
Abstract
Various factors of the tissue microenvironment such as the oxygen concentration influence the host–pathogen interaction. During the past decade, hypoxia-driven signaling via hypoxia-inducible factors (HIF) has emerged as an important factor that affects both the pathogen and the host. In this chapter, we will review the current knowledge of this complex interplay, with a particular emphasis given to the impact of hypoxia and HIF on the inflammatory and antimicrobial activity of myeloid cells, the bacterial responses to hypoxia and the containment of bacterial infections under oxygen-limited conditions. We will also summarize how low oxygen concentrations influence the metabolism of neutrophils, macrophages and dendritic cells. Finally, we will discuss the consequences of hypoxia and HIFα activation for the invading pathogen, with a focus on Pseudomonas aeruginosa, Mycobacterium tuberculosis, Coxiella burnetii, Salmonella enterica and Staphylococcus aureus. This includes a description of the mechanisms and microbial factors, which the pathogens use to sense and react to hypoxic conditions.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Myeloid cells are the first line of defense against bacterial infections. They are equipped with an arsenal of mechanisms to prevent spreading of the intruders, to alert the adaptive immune system, to prevent bacterial proliferation and to eliminate the pathogens without inducing immunopathology (reviewed: [1]). In recent years, it has become clear that the interplay between myeloid cells and the pathogen is strongly affected by the (patho)physiological conditions prevailing at the site of infection. Thus, tonicity, availability of nutrients and oxygen tension significantly influence the outcome of the host–pathogen interaction [2,3,4,5]. The oxygen availability is of particular interest in this context. Several important antimicrobial effector pathways require oxygen, such as the phagocyte NADPH-oxidase (PHOX), which generates reactive oxygen species (ROS), and the inducible or type 2 nitric oxide synthase (iNOS or NOS2), which produces high amounts of nitric oxide (NO) and leads to the formation of subsequent reactive nitrogen species (RNS). Both ROS and NO are capable of damaging and killing bacterial microorganisms and, therefore, are important to control the infection [6]. In addition, it is well established that oxygen levels differ in various organs (reviewed: [7]). Even under resting conditions, the oxygen level of the renal medulla, skin, and bone marrow are low [8,9,10]. In these organs, the availability of oxygen and, hence, the tissue oxygenation is thought to be largely dependent on the organ-specific vascular network. Not only vascularization and supply of oxygen, but also the consumption rate influences the oxygen level available within the tissue. Infiltration of immune cells in an organ increases the consumption of oxygen and as a consequence reduces the available oxygen level [11]. Similarly, the low oxygen levels found in epithelial layers facing the gastrointestinal lumen [12, 13] result from the metabolism of gastrointestinal microbiota [14] and the action of their products on the host epithelium [15]. These latter examples demonstrate that already under resting conditions, bacteria have an impact on the oxygen availability in host tissue. Importantly, infections with microbial pathogens lead to oxygen consumption in the affected tissues, which influences the host as well as the pathogen and their interplay and, thus, the outcome of infection.
In the following, we will review (i) the basic methodology to measure oxygen in tissues; (ii) the principle impact of infections on tissue oxygen levels; (iii) the metabolism of innate immune cells under hypoxic conditions; (iv) the role of hypoxia-inducible factors (HIF); (v) the functional regulation of myeloid cells by hypoxia and HIF; (vi) the sensing of hypoxia by bacteria and their reaction to oxygen deprivation; and (vii) the mechanisms of control of bacterial infections under hypoxic conditions.
Methods to quantify tissue oxygenation
Progress in our understanding of tissue oxygenation is limited by the fact that quantification of tissue oxygen is a difficult and tedious task (reviewed: [16, 17]). Over the last decades, different studies either employed the Clark polarographic electrode technique, used histochemical staining techniques to detect severely hypoxic regions with the help of 2-nitroimidazole derivatives (e. g., EF5, piminidazole, CCI-103F) or applied luminescence-based technologies to monitor tissue oxygen levels (reviewed: [16]). More recently, positron emission tomography (PET), single-photon emission computed tomography (SPECT) and magnetic resonance imaging (MRI)-based technologies have become available and offer new opportunities to assess oxygen levels in inflamed and infected tissues [18,19,20,21]. The advantage of these methods is that they are noninvasive and do not cause tissue injuries. PET/SPECT/MRI entered preclinical and clinical application in late 2000s/early 2010s for oxygen quantification. These methods rely on administration of various hypoxia tracers that enable in vivo oxygen measurement without tissue destruction (reviewed: [21]). Several studies were performed using [18F]fluoromisonidazole ([18F]FMISO), which is often referred to as a “gold standard” in PET/SPECT. However, several disadvantages of [18F]FMISO led to the development of novel tracers, such as [64Cu][Cu-diacetyl-bis(N(4)-methylthiosemicarbazone)] ([64Cu][Cu(ATSM)]), 68 Ga-labeled tracers, technetium-99 m ([99mTc]Tc-BRU59-21, [99mTc]Tc-EDTA-2-MN) or molybdenum-99 co-labeled nitroimidazole-containing or nitroimidazole-free compounds [22,23,24,25,26,27].
