gogo-B*07 alleles appear to be most similar to the orangutan B locus allele Popy-B*03:02 whereas there is sequence similarity to MHC-C alleles in the region encompassing exon 2 and 3 and its intervening intron. In exon 4 and intron 4, however, Gogo-B*07 alleles are related to other gorilla B locus alleles. The phylogenetic tree constructed from exon 5 to 8 sequences segregates Gogo-B*07:01 and -B*07:02 alleles together with Popy-B*03:02 distinct from Gogo-B*07:03 and -B*07:04 alleles which are most closely related to alleles of the ancient B lineage. Accordingly, the Gogo-B*07:03 and -B*07:04 alleles share the 3-bp insertion in exon 5 characteristic for the ancient B lineage suggesting that Gogo-B*07:03 and -B*07:04 acquired the 3′ region through interallelic gene conversion with Gogo-B alleles of the ancient B lineage. Thus, there is evidence to suggest that alleles of Gogo-B*07 represent the orthologue of Popy-B which can be found in multiple copies in individual orangutans (Chen et al. 1992; Adams et al. 1999; de Groot et al. 2016).
As described in detail above, some gorillas possess an additional Gogo-B gene which has no orthologue in the MHC of chimpanzee and human, respectively. Thus, for a comparable assessment of the MHC-B diversity in gorillas, chimpanzees, and humans, Gogo-B*07 alleles were excluded from sequence diversity analyses. Among the 33 unrelated gorillas, we identified a total of 14 different coding region sequences at Gogo-B. In contrast, a study of 24 chimpanzees revealed 23 Patr-B alleles (Table 3). Allelic variation at HLA-B is represented by 19 alleles which were characterized among 30 unrelated Yoruba individuals. Thus, based on the number of alleles, gorillas have lower variation at the orthologous MHC-B gene compared to chimpanzees and humans. As seen from the histograms in Fig. 4, the distribution of pairwise allelic differences is considerably different in gorillas as compared to chimpanzees and humans. This is predominantly due to the prevalence of divergent Gogo-B alleles, as reflected by phylogenetic patterns in Fig. 3. In contrast, distributions of pairwise allelic differences at Patr-B and HLA-B indicate that allelic diversity accumulated within MHC-B lineages of chimpanzees and humans, respectively. Consistent with the divergence of Gogo-B alleles, nucleotide diversity at MHC-B is higher in gorillas (π = 0.04667) than in chimpanzees (π = 0.04054) and humans (π = 0.03293) (Table 3). The numbers of variable sites within full-length coding region sequences of MHC-B alleles are 136, 122, and 97 for gorillas, chimpanzees, and humans, respectively. Within the ABS region, numbers of variable sites are 42, 36, and 31 for Gogo-B, Patr-B, and HLA-B, respectively. Calculation of mean Ka and Ks within the ABS shows significantly higher substitution rates in gorillas than in chimpanzees (Table 3). However, the higher Ka/Ks ratios at Patr-B is due to the significantly lower synonymous substitution rate suggesting that positive selection in chimpanzees is acting more effectively in altering amino acid residues than in gorillas. In contrast, Fisher’s exact test revealed no significant differences between synonymous (Ks) and nonsynonymous substitution rates at HLA-B (Table 3).
C gene in gorillas
Among the 35 gorillas analyzed, we identified a total of 10 Gogo-C full-length genomic sequences corresponding to nine different coding region sequences (Table 1). Comparison with known Gogo-C alleles showed that three coding region sequences were not described previously. The novel allele Gogo-C*01:01:03, which was only observed in the Cross River gorilla, differed from Gogo-C*01:01 by two synonymous substitutions (Lawlor et al. 1991). The novel allele Gogo-C*01:04 varied from Gogo-C*01:01 by one nonsynonymous and one synonymous substitution. Please also note that the previously described allele Gogo-C*01:02 was renamed to Gogo-C*01:01:02 because it varies from Gogo-C*01:01:01 at one synonymous nucleotide position (Lawlor et al. 1991). Differing by one nonsynonymous substitution, the novel allele Gogo-C*02:05 was most closely related to Gogo-C*02:04 which, however, has not been described in full-length (Urvater et al. 2001). As with the A and A-related loci, trans-species polymorphism of Gogo-C alleles was observed among the gorilla species and subspecies, all of which shared the allele Gogo-C*02:02 (Table 1). However, this allele has been designated as Gobe-C*02:01 in the eastern lowland gorilla. The nine different coding region sequences of Gogo-C alleles translated into seven unique amino acid sequences. Comparison of amino acid sequences revealed the existence of two Gogo-C lineages with intra-allelic variation mainly generated through point mutations (Fig. 1).
Within the phylogenetic tree in Fig. 3, coding region sequences of the MHC-C gene in human, chimpanzee, bonobo, gorilla, and orangutan clearly segregate within a distinct clade indicating their orthologous relationship. Besides species-specific clustering of alleles, phylogenetic patterns suggest the presence of conserved C lineages across species. As such, the gorilla alleles Gogo-C*02:01, -C*02:02, -C*02:03, -C*02:04, and -C*02:05 as well as certain chimpanzee and bonobo C alleles cluster together with HLA-C*07:01 (Zemmour and Parham 1992; Adams and Parham 2001). On the contrary, the gorilla alleles Gogo-C*01:01:01, -C*01:01:02, -C*01:01:03, -C*01:03, and -C*01:04 form a gorilla-specific branch within the C locus clade. However, the majority of Patr-C and HLA-C alleles segregate together into one allelic lineage which appears to be absent from gorillas.
Our study of 33 unrelated gorillas revealed nine different coding region sequences at the Gogo-C locus (Table 3). The study of 24 chimpanzees by Adams et al. (2000) characterized 16 different Patr-C alleles. Among the 30 unrelated individuals of the Yoruba population, a total of 12 HLA-C alleles were identified. Thus, gorillas appear to have lower diversity at the C locus with regard to number of alleles. The histogram of pairwise allelic differences at Gogo-C is substantially different from that of chimpanzees and humans (Fig. 4). As expected, the distribution of pairwise differences between Gogo-C alleles reflects the presence of only two divergent lineages with intra-allelic variation predominantly generated through point mutations. In contrast, the histograms of chimpanzees and humans reveal the presence of multiple lineages, as can be seen from the distribution of pairwise differences between Patr-C and HLA-C alleles. In accordance, nucleotide diversity is higher at Gogo-C (π = 0.03160) than at Patr-C (π = 0.02716) and HLA-C (π = 0.02752). However, the number of variable sites, both within coding regions and the ABS, is substantially lower in gorillas (66/9) compared to chimpanzees (86/16) and humans (97/12). Although Fisher’s exact test showed no significant differences between synonymous and nonsynonymous substitution rates in humans, chimpanzees and gorillas, respectively, it appears that gorillas have maintained diversity at functionally important ABS as suggested by an intermediate Ka/Ks ratio (Table 3).
Gogo-B and -C allotypes for KIR recognition
KIR recognition of MHC class I epitopes is determined by amino acid residues 76–83 in the alpha-1 domain. Lineage II KIR receptors interact with the Bw4 epitope expressed by MHC-B allotypes which is defined by arginine (R) at position 83 while expression of valine (V) at position 76 and asparagine (N) at position 80 defines the C1 epitope which is recognized by lineage III KIR receptors. MHC-B allotypes with other sequence motifs at positions 76–83 are not ligands for KIR receptors (Parham and Moffett 2013). In our study, 12 of the 18 Gogo-B allotypes are predicted to be ligands for KIR receptors. All of the Gogo-B*01 and -B*05 allotypes have the Bw4 epitope while Gogo-B*06:01 and Gobe-B*13:01 as well as all allotypes of Gogo-B*07 have the C1 epitope. The remaining six Gogo-B allotypes have neither the Bw4 epitope nor the C1 epitope (Fig. 5). In contrast, recognized by lineage III KIR receptors, MHC-C allotypes are distinguished by a dimorphism at amino acid position 80. The C1 epitope is carried by MHC-C allotypes that have asparagine (N) at position 80 while the C2 epitope is carried by MHC-C allotypes with lysine (K) at position 80 (Parham and Moffett 2013). Interestingly, all Gogo-C allotypes identified are predicted to have the motif which defines the C2 epitope (Fig. 5).

Amino acid sequences of Gogo-B and -C alleles determining the KIR-binding epitope. Identity to consensus sequence is denoted by a dash. For differences from the consensus sequence, the amino acid is shown. Positions highlighted in green, blue, and red define the Bw4 epitope, the C1 and the C2 epitopes, respectively
Discussion
In the present study, we studied 35 gorillas yielding 15 Gogo-A alleles, 4 Gogo-Oko, 21 Gogo-B alleles, and 10 Gogo-C alleles. We identified two previously undetected genes related to Gogo-A and Gogo-B in some of the individuals. Data obtained in gorillas were analyzed in the context of other available information from humans, chimpanzees, bonobos, and orangutans. Our results provide evidence for gorillas having a comparatively complex MHC class I haplotype structure but the overall MHC class I variation appears to be low.
The previous, albeit small, sampling of Gogo class I alleles showed that Gogo-A alleles segregate only into the A2 lineage suggesting that A3 lineage alleles are lacking in gorillas (Lawlor et al. 1991; Watkins et al. 1991; Cadavid and Watkins 1997; Adams and Parham 2001). Indeed, among the 35 gorillas analyzed, we identified only Gogo-A alleles of the A2 lineage (Fig. 2). Whereas in humans, the A2 lineage has diversified considerably and now includes the three divergent families HLA-A2, -A10, and -A19; gorillas have only equivalents of the HLA-A19 family (Fig. 2) (Lawlor et al. 1990). Equally diverse in humans is the A3 lineage with the three families HLA-A1/A3/A11, -A9, and -A80 whereas the A locus alleles of chimpanzees and bonobos, Patr-A and Papa-A, segregate only into the HLA-A1/A3/A11 family (Parham et al. 1989; Lawlor et al. 1990, 1995; Domena et al. 1993; McAdam et al. 1995; Adams et al. 2000; de Groot et al. 2000). This reduction of the chimpanzee and bonobo MHC class I repertoire has been attributed to a pathogen-mediated selective sweep which caused the extinction of A2 lineage alleles in the ancestral Pan population (de Groot et al. 2002, 2008). However, whether the A3 lineage emerged after the split of humans and chimpanzees from gorillas or was present in their common ancestor and subsequently lost during gorilla evolution remains to be elucidated. In this context, previous studies revealed that gorillas show comparatively little variability at MHC class II genes as well as a marked reduction of genetic diversity in regions flanking the MHC indicative of a recent selective sweep (Scally et al. 2013; Hans et al. 2015; Xue et al. 2015). Thus, our findings further support the hypothesis that gorillas might have experienced a reduction of their MHC diversity. Indeed, the small number of high-frequency alleles associated with an increased homozygosity suggests that the reduction at Gogo-A has occurred quite recently (Tables 1 and 2). In accordance, gorillas appear to have accumulated only limited variation both within coding and non-coding region sequences, as reflected by an interspecies comparison of allelic variation (Table 3 and Fig. 4). Thus, our findings strongly suggest that gorillas recently underwent an episode of increased selective pressure which might have resulted in the low Gogo-A diversity.
Interestingly, some gorillas possess an additional and putatively functional A-related locus, designated Gogo-Oko, which has been shown to be located in the same genomic position as Patr-AL (Lawlor et al. 1991; Watkins et al. 1991; Gleimer et al. 2011). However, distinguishing Gogo-Oko is the unusual structure of its corresponding gorilla MHC haplotype. Whereas Patr-AL co-segregates with Patr-A, the gorilla MHC haplotype with Gogo-Oko is characterized by the absence of the genomic block containing the A gene (Adams et al. 2001; Geller et al. 2002; Gleimer et al. 2011). In accordance, we identified three individuals homozygous for Gogo-Oko (Table 2). Besides the sole previously described Gogo-Oko allele, we characterized three novel alleles suggesting that Gogo-Oko evolved to become a classical MHC class I gene. Indeed, evidence that Gogo-Oko gene products are involved in the antigen presentation comes from the study by Watkins et al. (1991), which demonstrated its expression in 7 of 13 gorillas (53.8%). We observed a similar distribution with 16 of 35 gorillas (45.7%) possessing Gogo-Oko suggestive of balancing selection maintaining this haplotype at intermediate frequencies in the population. We thus propose that Gogo-Oko, at least in part, compensates for the low diversity at the Gogo-A locus.