Infection triggers low tissue oxygen levels: underlying mechanisms
There is substantial evidence that inflamed and infected tissue displays low oxygen levels (reviewed: [16]). However, only recently, studies on the mechanisms that account for the reduced oxygen levels in infected and inflamed tissues were conducted. In a mouse model of pyelonephritis, inflammation-induced clotting contributed to a low oxygen microenvironment in the kidney [28]. Moreover, inflammation was able to trigger clotting processes and vice versa (reviewed: [29]). Clotting of vessels in infected tissues can help to sequester and compartmentalize infections [30], but will result in reduced oxygen levels in the afflicted tissues. Thus, induction of low tissue oxygenation might be a side effect of host efforts to inhibit bacterial spreading.
The influx of neutrophils also plays a role in mediating low tissue oxygenation upon an infection. In a seminal study, Campbell et al. demonstrated in a model of dextran sulfate sodium (DSS)-induced colitis that the influx of polymorphonuclear neutrophils (PMN) and their NADPH oxidase activity caused increased oxygen consumption and ultimately low mucosal tissue oxygenation [11]. As Campbell et al. used DSS to induce colitis and not a specific pathogen, it is formally unclear whether infections with intestinal pathogens induce low oxygen levels in mouse mucosal gut tissue. However, infections with enteropathogenic bacteria such as Salmonella and Shigella were shown to trigger low oxygen levels in infected lamina propria and serosal gut tissue [31,32,33,34]. The exact mechanisms that lead to low oxygen levels in Salmonella-infected tissues are still unknown. It is possible that Salmonella inhibits inflammation-triggered de novo formation of blood vessels by limiting vascular endothelial growth factor (VEGF)-driven angiogenesis [35]. In a Shigella infection model, the relative contribution of the oxygen consumption induced by enteropathogens and/or the infection-triggered inflammation was investigated. Tinevez and coworkers found that, unlike the above-mentioned Salmonella studies, the pathogen themselves, but not the infiltrating PMN, were largely responsible for the low oxygen conditions in the gut mucosa [34].
Nonetheless, in addition to the above-mentioned study, analyses in other models confirmed that PMN critically contributes to low tissue oxygen levels. The influx of PMN reduced local oxygen levels in a preclinical model of Herpes simplex virus 1 (HSV-1) keratitis [36]. Similarly, accumulation of PMN into a Candida albicans-induced subdermal abscess triggered a hypoxic microenvironment [37]. In addition, interleukin 1 (IL-1)-dependent signaling elicited low O2 levels in a preclinical model of pulmonary aspergillosis [38]. These findings suggest that the inflammatory response of the host and especially the activities of neutrophils are the main drivers of infection-triggered low tissue oxygenation .
Thus, from a mechanistic point of view, the reduced oxygen levels in infected tissues can result from several, presumably overlapping processes: (i) oxygen consumption by the invading immune cells; (ii) oxygen consumption by the invading pathogens; (iii) induction of host cell signaling cascades that control oxygen availability, by bacterial compounds and/or by host cell products; (iv) alterations of the microenvironment during the infection that result in changes in metabolism and thereby oxygen availability.
Metabolic response of innate immune cells to hypoxia
Similar to other immune cells, myeloid cells require various cellular metabolic pathways to function and respond properly (reviewed: [39], Fig. 1). The prevailing metabolic pathways vary between different types and activation statuses of cells. Moreover, depending on the local microenvironment in which the cells reside, they must adapt their cellular metabolism (reviewed: [40]).

Cellular metabolism differences under normoxia and hypoxia or between resting or activated profiles of immune cells. Regardless of the oxygen or activation status, neutrophils witness increased glycolysis and decreased tricarboxylic acid (TCA) cycle and oxidative phosphorylation (OXPHOS) activity, with an augmentation of glycolysis under hypoxia or upon activation. Resting or normoxic macrophages/dendritic cells, however, depend on the TCA cycle to ensure longevity and biomass. Upon exposure to hypoxia or pro-inflammatory signals, they switch to heavy glycolysis and hamper the TCA cycle activity as well as OXPHOS. Macrophages, which have encountered anti-inflammatory signals, on the other hand, depend greatly on the TCA cycle activity and OXPHOS
As discussed above, oxygen is limited in inflamed and infected tissues. Therefore, immune cells have to operate under hypoxic conditions and, consequently, switch to anaerobic glycolysis (reviewed: [41]), while the oxygen-dependent energy generation via the tricarboxylic acid (TCA) cycle is suppressed (reviewed: [39, 42,43,44,45,46,47]).