Besides Gogo-Oko, we here show that gorillas possess another A-related gene which is represented by Gogo-A*05 alleles. Despite previous suggestions, alleles of Gogo-A*05 cannot represent a divergent Gogo-A lineage because we identified individual gorillas which have Gogo-A*05 alleles in addition to two Gogo-A alleles showing that these are definitely separate genes (Table 2) (Lawlor et al. 1991; Coquillard et al. 2004). Domain-by-domain phylogenetic analyses clearly demonstrate that Gogo-A*05 is the equivalent of the human pseudogene HLA-Y and our findings suggest that Gogo-A*05 likewise represents a pseudogene in the gorillas analyzed (Online Resource 4) (Williams et al. 1999; Coquillard et al. 2004; Marsh et al. 2010). A nonsense mutation in exon 4 argues that the Gogo-A*05 alleles identified are nonfunctional. Similarly, the HLA-Y alleles, -COQ, and -DEL, are nonfunctional due to nonsense mutations in exon 4 whereas HLA-Y, also designated as HLA-BEL, lacks the exon 3 in addition to a dual-cytosine insertion in exon 4 causing a frameshift and premature termination (Williams et al. 1999; Coquillard et al. 2004). Due to their structural and functional similarities, we propose to rename its gorilla equivalent as Gogo-Y.
Our analyses further show that Gogo-Y is orthologous to the 5′ part of the functional A-related genes Patr-AL in chimpanzees and Popy-A in orangutans indicating that these genes shared a common ancestor before the hominid diversification (Online Resource 4) (Adams et al. 2001; Gleimer et al. 2011; Goyos et al. 2015). That the orthologous A-related (pseudo)genes in human and gorilla independently acquired nonsense mutations argues for selection that has led to their inactivation, possibly associated with disease susceptibility. However, Gogo-Y is also orthologous to the 5′ flanking region of Gogo-Oko suggesting that the ancestral A-related gene in gorillas evolved differently to become nonfunctional and classical (Online Resource 4). As is the case for Gogo-Oko, our findings suggest that Gogo-Y is not present on all gorilla MHC haplotypes although linkage disequilibrium to Gogo-A demonstrates its location within the MHC class I region, as determined through patterns of allelic inheritance among related individuals (Table 2). Similarly, HLA-Y and Patr-AL are present only on a subset of the human and chimpanzee MHC haplotypes despite their co-segregation with the orthologous A locus (Fig. 6) (Williams et al. 1999; Adams et al. 2001; Geller et al. 2002; Coquillard et al. 2004; Gleimer et al. 2011). Interestingly, the presence of HLA-Y is predominantly associated with alleles of the HLA-A19 family and it is this family to which the orthologous Gogo-A alleles are most closely related (Williams et al. 1999; Coquillard et al. 2004; Gleimer et al. 2011). Thus, it appears that this MHC haplotype has remained stable since humans and gorillas last shared a common ancestor. In nucleotide sequence, both HLA-Y and Gogo-Y share sequence similarity with A2 lineage alleles whereas Gogo-Oko through recombination obtained segments from H and other primate A locus alleles (Online Resource 4) (Lawlor et al. 1991; Watkins et al. 1991; Adams and Parham 2001; Gleimer et al. 2011). In contrast, Patr-AL is most closely related to Popy-A, and for that reason it has been suggested that true orthologues of the A locus are present only in humans (HLA-A) and the African great apes, chimpanzees (Patr-A), bonobos (Papa-A), and gorillas (Gogo-A), but not orangutans (Fig. 6) (Adams et al. 2001; Gleimer et al. 2011). Consistent with our findings in gorillas, an evolutionary model has been proposed in which the A and A-related genes descended from a common ancestor by two successive duplications followed by partial deletions forming the extant hominid MHC haplotypes (Gleimer et al. 2011). As such, the deletion of the genomic block with the A-related gene produced the A haplotype which now can be found in humans, chimpanzees, bonobos and gorillas (Fig. 6) (Watanabe et al. 1997; Gleimer et al. 2011). On the contrary, deletion of the genomic block with the A locus formed the A-related haplotype represented only by Gogo-Oko and absent from humans and the other hominid species (Fig. 6) (Gleimer et al. 2011). Thus, our results in combination with previous findings show that gorillas have a comparatively complex MHC class I region although the overall functional diversity appears to be low at the A and A-related genes.

Simplified MHC class I A region haplotypes of humans and the great apes. White boxes represent the orthologous MHC-A gene whereas gray boxes represent the orthologous MHC-A-related gene
In contrast, the previous characterization of 18 alleles of Gogo-B, the gorilla orthologue of HLA-B, suggested a high degree of polymorphism at this MHC class I locus (Lawlor et al. 1991; Urvater et al. 2001; Martínez-Laso et al. 2006; Abi-Rached et al. 2011). Our study of 35 gorillas identified a total of 17 full-length Gogo-B alleles, five of which had novel coding region sequences (Table 1). Phylogenetically, Gogo-B alleles group into two distinct clusters whereas the HLA-B and Patr-B alleles show a dispersed distribution with multiple species-specific branches (Fig. 3). These patterns are also evident from the distributions of pairwise allelic differences reflecting the prevalence of divergent alleles in gorillas as compared to chimpanzees and humans (Fig. 4). The majority of Gogo-B alleles segregate with certain Patr-B alleles and the HLA-B*73:01 allele forming an ancestral B lineage shared between humans, chimpanzees and gorillas while the remaining Gogo-B alleles group phylogenetically into a gorilla-specific branch of the clade (Parham et al. 1994; Abi-Rached et al. 2011; Yasukochi and Ohashi 2016). Included in the latter are alleles of Gogo-B*01 which were identified in 20 of the 35 gorillas analyzed (57.1%). In this context, Gogo-B*01 alleles have been suggested to be responsible for the high prevalence of spondyloarthropathy (SpA) in gorillas, an inflammatory disorder primarily affecting the spine (Rothschild and Woods 1989; Urvater et al. 2001). Indeed, although divergent in their nucleotide sequences, Gogo-B*01 molecules have peptide-binding repertoires similar to HLA-B*27 which is a strong risk factor for SpA in humans (Urvater et al. 2001). Molecules of HLA-B*27 and -B*57 have also been shown to give a protective effect in HIV-1 infection (Kiepiela et al. 2004; Fellay et al. 2007). In particular, HLA-B*27 and -B*57 alleles expressing the Bw4 epitope and isoleucine (I) at amino acid residue 80 in combination with the activating KIR II lineage allele KIR3DS1 are associated with a delayed disease progression to AIDS (Martin et al. 2002). Interestingly, like the protective HLA-B allotypes, Gogo-B*01 and -B*05 alleles are predicted to express the Bw4 epitope and isoleucine at position 80 (Fig. 5). That two groups of HIV-1 originated through cross-species transmission of SIV from gorillas (SIVgor) to humans highlights the possibility of gorillas having MHC class I molecules which provide resistance to lentiviral infections (Van Heuverswyn et al. 2006; Plantier et al. 2009; D’arc et al. 2015). However, it is unknown whether SIVgor infections are pathogenic in its gorilla host (Sharp and Hahn 2011; but see Moeller et al. 2015). The other two groups of HIV-1, including its fatal form which develops into AIDS in humans, descended from SIV in chimpanzees (SIVcpz) (Keele et al. 2006). However, in contrast to humans, chimpanzees naturally infected with SIVcpz are less likely to develop AIDS-like symptoms (reviewed in Sharp and Hahn 2011; but see Pandrea et al. 2009; Keele et al. 2009). This relative resistance of chimpanzees has been attributed to a selective sweep which was caused by an AIDS-like pandemic in the ancestral Pan population (de Groot et al. 2002). As a consequence, AIDS-protective MHC class I alleles were selected and increased in frequency in modern chimpanzees, a process that was, however, accompanied by a general reduction of the Patr class I repertoire (de Groot et al. 2002, 2008; de Groot and Bontrop 2013). Indeed, it has been shown that the most frequent Patr class I molecules recognize similar HIV/SIVcpz epitopes as the AIDS-protective HLA-B*27/B*57 in humans (de Groot et al. 2010; see also Wroblewski et al. 2015). Thus, there is evidence to suggest that in gorillas, the comparatively little MHC class I diversity likewise resulted from a pathogen-mediated selective sweep.
Despite the apparent low Gogo-B diversity, we show that certain gorillas possess an additional B locus because we identified seven individuals which have three Gogo-B alleles (Table 2). Divergent from the other gorilla B locus alleles, Gogo-B*07 alleles fall outside of the orthologous B clade shared between humans, chimpanzees, bonobos, and gorillas and appear to be most closely related to Popy-B which exhibits diverse copy number variation in orangutans (Fig. 3) (Chen et al. 1992; Adams et al. 1999; de Groot et al. 2016). Indeed, domain-by-domain phylogenetic analyses show that the newly identified Gogo-B*07 gene shares partial sequence similarity with Popy-B suggesting that Gogo-B*07 represents an ancestral B lineage predating the hominid divergence (Online Resource 5). In contrast to most other Gogo-B alleles which either have the Bw4 epitope or do not interact with KIR receptors, alleles of Gogo-B*07 are predicted to carry the C1 epitope (Fig. 5). Thus, it appears that gorillas have maintained the putative ancestral B locus on certain MHC haplotypes due to its important function in controlling NK cell activity via KIR receptors.
However, the dominant ligand for KIR receptors in humans is MHC-C and its comparatively low polymorphism has been attributed to the coevolution with KIR receptors (Adams and Parham 2001; Vilches and Parham 2002; Single et al. 2007; Older Aguilar et al. 2010). In humans and chimpanzees, all MHC haplotypes have the MHC-C gene whereas in orangutans, it is present only on some of the MHC haplotypes (Adams et al. 1999; de Groot et al. 2016). Similar to its fixation in humans and chimpanzees, our findings suggest that Gogo-C, the gorilla orthologue of HLA-C, is present on all gorilla MHC haplotypes (Table 2). Among the 35 gorillas, we identified a total of 10 full-length Gogo-C alleles including three coding region sequences that have not been previously described (Table 1). Phylogenetically, Gogo-C alleles clearly segregate with C locus alleles of human, chimpanzee, and bonobo as well as orangutan indicating their orthologous relationship (Fig. 3). However, Gogo-C alleles group phylogenetically only into two lineages with polymorphism mainly generated through point mutations as evidenced by the distribution of pairwise allelic differences, thus suggesting low functional variation (Figs. 3 and 4). Indeed, Gogo-C allotypes are predicted to express only one of the two epitopes that are recognized by lineage III KIR receptors, the C2 epitope, whereas in humans and chimpanzees MHC-C allotypes with both the C1 and C2 epitopes have been identified (Fig. 5) (Adams and Parham 2001; Moesta et al. 2009). In contrast, orangutan MHC-C allotypes carry only the C1 epitope indicating that it was present in the common ancestor of humans and the great apes (Guethlein et al. 2002, 2007). The C2 epitope, however, emerged with the fixation of the MHC-C gene, and in accordance, is carried by MHC-C allotypes of gorillas, chimpanzees, and humans but not orangutans (Parham and Moffett 2013; Guethlein et al. 2015). Thus, there is evidence to suggest that gorillas have lost the C1-bearing allotypes of MHC-C. The C1 epitope is also carried by some orangutan, chimpanzee, and human MHC-B allotypes indicating that it was also present in the ancestral MHC-B-like gene that duplicated to produce two paralogous MHC class I genes, now recognized as MHC-B and MHC-C (Fukami-Kobayashi et al. 2005). In gorillas, the C1 epitope is predominantly carried by the newly identified Gogo-B*07 gene. Interestingly, exons 2 and 3 and the intervening intron of Gogo-B*07 are most closely related to MHC-C which suggests that Gogo-B*07 represents the remnant of the MHC-B-like ancestor that was maintained by selection on certain gorilla and orangutan MHC haplotypes but lost in the common ancestor of Pan and humans (Fig. 7). Taken together, there is evidence for a complex history of the hominid MHC-B and MHC-C genes which cannot be explained by an evolutionary model involving a single duplication event.

Simplified MHC class I B and C region haplotypes of humans and the great apes. White boxes represent the orthologous MHC-C gene whereas light gray boxes represent the orthologous MHC-B gene. Dark gray boxes represent the orthologous MHC-B gene present only in gorillas and orangutans. Copy number variation at the MHC-B gene in orangutans is depicted by a question mark
In sum, our study demonstrates the utility of characterizing a larger sample of gorillas for a more complete understanding of the MHC class I gene variability. We identified that gorillas have a comparatively complex MHC class I haplotype structure with characteristics shared between humans and chimpanzees as well as orangutans, thereby providing new insights into the evolution of the MHC class I genes in humans and the great apes. However, the apparently low diversity suggests that gorillas might have experienced a reduction of their MHC class I repertoire, presumably through a selective sweep, which should be investigated in more detail in future studies.