However, not only hypoxic immune cells rely on glycolysis for energy generation, but in general, activated or proliferating immune cells also shift their metabolism to aerobic glycolysis, which resembles a metabolic profile observed by Otto Warburg in tumor cells (Warburg effect) (reviewed: [41, 42]). Instead of using oxidative phosphorylation (OXPHOS) for highly efficient adenosine triphosphate (ATP) production, proliferating cells tend to increase their glycolysis rate, which, however, only generates two ATP molecules per round of glycolysis (reviewed: [48]). To compensate for the reduced or absent mitochondrial OXPHOS, proliferating cells need to increase their glucose uptake and boost their glycolytic activity to meet the energy demand of proliferating cells (reviewed: [41, 48]). Since activation of immune cells under normoxic conditions already triggers a switch to glycolytic energy generation, these cells are well prepared to operate in inflamed areas, which are very likely to display low oxygen levels. The hypoxia-inducible factor (HIF) plays an important role not only for the adaptation of cells to oxygen-poor environments (reviewed: [49, 50]), but also under normoxic conditions (reviewed: [39, 42, 44,45,46, 51]). Therefore, we will very briefly review the molecular mechanisms that result in HIF stabilization in myeloid cells and we will describe the role of HIF in the immunobiology of neutrophils, macrophages, and dendritic cells.
Hypoxia and inflammation-/infection-triggered HIF stabilization
HIF, initially discovered as oxygen-dependent complex for erythropoietin induction in the liver and kidney, is part of the PER-ARNT-SIM (PAS) protein subfamily of the basic helix-loop-helix (bHLH) family (reviewed: [52]). This dimeric transcription factor consists of two subunits (HIFα and HIFβ), which have to dimerize to attach to the promoter region of target genes harboring the hypoxia response element (HRE). There is only one HIFα isoform, but there are three closely related HIFß isoforms, HIF1α, HIF2α, and HIF3α (reviewed: [53]). HIF1α is considered a ubiquitous transcription factor. According to the Immgen Database (www.immgen.org), all myeloid cells are able to express HIF1α mRNA at medium to high range level [54]. In contrast, the expression of HIF2α is much more restricted. Thus, HIF2α is expressed in endothelial cells [55, 56], but is also present in some immune cells [57, 58]. It is known that various cells express HIF3α [59], but there is only very limited data on its expression in innate immune cells. While thioglycolate-elicited peritoneal macrophages express prominent amounts of HIF2α mRNA, the mRNA expression of both HIF2α and HIF3α in other myeloid cells is low [54].
Hypoxic HIF stabilization
HIF activity is regulated by modulating the stability of its α-subunit. Under conditions of ample oxygen, HIF1α is readily hydroxylated at proline residues located in the oxygen-dependent degradation domain, which is present in all three HIFα isoforms. Hydroxylation of the HIFα isoforms are governed by a class of enzymes called prolyl hydroxylases (PHDs), which serve as cellular oxygen sensors. PHD-mediated HIF1α-hydroxylation is highly specific [60] and leads to ubiquitination of HIF1α by von Hippel–Lindau (VHL) E3 ubiquitin ligase, which ultimately targets HIF1α for proteasomal degradation. Of note, PHD requires the presence of its cofactor Fe2+ and the co-substrate 2-oxoglutarate for HIF1α hydroxylation (reviewed: [61]). In the absence of oxygen, its co-substrate or co-factors, the PHD activity is suppressed, which in turn triggers subsequent HIF1α activation (reviewed: [49, 50, 52, 61]).
HIFα governs the expression of a plethora of different genes involved in metabolism, immune system regulation, and in general cellular functions in response to hypoxia (reviewed: [39, 42, 44,45,46, 51]). A detailed global assessment of HIF1α and HIF2α binding sites in MCF7 breast cancer cells, for instance, revealed that many of these sites bound HIF1α and HIF2α equally well, while there were only very few sites that bound HIF2α exclusively [62]. It will require further endeavors to understand differential regulation of target genes by HIFα isoforms in various cell types [63], including immune cells.
Inflammatory and infectious HIFα stabilization
In addition to hypoxia, other non-hypoxic stimuli are able to induce HIFα accumulation. This holds especially true for immune cells in which several inflammatory stimuli and cytokines are known to trigger HIFα accumulation (reviewed: [2, 7, 16, 64,65,66]. In line with this, there is evidence that a large array of human pathogens or microbial products are able to induce HIF1α accumulation even in the presence of O2 [67]. The most prominent and best studied pathogen-associated molecular pattern in this respect is lipopolysaccharide (LPS), a component of the cell wall of Gram-negative bacteria [68]. Under normoxic conditions, the LPS-triggered HIFα accumulation depends on nuclear factor (NF)-κB- and p42/44 MAPK-dependent signal transduction [69, 70]. Ultimately, this results in PHD inhibition via (i) enhanced ROS and RNS production [71,72,73,74], (ii) metabolic inhibition of PHD by succinate accumulation [75], and/ or (iii) decrease of availability of the PHD cofactor Fe2+ [3]. Adding another layer of complexity, HIF activation induced by LPS leads to a distinct and different response of myeloid cells compared to hypoxia-driven HIF activation [76]. The mechanisms that underlie this divergent response are still unclear and warrant further investigation (reviewed: [16]).