References
Abi-Rached L, Jobin MJ, Kulkarni S, McWhinnie A, Dalva K, Gragert L, Babrzadeh F, Gharizadeh B, Luo M, Plummer FA, Kimani J, Carrington M, Middleton D, Rajalingam R, Beksac M, Marsh SG, Maiers M, Guethlein LA, Tavoularis S, Little AM, Green RE, Norman PJ, Parham P (2011) The shaping of modern human immune systems by multiregional admixture with archaic humans. Science 334:89–94. doi:10.1126/science.1209202
Adams EJ, Parham P (2001) Species-specific evolution of MHC class I genes in the higher primates. Immunol Rev 183:41–64
Adams EJ, Thomson G, Parham P (1999) Evidence for an HLA-C-like locus in the orangutan Pongo pygmaeus. Immunogenetics 49:865–871
Adams EJ, Cooper S, Thomson G, Parham P (2000) Common chimpanzees have greater diversity than humans at two of the three highly polymorphic MHC class I genes. Immunogenetics 51:410–424
Adams EJ, Cooper S, Parham P (2001) A novel, nonclassical MHC class I molecule specific to the common chimpanzee. J Immunol 167:3858–3869
Cadavid LF, Watkins DI (1997) MHC class I genes in nonhuman primates. In: Blancher A, Klein J, Socha WW (eds) Molecular biology and evolution of blood group and MHC antigens in primates. Springer, Berlin, pp 339–357
Chen ZW, McAdam SN, Hughes AL, Dogon AL, Letvin NL, Watkins DI (1992) Molecular cloning of orangutan and gibbon MHC class I cDNA. The HLA-A and -B loci diverged over 30 million years ago. J Immunol 148:2547–2554
ConsensusTools v2.3.0 Documentation (2014) GitHub, Inc. https://github.com/PacificBiosciences/SMRT-Analysis/wiki/ConsensusTools-v2.3.0-Documentation. Assessed 19 Apr 2016
Coquillard G, Lau M, Kletzel M, Rodriguez-Marino SG (2004) Identification of two pseudogenes with sequence homology to human and gorilla MHC class IA genes: ancestral haplotype in the Filipino population. Hum Immunol 65:665–673
D’arc M, Ayouba A, Esteban A, Learn GH, Boué V, Liegeois F, Etienne L, Tagg N, Leendertz FH, Boesch C, Madinda NF, Robbins MM, Gray M, Cournil A, Ooms M, Letko M, Simon VA, Sharp PM, Hahn BH, Delaporte E, Mpoudi Ngole E, Peeters M (2015) Origin of the HIV-1 group O epidemic in western lowland gorillas. Proc Natl Acad Sci U S A 112:E1343–E1352. doi:10.1073/pnas.1502022112
de Bakker PI, McVean G, Sabeti PC, Miretti MM, Green T, Marchini J, Ke X, Monsuur AJ, Whittaker P, Delgado M, Morrison J, Richardson A, Walsh EC, Gao X, Galver L, Hart J, Hafler DA, Pericak-Vance M, Todd JA, Daly MJ, Trowsdale J, Wijmenga C, Vyse TJ, Beck S, Murray SS, Carrington M, Gregory S, Deloukas P, Rioux JD (2006) A high-resolution HLA and SNP haplotype map for disease association studies in the extended human MHC. Nat Genet 38:1166–1172. doi:10.1038/ng1885
de Groot NG, Otting N, Argüello R, Watkins DI, Doxiadis GG, Madrigal JA, Bontrop RE (2000) Major histocompatibility complex class I diversity in a West African chimpanzee population: implications for HIV research. Immunogenetics 51:398–409
de Groot NG, Otting N, Doxiadis GG, Balla-Jhagjhoorsingh SS, Heeney JL, van Rood JJ, Gagneux P, Bontrop RE (2002) Evidence for an ancient selective sweep in the MHC class I gene repertoire of chimpanzees. Proc Natl Acad Sci U S A 99:11748–11753. doi:10.1073/pnas.182420799
de Groot NG, Heijmans CM, de Groot N, Otting N, de Vos-Rouweller AJ, Remarque EJ, Bonhomme M, Doxiadis GG, Crouau-Roy B, Bontrop RE (2008) Pinpointing a selective sweep to the chimpanzee MHC class I region by comparative genomics. Mol Ecol 17:2074–2088. doi:10.1111/j.1365-294X.2008.03716.x
de Groot NG, Heijmans CM, Zoet YM, de Ru AH, Verreck FA, van Veelen PA, Drijfhout JW, Doxiadis GG, Remarque EJ, Doxiadis II, van Rood JJ, Koning F, Bontrop RE (2010) AIDS-protective HLA-B*27/B*57 and chimpanzee MHC class I molecules target analogous conserved areas of HIV-1/SIVcpz. Proc Natl Acad Sci U S A 107:15175–15180. doi:10.1073/pnas.1009136107
de Groot NG, Otting N, Robinson J, Blancher A, Lafont BA, Marsh SG, O’Connor DH, Shiina T, Walter L, Watkins DI, Bontrop RE (2012) Nomenclature report on the major histocompatibility complex genes and alleles of great ape, old and new world monkey species. Immunogenetics 64:615–631. doi:10.1007/s00251-012-0617-1
de Groot NG, Bontrop RE (2013) The HIV-1 pandemic: does the selective sweep in chimpanzees mirror humankind’s future? Retrovirology 10:53. doi:10.1186/1742-4690-10-53
de Groot NG, Heijmans CM, van der Wiel MK, Blokhuis JH, Mulder A, Guethlein LA, Doxiadis GG, Claas FH, Parham P, Bontrop RE (2016) Complex MHC class I gene transcription profiles and their functional impact in orangutans. J Immunol 196:750–758. doi:10.4049/jimmunol.1500820
Domena JD, Hildebrand WH, Bias WB, Parham P (1993) A sixth family of HLA-A alleles defined by HLA-A*8001. Tissue Antigens 42:156–159
Fellay J, Shianna KV, Ge D, Colombo S, Ledergerber B, Weale M, Zhang K, Gumbs C, Castagna A, Cossarizza A, Cozzi-Lepri A, De Luca A, Easterbrook P, Francioli P, Mallal S, Martinez-Picado J, Miro JM, Obel N, Smith JP, Wyniger J, Descombes P, Antonarakis SE, Letvin NL, McMichael AJ, Haynes BF, Telenti A, Goldstein DB (2007) A whole-genome association study of major determinants for host control of HIV-1. Science 317:944–947. doi:10.1126/science.1143767
Fukami-Kobayashi K, Shiina T, Anzai T, Sano K, Yamazaki M, Inoko H, Tateno Y (2005) Genomic evolution of MHC class I region in primates. Proc Natl Acad Sci U S A 102:9230–9234. doi:10.1073/pnas.0500770102
Geller R, Adams EJ, Guethlein LA, Little AM, Madrigal JA, Parham P (2002) Linkage of Patr-AL to Patr-A and- B in the major histocompatibility complex of the common chimpanzee (Pan troglodytes). Immunogenetics 54:212–215. doi:10.1007/s00251-002-0452-x
Gleimer M, Wahl AR, Hickman HD, Abi-Rached L, Norman PJ, Guethlein LA, Hammond JA, Draghi M, Adams EJ, Juo S, Jalili R, Gharizadeh B, Ronaghi M, Garcia KC, Hildebrand WH, Parham P (2011) Although divergent in residues of the peptide binding site, conserved chimpanzee Patr-AL and polymorphic human HLA-A*02 have overlapping peptide-binding repertoires. J Immunol 186:1575–1588. doi:10.4049/jimmunol.1002990
Goyos A, Guethlein LA, Horowitz A, Hilton HG, Gleimer M, Brodsky FM, Parham P (2015) A distinctive cytoplasmic tail contributes to low surface expression and intracellular retention of the Patr-AL MHC class I molecule. J Immunol 195:3725–3736. doi:10.4049/jimmunol.1500397
Groves C (2001) Primate taxonomy. Smithsonian Institution Press, Washington DC
Guethlein LA, Flodin LR, Adams EJ, Parham P (2002) NK cell receptors of the orangutan (Pongo pygmaeus): a pivotal species for tracking the coevolution of killer cell Ig-like receptors with MHC-C. J Immunol 169:220–229
Guethlein LA, Older Aguilar AM, Abi-Rached L, Parham P (2007) Evolution of killer cell Ig-like receptor (KIR) genes: definition of an orangutan KIR haplotype reveals expansion of lineage III KIR associated with the emergence of MHC-C. J Immunol 179:491–504
Guethlein LA, Norman PJ, Hilton HH, Parham P (2015) Co-evolution of MHC class I and variable NK cell receptors in placental mammals. Immunol Rev 267:259–282. doi:10.1111/imr.12326
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl Acids Symp Ser 41:95–98
Hans JB, Haubner A, Arandjelovic M, Bergl RA, Fünfstück T, Gray M, Morgan DB, Robbins MM, Sanz C, Vigilant L (2015) Characterization of MHC class II B polymorphism in multiple populations of wild gorillas using non-invasive samples and next-generation sequencing. Am J Primatol 77:1193–1206. doi:10.1002/ajp.22458
Hedrick PW (1994) Evolutionary genetics of the major histocompatibility complex. Am Naturalist 143:945–964
Hoffmann HJ, Kristensen TJ, Jensen TG, Graugaard B, Lamm LU (1995) Antigenic characteristics and cDNA sequences of HLA-B73. Eur J Immunogenet 22:231–240
Hughes AL, Hughes MK (1995) Natural selection on the peptide-binding regions of major histocompatibility complex molecules. Immunogenetics 42:233–243
Ishikawa Y, Tokunaga K, Tanaka H, Nishimura M, Muraoka M, Fujii Y, Akaza T, Tadokoro K, Juji T (1996) HLA-A null allele with a stop codon, HLA-A*0215 N, identified in a homozygous state in a healthy adult. Immunogenetics 43:1–5
Jakobsen IB, Wilson SR, Easteal S (1998) Patterns of reticulate evolution for the classical class I and II HLA loci. Immunogenetics 48:312–323
Katoh K, Misawa K, Kuma K, Miyata T (2002) MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30:3059–3066
Keele BF, Van Heuverswyn F, Li Y, Bailes E, Takehisa J, Santiago ML, Bibollet-Ruche F, Chen Y, Wain LV, Liegeois F, Loul S, Ngole EM, Bienvenue Y, Delaporte E, Brookfield JF, Sharp PM, Shaw GM, Peeters M, Hahn BH (2006) Chimpanzee reservoirs of pandemic and nonpandemic HIV-1. Science 313:523–526. doi:10.1126/science.1126531
Keele BF, Jones JH, Terio KA, Estes JD, Rudicell RS, Wilson ML, Li Y, Learn GH, Beasley TM, Schumacher-Stankey J, Wroblewski E, Mosser A, Raphael J, Kamenya S, Lonsdorf EV, Travis DA, Mlengeya T, Kinsel MJ, Else JG, Silvestri G, Goodall J, Sharp PM, Shaw GM, Pusey AE, Hahn BH (2009) Increased mortality and AIDS-like immunopathology in wild chimpanzees infected with SIVcpz. Nature 460:515–519. doi:10.1038/nature08200
Kelley J, Walter L, Trowsdale J (2005) Comparative genomics of major histocompatibility complexes. Immunogenetics 56:683–695. doi:10.1007/s00251-004-0717-7
Kibbe WA (2007) OligoCalc: an online oligonucleotide properties calculator. Nucleic Acids Res 35:W43–W46. doi:10.1093/nar/gkm234
Kiepiela P, Leslie AJ, Honeyborne I, Ramduth D, Thobakgale C, Chetty S, Rathnavalu P, Moore C, Pfafferott KJ, Hilton L, Zimbwa P, Moore S, Allen T, Brander C, Addo MM, Altfeld M, James I, Mallal S, Bunce M, Barber LD, Szinger J, Day C, Klenerman P, Mullins J, Korber B, Coovadia HM, Walker BD, Goulder PJ (2004) Dominant influence of HLA-B in mediating the potential co-evolution of HIV and HLA. Nature 432:769–775. doi:10.1038/nature03113
Klein J, Figueroa F (1986) Evolution of the major histocompatibility complex. Crit Rev Immunol 6:295–386
Klein J, Bontrop RE, Dawkins RL, Erlich HA, Gyllensten UB, Heise ER, Jones PP, Parham P, Wakeland EK, Watkins DI (1990) Nomenclature for the major histocompatibility complexes of different species: a proposal. Immunogenetics 31:217–219
Klein J, Sato A, Nagl S, O’hUigín C (1998) Molecular trans-species polymorphism. Ann Rev Ecol Syst 29:1–21
Kulski JK, Shiina T, Anzai T, Kohara S, Inoko H (2002) Comparative genomic analysis of the MHC: the evolution of class I duplication blocks, diversity and complexity from shark to man. Immunol Rev 190:95–122
Lanier LL (2005) NK cell recognition. Annu Rev Immunol 23:225–274. doi:10.1146/annurev.immunol.23.021704.115526
Lawlor DA, Warren E, Ward FE, Parham P (1990) Comparison of class I MHC alleles in humans and apes. Immunol Rev 113:147–185
Lawlor DA, Warren E, Taylor P, Parham P (1991) Gorilla class I major histocompatibility complex alleles: comparison to human and chimpanzee class I. J Exp Med 174:1491–1509