Recently, Solis et al. discovered a novel mechanism leading to HIF1α stabilization in innate immunity, which turned out to be very different from hypoxic or inflammatory HIF1α stabilization [77]. The authors studied how mechanosensation activates innate immunity. Immune cells-infiltrating sites of infection in the lung faced cyclical hydrostatic pressure [77, 78]. Monocytes detected this mechanical force via PIEZO1, a mechanically activated ion channel. Ca2+-influx by PIEZO1 subsequently triggered activating protein-1 (AP-1), which led to the transcription of endothelin-1 (Edn1). EDN1, in turn, caused HIF1α stabilization that upregulated pro-inflammatory genes and thereby facilitated monocyte-driven pathogen clearance [77].
In addition, bacterial virulence factors such as the Bartonella henselae adhesion A (BadA) and bacterial siderophores (Fe-chelating agents) are able to trigger normoxic HIF1α stabilization ([79,80,81]; Table 1).
An unexpected observation was that certain pathogens promoted the degradation of HIF1α rather than causing HIF1α stabilization (Table 1). For instance, Salmonella interfered with HIF1α accumulation [35]. Moreover, Pseudomonas aeruginosa 2-alkyl-4-quinolone (AQ) quorum sensing signaling molecule directly targeted HIF1α for proteasomal degradation independently of PHDs [82]. Similarly, HIF1α was degraded during the late phases of intracellular chlamydial replication. In contrast, during the early phase of infection, C. trachomatis enhanced HIF1α stabilization [83].
Altogether, these findings already suggest that HIF1α is not only required for the cellular adaptation to hypoxia, but also for the immune response to infection under normoxic conditions. Therefore, for studying HIFα-responses in the context of infections, we need to take into account that the HIFα response may be triggered and manipulated by the pathogens itself (Table 1) and/or the low oxygen environment induced by the infection.
Role of hypoxia and HIF in the immunobiology of neutrophils, macrophages, and dendritic cells
Hypoxia and neutrophils
Both inflammatory and hypoxic HIF1α stabilization play a major role in activated neutrophils [84]. Since neutrophils require high ATP to combat infections, they depend on increased glycolysis to meet their energetic needs. Interestingly, neutrophils possess only a limited number of mitochondria, which, in addition, do not participate in the production of ATP, but are rather involved in regulating cell death decisions [42, 85]. Thus, neutrophils most likely depend on glycolysis for ATP-production. This assumption is supported by the observation that ATP-production was reduced in neutrophils treated with an inhibitor of glycolysis (2-deoxyglucose). In 2003, Cramer et al. showed that peritoneal neutrophils lacking HIF1α produced 40% less ATP than wild-type controls [86]. The fact that HIF1α, induced by hypoxia or by LPS stimulation, increases the expression of glycolytic target genes, including pyruvate kinase M2 (Pkm2), phosphoglycerate kinase (Pgk), glyceraldehyde 3-phosphate dehydrogenase (Gapdh), and triosephosphate isomerse-1 (Tpi1) [87,88,89], offers an explanation for the crucial role of HIF1α in ATP production.
Stabilization of HIFα in neutrophils using a PHD2-deficient mouse model revealed that HIFα is critically involved in augmented inflammatory and antimicrobial responses against Streptococcus pneumoniae through rapid recruitment, enhanced chemotaxis, and prolonged survival of neutrophils. Of note, stabilization of HIFα by interfering with PHD activity in neutrophils did not trigger changes in respiratory burst or in inner mitochondrial membrane potential [89]. However, hypoxic HIF1α enhanced production of granule proteases (neutrophil elastase and cathepsin G) and antimicrobial peptides (cathelicidin) in neutrophils [90]. In addition, HIF1α (but not hypoxia; see “Hypoxia-mediated containment of bacterial infections”) promoted the formation of neutrophil extracellular traps (NET) [91]. Normoxic [92] and hypoxic HIF1α also increased the lifespan of the otherwise short-lived neutrophils by inhibiting apoptosis via activating the NF-κB pathway [87]. This was accompanied by the hypoxia-induced release of the macrophage inflammatory protein-1β (MIP-1β) enhancing the survival effect of neutrophils under hypoxia [87].
Similar to HIF1α, HIF2α was able to prolong longevity of granulocytes as well, while having little impact on phagocytosis of bacteria [58]. Overall, these findings clearly demonstrate that HIFα is not only critical for maintaining the energy homeostasis of neutrophils, but, in addition, is critical for maintaining the longevity and inflammatory activity of neutrophils.
Macrophages and hypoxia
Similar to neutrophils, macrophages also increase their glycolytic activity upon exposure to hypoxia and/ or inflammatory/ infectious conditions. In macrophages, HIF1α forms a complex with the pyruvate kinase M2 (PKM2) and thereby contributes to the upregulation of glycolytic activity of macrophages ( [75, 93], Fig. 1). Interestingly, HIF1α was not only involved in fostering metabolic reprogramming, but regulated the expression of pathogen-recognition molecules (e.g., toll-like receptor 4) [94], antimicrobial peptides [90], and of inducible NO synthase (NOS2) [90]. In addition, HIF1α activity was negatively correlated with IL-10 production [74, 95] and promoted the expression of IL-1β [75, 93], which ultimately helped fighting invading intruders [75, 86, 90, 93, 96,97,98].