Lawlor DA, Edelson BT, Parham P (1995) Mhc-a locus molecules in pygmy chimpanzees: conservation of peptide pockets. Immunogenetics 42:291–295
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452. doi:10.1093/bioinformatics/btp187
Maccari G, Robinson J, Ballingall K, Guethlein LA, Grimholt U, Kaufman J, Ho CS, de Groot NG, Flicek P, Bontrop RE, Hammond JA, Marsh SGE (2017) IPD-MHC 2.0: an improved inter-species database for the study of the major histocompatibility complex. Nucleic Acids Res in press
Marsh SG, Albert ED, Bodmer WF, Bontrop RE, Dupont B, Erlich HA, Fernández-Viña M, Geraghty DE, Holdsworth R, Hurley CK, Lau M, Lee KW, Mach B, Maiers M, Mayr WR, Müller CR, Parham P, Petersdorf EW, Sasazuki T, Strominger JL, Svejgaard A, Terasaki PI, Tiercy JM, Trowsdale J (2010) Nomenclature for factors of the HLA system, 2010. Tissue Antigens 75:291–455. doi:10.1111/j.1399-0039.2010.01466.x
Martin MP, Gao X, Lee JH, Nelson GW, Detels R, Goedert JJ, Buchbinder S, Hoots K, Vlahov D, Trowsdale J, Wilson M, O’Brien SJ, Carrington M (2002) Epistatic interaction between KIR3DS1 and HLA-B delays the progression to AIDS. Nat Genet 31:429–434. doi:10.1038/ng934
Martin DP, Murrell B, Golden M, Khoosal A, Muhire B (2015) RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evol 1:vev003. doi:10.1093/ve/vev003
Martínez-Laso J, Gómez-Casado E, Arnaiz-Villena A (2006) Description of seven new non-human primate MHC-B alleles. Tissue Antigens 67:85–88. doi:10.1111/j.1399-0039.2005.00496.x
Mayor NP, Robinson J, McWhinnie AJ, Ranade S, Eng K, Midwinter W, Bultitude WP, Chin CS, Bowman B, Marks P, Braund H, Madrigal JA, Latham K, Marsh SG (2015) HLA typing for the next generation. PLoS One 10:e0127153. doi:10.1371/journal.pone.0127153
McAdam SN, Boyson JE, Liu X, Garber TL, Hughes AL, Bontrop RE, Watkins DI (1995) Chimpanzee MHC class I a locus alleles are related to only one of the six families of human a locus alleles. J Immunol 154:6421–6429
Moeller AH, Peeters M, Ayouba A, Ngole EM, Esteban A, Hahn BH, Ochman H (2015) Stability of the gorilla microbiome despite simian immunodeficiency virus infection. Mol Ecol 24:690–697. doi:10.1111/mec.13057
Moesta AK, Abi-Rached L, Norman PJ, Parham P (2009) Chimpanzees use more varied receptors and ligands than humans for inhibitory killer cell Ig-like receptor recognition of the MHC-C1 and MHC-C2 epitopes. J Immunol 182:3628–3637. doi:10.4049/jimmunol.0803401
Ohta T (1983) On the evolution of multigene families. Theor Popul Biol 23:216–240
Older Aguilar AM, Guethlein LA, Adams EJ, Abi-Rached L, Moesta AK, Parham P (2010) Coevolution of killer cell Ig-like receptors with HLA-C to become the major variable regulators of human NK cells. J Immunol 185:4238–4251. doi:10.4049/jimmunol.1001494
Pandrea I, Silvestri G, Apetrei C (2009) AIDS in African nonhuman primate hosts of SIVs: a new paradigm of SIV infection. Curr HIV Res 7:57–72
Parham P, Ohta T (1996) Population biology of antigen presentation by MHC class I molecules. Science 272:67–74
Parham P, Moffett A (2013) Variable NK cell receptors and their MHC class I ligands in immunity, reproduction and human evolution. Nat Rev Immunol 13:133–144. doi:10.1038/nri3370
Parham P, Lomen CE, Lawlor DA, Ways JP, Holmes N, Coppin HL, Salter RD, Wan AM, Ennis PD (1989) Nature of polymorphism in HLA-A, -B, and -C molecules. Proc Natl Acad Sci U S A 85:4005–4009
Parham P, Arnett KL, Adams EJ, Barber LD, Domena JD, Stewart D, Hildebrand WH, Little AM (1994) The HLA-B73 antigen has a most unusual structure that defines a second lineage of HLA-B alleles. Tissue Antigens 43:302–313
Plantier JC, Leoz M, Dickerson JE, De Oliveira F, Cordonnier F, Lemée V, Damond F, Robertson DL, Simon F (2009) A new human immunodeficiency virus derived from gorillas. Nat Med 15:871–872. doi:10.1038/nm.2016
Robinson J, Halliwell JA, Hayhurst JH, Flicek P, Parham P, Marsh SGE (2015) The IPD and IMGT/HLA database: allele variant databases. Nucleic Acids Res 43:D423–D431. doi:10.1093/nar/gku1161
Ross MG, Russ C, Costello M, Hollinger A, Lennon NJ, Hegarty R, Nusbaum C, Jaffe DB (2013) Characterizing and measuring bias in sequence data. Genome Biol 14:R51. doi:10.1186/gb-2013-14-5-r51
Rothschild BM, Woods RJ (1989) Spondyloarthropathy in gorillas. Semin Arthritis Rheum 18:267–276
Ruvolo M, Disotell TR, Allard MW, Brown WM, Honeycutt RL (1991) Resolution of the African hominoid trichotomy by use of a mitochondrial gene sequence. Proc Natl Acad Sci U S A 88:1570–1574
SantaLucia J Jr (1998) A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proc Natl Acad Sci U S A 95:1460–1465
Scally A, Dutheil JY, Hillier LW, Jordan GE, Goodhead I, Herrero J, Hobolth A, Lappalainen T, Mailund T, Marques-Bonet T, McCarthy S, Montgomery SH, Schwalie PC, Tang YA, Ward MC, Xue Y, Yngvadottir B, Alkan C, Andersen LN, Ayub Q, Ball EV, Beal K, Bradley BJ, Chen Y, Clee CM, Fitzgerald S, Graves TA, Gu Y, Heath P, Heger A, Karakoc E, Kolb-Kokocinski A, Laird GK, Lunter G, Meader S, Mort M, Mullikin JC, Munch K, O’Connor TD, Phillips AD, Prado-Martinez J, Rogers AS, Sajjadian S, Schmidt D, Shaw K, Simpson JT, Stenson PD, Turner DJ, Vigilant L, Vilella AJ, Whitener W, Zhu B, Cooper DN, de Jong P, Dermitzakis ET, Eichler EE, Flicek P, Goldman N, Mundy NI, Ning Z, Odom DT, Ponting CP, Quail MA, Ryder OA, Searle SM, Warren WC, Wilson RK, Schierup MH, Rogers J, Tyler-Smith C, Durbin R (2012) Insights into hominid evolution from the gorilla genome sequence. Nature 483:169–175. doi:10.1038/nature10842