In contrast to HIF1α, much less is known about the role of HIF2α in macrophages. Similar to HIF1α-knockout macrophages, HIF2α-deficient macrophages displayed reduced inflammatory responses compared to controls [57]. In contrast to HIF1α, however, HIF2α appears to be particularly important for the induction of regulatory and/or anti-inflammatory cascades. For instance, IL-4-activated macrophages expressed HIF2α, which induced arginase 1 and suppressed NO synthesis, while in classically activated macrophages, LPS-induced HIF1α raised the expression of NOS2 and thereby the generation of NO [99].
Dendritic cells and hypoxia
As demonstrated earlier for neutrophils and macrophages, HIF1α stabilization contributed to increased glycolytic activities in activated mouse and human dendritic cells (DCs) [44, 100,101,102,103]. This supported the migration of DCs to lymph nodes to stimulate an immune response, which involves C–C chemokine receptor type 7 (CCR7) upregulation [104]. Inhibition of HIF1α [105] and HIF1α-dependent glycolytic activity impaired the migratory capacity of DC [106]. In addition, HIF1α was required for antigen presentation [107] and maturation [107, 108] and promoted DC-dependent T cell proliferation/activation [107,108,109]. Interestingly, an increase in glycolytic activity or stabilization of HIF1α alone was not sufficient to drive inflammatory outputs [108, 110]. However, under inflammatory conditions, HIF1α upregulated, in addition to glycolytic genes, a set of inflammatory genes, such as prostaglandin-endoperoxide synthase 2 (Ptgs2) and NOS2, which, in addition to HIF1α, co-depended on NF-κB [76]. Thus, the context of HIF1α activation is of critical relevance (reviewed: [16]). Recently, evidence was provided that the strength of the signal used to trigger inflammatory DC activation influences DC metabolism and their inflammatory output [104]. In contrast to weak DC activators such as “house dust mite-derived allergens”, a potent pro-inflammatory stimulus, such as LPS, led to a strong HIF1α-dependent pro-inflammatory phenotype, an increase in glycolysis and cessation of mitochondrial respiration. This is in line with earlier findings that the long-term commitment to glycolysis in activated DCs was indirectly regulated by PI(3)K-Akt-mTORC1 upregulation of NOS2 and HIF1α [111]. Moreover, conditions that resulted in excess glycolytic activity of DC even limited the ability of DC to induce T cell responses [73]. Weakly activated DCs, in contrast, showed no significant HIF1α accumulation, along with decreased pro-inflammation and absent glycolytic reprogramming [104]. Collectively, these findings suggest that fine-tuning of the immune-metabolism via HIF1α holds great potential in modifying DC immunobiology.
HIF1α is not only important for acute innate immune responses, but is also an important factor in trained immunity [112]. Trained immunity is a term for the memory-like response that is generated in innate immune cells due to a previous inflammatory stimulus which enables innate immune cells to respond more vigorously to a second insult in a nonspecific manner [113,114,115]. Trained immunity requires Akt-mTOR-HIF1α-dependent glycolytic reprogramming [114, 116].
However, boosting of HIF1α activity to fight infections bears risks as well. Recent evidence demonstrates that increasing HIF1α responses by subjecting mice to acute low oxygen environments aggravated inflammatory responses and can be detrimental. In contrast, long-term exposure to low oxygen environments dampened HIF1α activation, glycolysis and decreased overall pathology [117]. Further studies are required to understand the differences of HIF1α signaling in hypoxic preconditioning versus its role in trained immunity.
Collectively, neutrophils, macrophages and DCs are adept in accommodating to hypoxic niches by adjusting their inflammatory and metabolic activity (Fig. 2). HIF1α plays a key role in the regulation of this bidirectional interplay between metabolism and inflammation.

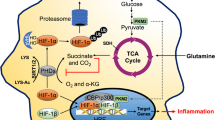
Antimicrobial mechanisms of macrophages under hypoxia. a Prolyl hydroxlases (PHD) are inactive in the absence of oxygen and thus, HIF1α is stabilized. HIF1α translocates into the nucleus, where it dimerizes with HIF1β. The dimer binds to the hypoxia responsive elements (HRE) and induces the transcription of target genes. b Hypoxia induces increased toll-like receptor 4 (TLR-4) expression. c Under hypoxia, the expression of glucose transporter GLUT1 is enhanced, which leads to increased glucose uptake into the cell. Hypoxia also enhances glycolysis. The end product of glycolysis, pyruvate, is metabolized into lactate. Due to the inhibition of the TCA cycle, less citrate and itaconate (the antimicrobial metabolite) are generated. Furthermore, hypoxia impairs OXPHOS. d Cathelicidin, the antimicrobial peptide, is also augmented under hypoxia. Cathelicidin can then be transported into the phagolysosome to eliminate pathogens. e The phagocytic uptake of macrophages is enhanced under hypoxia. f The oxygen-dependent effectors, PHOX and NOS2, are impaired under hypoxia. Thus, less ROS and RNS production is evident. g The process of autophagy is also increased under hypoxia
Bacterial sensing of and reaction to hypoxia
Bacterial oxygen sensing
Not only does the host sense hypoxia and react to it accordingly, but also the pathogen perceives and responds to drops in oxygen levels (Table 1). The bacterial response to hypoxia is species specific and depends on the genetic configuration. Enteric pathogens, for example, are capable of growing under low oxygen conditions (reviewed: [118]). However, even the obligative aerobic species of Pseudomonas (P.) aeruginosa is able to adapt to low oxygen levels and persist under these conditions [119]. To change transcription and translation, the pathogen has to be able to sense the altered environmental conditions. Thus, P. aeruginosa contains a homolog to the eukaryotic 2-oxoglutarate-dependent prolyl hydroxylases that are key oxygen sensors. This homolog was termed Pseudomonas prolyl-hydroxylase domain-containing protein (PPHD) and has been implicated in oxygen sensing [120, 121]. In addition, PPDH induces the expression of several virulence factors, and pharmacological repression of PPDH reduces the pathogenicity of P. aeruginosa in a mouse model of pneumonia [121]. The fact that PPDH also influences antibiotic susceptibility, expression of efflux pumps, motility and biofilm formation [122] demonstrates the importance of this oxygen sensor. P. aeruginosa encodes for additional oxygen sensors. One of them, the response regulator AtvR, seems to be important for survival under hypoxic conditions within the host [123]. Hypoxia influences P. aeruginosa transcription, which allows the pathogen to adapt metabolically [124].
Similarly to P. aeruginosa, M. tuberculosis grows better under normoxic conditions than in low oxygen environments [125]. Nonetheless, M. tuberculosis is capable of adapting to hypoxic environments. M. tuberculosis encodes for several two-component systems that allow the pathogen to respond to environmental cues, including oxygen. Two-component systems are composed of a sensor kinase and a cognate response regulator (reviewed: [126,127,128]). Recently, it was reported that an M. tuberculosis strain lacking the M. tuberculosis two-component regulatory system MtrA/MtrB (MtrAB) showed a decreased viability under hypoxia. MtrAB interacts with the non-cognate response regulator dormancy survival regulator (Dos)R and induces DosR-mediated gene expression. As DosR is part of the oxygen- and redox-sensing two-component system DosR/DosT [129], it allows adaptation to the hypoxic environment [130]. In detail, under hypoxia, DosR is acetylated at lysine 182 by the M. tuberculosis acetyltransferase Rv0998. This increases the DNA-binding ability of DosR to promote transcription of genes, which allows the adaptation to hypoxia [131]. Other bacterial transcription factors are also involved in hypoxic gene regulation [132, 133], from which Rv0081 seems to form the largest hub [134]. The DosR/DosT regulon not only helps the pathogen to adapt to the hypoxic environment, but it also modulates the host cell response to infection. The rv2626c gene, one of the most prominently induced genes of DosR/DosT regulon, is involved in inducing a pro-inflammatory host cell response and necrotic cell death [135]. As the DosR/DosT regulon is important for adaptation and survival of M. tuberculosis under hypoxic conditions, it might be a good target for new therapeutics. One promising inhibitor is artemisinin that disables the heme-base DosT sensor kinase [136]; further inhibitors with multiple distinct mechanisms have been identified [137].
Bacterial responses to hypoxia: modulation of the host tissue response
Tobin and coworkers used a zebrafish model to investigate the role of tissue oxygen in the Mycobacterium–host interplay. They showed that mycobacterial granuloma in zebrafish also become hypoxic. In addition, they discovered that M. marinum-infected macrophages trigger a vascular endothelial angiogenic program in granulomas. This response was absent when a replication-deficient M. marinum strain was used that was lacking ESX1 protein export systems. These findings indicate that Mycobacteria manipulate angiogenesis of the host to generate an at least partially vascularized and hence oxygenated microenvironment that is required to allow mycobacterial replication [138]. Therefore, M. marinum is able to counteract the efforts of the immune system to withhold oxygen from the invading pathogen. Although M. marinum infection in zebrafish is only a surrogate model mimicking immune responses in tuberculosis, it has offered substantial insights into mycobacterial pathogenesis over the last decades [139]. The question how angiogenesis and tissue oxygenation are regulated is certainly also of central relevance for M. tuberculosis infections.