Scally A, Yngvadottir B, Xue Y, Ayub Q, Durbin R, Tyler-Smith C (2013) A genome-wide survey of genetic variation in gorillas using reduced representation sequencing. PLoS One 8:e65066. doi:10.1371/journal.pone.0065066
Sharp PM, Hahn BH (2011) Origins of HIV and the AIDS pandemic. Cold Spring Harb Perspect Med 1:a006841. doi:10.1101/cshperspect.a006841
Shiina T, Ota M, Shimizu S, Katsuyama Y, Hashimoto N, Takasu M, Anzai T, Kulski JK, Kikkawa E, Naruse T, Kimura N, Yanagiya K, Watanabe A, Hosomichi K, Kohara S, Iwamoto C, Umehara Y, Meyer A, Wanner V, Sano K, Macquin C, Ikeo K, Tokunaga K, Gojobori T, Inoko H, Bahram S (2006) Rapid evolution of major histocompatibility complex class I genes in primates generates new disease alleles in humans via hitchhiking diversity. Genetics 173:1555–1570. doi:10.1534/genetics.106.057034
Shimizu M, Nakajima F, Okazaki H, Satake M, Tadokoro K (2013) Two novel null HLA-A alleles with identical exon 4 nonsense mutations: HLA-A*24:183 N and a*02:356 N. Tissue Antigens 82:136–137. doi:10.1111/tan.12139
Single RM, Martin MP, Gao X, Meyer D, Yeager M, Kidd JR, Kidd KK, Carrington M (2007) Global diversity and evidence for coevolution of KIR and HLA. Nat Genet 39:1114–1119. doi:10.1038/ng2077
Sommer S (2005) The importance of immune gene variability (MHC) in evolutionary ecology and conservation. Front Zool 2:16. doi:10.1186/1742-9994-2-16
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729. doi:10.1093/molbev/mst197
Thalmann O, Fischer A, Lankester F, Pääbo S, Vigilant L (2007) The complex evolutionary history of gorillas: insights from genomic data. Mol Biol Evol 24:146–158. doi:10.1093/molbev/msl160
Urvater JA, Hickman H, Dzuris JL, Prilliman K, Allen TM, Schwartz KJ, Lorentzen D, Shufflebotham C, Collins EJ, Neiffer DL, Raphael B, Hildebrand W, Sette A, Watkins DI (2001) Gorillas with spondyloarthropathies express an MHC class I molecule with only limited sequence similarity to HLA-B27 that binds peptides with arginine at P2. J Immunol 166:3334–3344
Van Heuverswyn F, Li Y, Neel C, Bailes E, Keele BF, Liu W, Loul S, Butel C, Liegeois F, Bienvenue Y, Ngolle EM, Sharp PM, Shaw GM, Delaporte E, Hahn BH, Peeters M (2006) Human immunodeficiency viruses: SIV infection in wild gorillas. Nature 444:164. doi:10.1038/444164a
Venditti CP, Lawlor DA, Sharma P, Chorney MJ (1996) Structure and content of the major histocompatibility complex (MHC) class I regions of the great anthropoid apes. Hum Immunol 49:71–84
Watanabe Y, Tokunaga K, Geraghty D, Tadokoro K, Juji T (1997) Large-scale comparative mapping of the MHC class I region of predominant haplotypes in Japanese. Immunogenetics 46:135–141. doi:10.1007/s002510050252
Watkins DI, Chen ZW, Garber TL, Hughes AL, Letvin NL (1991) Segmental exchange between MHC class I genes in a higher primate: recombination in the gorilla between the ancestor of a human non-functional gene and an a locus gene. Immunogenetics 34:185–191
Wroblewski EE, Norman PJ, Guethlein LA, Rudicell RS, Ramirez MA, Li Y, Hahn BH, Pusey AE, Parham P (2015) Signature patterns of MHC diversity in three Gombe communities of wild chimpanzees reflect fitness in reproduction and immune defense against SIVcpz. PLoS Biol 13(5):e1002144. doi:10.1371/journal.pbio.1002144
Vilches C, Parham P (2002) KIR: diverse, rapidly evolving receptors of innate and adaptive immunity. Annu Rev Immunol 20:217–251. doi:10.1146/annurev.immunol.20.092501.134942
Williams F, Curran MD, Middleton D (1999) Characterisation of a novel HLA-A pseudogene, HLA-BEL, with significant sequence identity with a gorilla MHC class I gene. Tissue Antigens 54:360–369
Xue Y, Prado-Martinez J, Sudmant PH, Narasimhan V, Ayub Q, Szpak M, Frandsen P, Chen Y, Yngvadottir B, Cooper DN, de Manuel M, Hernandez-Rodriguez J, Lobon I, Siegismund HR, Pagani L, Quail MA, Hvilsom C, Mudakikwa A, Eichler EE, Cranfield MR, Marques-Bonet T, Tyler-Smith C, Scally A (2015) Mountain gorilla genomes reveal the impact of long-term population decline and inbreeding. Science 348:242–245. doi:10.1126/science.aaa3952
Yasukochi Y, Ohashi J (2016) Elucidating the origin of HLA-B*73 allelic lineage: did modern humans benefit by archaic introgression? Immunogenetics. doi:10.1007/s00251-016-0952-8
Zemmour J, Parham P (1992) Distinctive polymorphism at the HLA-C locus: implications for the expression of HLA-C. J Exp Med 176:937–950
Acknowledgements
Open access funding provided by Max Planck Society. This work was funded by Max Planck Society. We thank Christoph König and Philip Lobb from Pacific Biosciences, Janet Kelso and the Sequencing group, as well as Montgomery Slatkin for helpful discussions. Samples used in this study were previously collected by the veterinarians and staff of the following zoos and we are grateful for their assistance: North Carolina Zoo (Asheboro, USA), Zoo Atlanta (USA), Brookfield Zoo (Chicago, USA), and Lincoln Park Zoo (Chicago, USA). We also thank Kathrin Köhler and Birgit Nickel for assistance with archived samples.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Hans, J.B., Bergl, R.A. & Vigilant, L. Gorilla MHC class I gene and sequence variation in a comparative context. Immunogenetics 69, 303–323 (2017). https://doi.org/10.1007/s00251-017-0974-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00251-017-0974-x