Bacterial response to hypoxia: changes of the bacterial metabolism
Because the host metabolism changes with the level of oxygen, the pathogens also have to adapt to the altered metabolic environment (Table 1). This is especially important for pathogens that utilize host metabolites [140]. A recent report demonstrated that P. aeruginosa can grow on ethanol, produced by many other microbes, including Klebsiella pneumoniae [141, 142], as a sole carbon source in hypoxic settings. Accordingly, under hypoxic conditions, P. aeruginosa upregulates the NAD-linked alcohol dehydrogenase AdhA, which enables the pathogen to catabolize ethanol [143]. In addition, P. aeruginosa can respire nitrate and utilize pyruvate when oxygen is limited [144,145,146]. These changes in metabolism allow the pathogen to grow even under unfavorable conditions.
Bacterial response to hypoxia: induction of bacterial dormancy
Mycobacterium tuberculosis is able to survive in a low oxygen environment by inducing a state of dormancy that prevents sterile immunity [147]. For the induction of the dormancy survival program, the DosR/DosT regulon is essential [148]. Importantly, persistent bacteria develop a thick outer layer that helps to restrict entry of the antibiotic rifampicin [149] and confers antibiotic resistance. This is in agreement with other findings showing reduced antibiotic sensitivity of hypoxia-induced persistent M. tuberculosis [150]. Additionally, hypoxia might induce an internal bacterial program that alters the composition and function of multidrug efflux pumps resulting in antibiotic resistance, as reported for Pseudomonas aeruginosa [151]. During C. burnetii infection, the lack of oxygen also seems to induce a state of dormancy [95], but this has to be studied in more detail. It is clear that hypoxia allows the initial containment of oxygen-dependent pathogens, but triggers the development of bacterial persistence and dormancy that impairs pathogen elimination and prepares the ground for chronic latent and eventually recrudescing infections.
Bacteria might re-encounter atmospheric oxygen levels and re-enter into replication mode. How this is controlled and regulated is not completely understood. In the case of M. tuberculosis, global transcriptional and physiological changes are required [152, 153]. The pathogen mounts a metabolic shift under hypoxic conditions, which allows accumulation of metabolites that can be used for growth after re-aeration [154].
Hypoxia-mediated containment of bacterial infections
The antimicrobial and immunoregulatory enzyme NOS2, which produces high levels of RNS in macrophages [155], contributes to the control of M. tuberculosis [155, 156]. However, NOS2 requires oxygen as a substrate (reviewed: [16]) and loses its efficiency under hypoxic conditions (reviewed: [157]). Thus, hypoxia inhibits this important antimicrobial defense mechanism. In addition, low concentration of RNS might even support bacterial survival and adaptation. Nitrite was found to induce transcriptional alterations in M. tuberculosis that allowed the pathogen to withstand stress conditions [158].
Hypoxia does not only impair the antimicrobial activity of macrophages directed against S. aureus, but also blocks PHOX-dependent antimicrobial activity of granulocytes [159], such as NET formation [160] and degranulation of mast cells [161]. In the case of S. aureus infections, hypoxia is able to boost the virulence of S. aureus via the two-component system SrrAB ([162]; Fig. 3). In line with this, acute systemic hypoxia results in impaired antimicrobial response of infected mice kept under conditions of low oxygen [117, 163].

Host–pathogen interaction in macrophages under hypoxia. Mycobacteria replication is inhibited under hypoxia. This is due to HIF1α-mediated increase in lactate dehydrogenase (LDH-A), which catalyzes the conversion of the carbon source of Mycobacteria, pyruvate into lactate. Depletion of pyruvate results in reduced replication of Mycobacteria. HIF1α-induced IL-1β generation also limits Mycobacteria replication. Itaconate, an important antimicrobial effector, is decreased under hypoxia, due to hypoxia-mediated reduction of the TCA cycle. The DosR regulon is activated under hypoxia; this allows Mycobacteria to survive in those harsh conditions. Coxiella replication is also impaired under hypoxia. This is mainly due to reduced citrate levels. HIF1α-mediated increase in IFNγ results in augmentation of IDO, which catalyzes the transformation of tryptophan to kyurenines. This exhaustion of tryptophan limits its uptake by Coxiella, which is tryptophan auxotroph and, thus, replication is prevented. Yet, IDO is inhibited by hypoxia. Salmonella and Staphylococcus, however, are characterized by replicating under hypoxia. This is due to the inhibition of the antimicrobial effector enzymes, PHOX and NOS2, which leads to less ROS and RNS and, thus, aids replication. Salmonella’s virulence is enhanced under reduced oxygen levels, through increasing T3SS-dependent secretion of effector proteins; and Staphylococcus increases its two-component system SrrAB
Therefore, the question arises how myeloid cells control bacterial infection under oxygen limiting conditions. For aerobic bacteria, the hypoxic microenvironment itself already impedes bacterial replication and, thereby, helps to control disease. In addition, the following mechanisms contribute to the control of bacterial infection under hypoxia: (i) induction of antimicrobial peptides; (ii) depletion of essential metabolites and (iii) alterations of the defense mechanisms for the benefit of the host (Table 1).
Induction of antimicrobial peptides
Preclinical studies demonstrated that hypoxia was able to inhibit mycobacterial growth [164], as it caused the expression of antimicrobial molecules, like granulysin [165]. In line with this, the stabilization of HIF1α in bacterial dermatitis via lack of oxygen and/or infection itself resulted in the production and secretion of antibacterial peptides and pro-inflammatory cytokines (reviewed: [166]).
Hypoxia also helped to control infections with Pseudomonas aeruginosa. Increased bactericidal activities were observed in vitro and in vivo in a murine infection model [167]. HIF1α is partly responsible of this control, as demonstrated by the control of an ocular infection with P. aeruginosa, which might be dependent on NO and antimicrobial peptide production [168]. In a Caenorhabditis elegans infection model, loss of HIF1α enhanced the susceptibility of the nematode to P. aeruginosa [169]. Although HIF1α turned out to be important for the control of P. aeruginosa infections, the siderophore pyoverdin, an essential virulence factor of P. aeruginosa [170], unexpectedly induced HIF1α stabilization [169]. However, under hypoxic conditions the expression of pyoverdin was limited [121]. Together, these data suggest that HIF1α stabilization is a relevant component of the host defense against P. aeruginosa (Table 2).
Depletion of essential metabolites
Increased HIF1α levels correlated with bacterial killing in a zebrafish model of M. avium infections [96]. How the containment of mycobacteria is achieved via HIF1α accumulation is not yet clear. The underlying mechanism might involve HIF1α-mediated expression of the lactate dehydrogenase-A (LDH-A), which converts pyruvate to lactate. As M. tuberculosis can use pyruvate as a carbon source for intracellular replication, depletion of pyruvate might help to control mycobacteria ([171]; Fig. 3; Table 2).
The cytosolic conversion of citrate to acetyl-CoA is required for fatty acid biosynthesis and the synthesis of pro-inflammatory mediators [172,173,174]. Infection with C. burnetii, L. pneumophila [95] and C. trachomatis [175] induced an upregulation of citrate levels, which might result in a pro-inflammatory environment to fight the infection. However, C. burnetii only replicated in the presence of citrate ([95]; Fig. 3; Table 2), demonstrating that the increased availability of citrate in an inflammatory environment is exploited by the pathogen for its own purposes. Why C. burnetii requires citrate is unknown. It might need citrate for its energy metabolism. Alternatively, C. burnetii might sense host cell-derived citrate allowing the pathogen to adjust to the microenvironment and/or to express virulence genes. Importantly, citrate levels were markedly reduced under hypoxia, which resulted in impaired C. burnetii replication [95]. Thus, hypoxia-mediated restriction of citrate functions as a nutritional antibacterial effector mechanism.
Modulation of host defense mechanisms: induction of pro-inflammatory cytokines
HIF1α stabilization drives expression of pro-inflammatory cytokines [75, 93]. In addition, HIF1α stabilization by oxygen deficiency or infection might enhance wound healing and tissue repair [166]. Experiments with mice deficient in HIF1α in the myeloid lineage support the assumption that HIF1α is required to prevent immuno-pathological consequences for the host. Thus, mice lacking HIF1α in myeloid cells showed a stronger inflammatory response and died earlier than wild-type mice during chronic M. tuberculosis infection [176]. HIF1α not only prevents immuno-pathology during infection, but also protects against M. marinum infection by inducing the pro-inflammatory cytokine IL-1β ( [177]; Fig. 3; Table 2).
Interferon γ (IFNγ) is essential for the activation of macrophages and the control of many intracellular pathogens, including M. tuberculosis [178,179,180]. HIF1α regulates ~ 50% of all IFNγ-inducible genes during M. tuberculosis infection [181]. In uninfected dendritic cells hypoxia enhanced the IFNγ-induced mRNA expression of indoleamine 2,3-dioxygenase (IDO), which converts tryptophan into kynurenines [182]. IDO is known to suppress proliferation and survival of lymphocytes under normoxia [183]. Reports about the function of IDO during infections mainly concentrated on IDO-mediated depletion of tryptophan, which impaired the replication of tryptophan auxotrophic pathogens such as Chlamydia species, C. burnetii and Toxoplasma gondii ([184,185,186]; Fig. 3). However, unlike the findings with non-infected dendritic cells [182], IDO mRNA expression and activity were diminished in IFNγ-stimulated C. trachomatis-infected HEp2 cells under hypoxia due to an impaired IFNγ-STAT1 signaling [187]. Thus, the definitive role of IDO for the control of infections in a hypoxic microenvironment requires further studies.
Modulation of host cell defense mechanisms: phagocytosis and phagosome maturation
Previous studies have found that hypoxia increased the phagocytic activity of macrophages in an HIF1α-dependent manner [188]. Exposure of mice to hypoxia improved the uptake of E. coli by peritoneal macrophages [188].
HIF1α influences autophagy [189, 190]. Autophagy is an important process to sequester damaged organelles, protein aggregates, or bacteria in a double-membrane-bound vesicle, the autophagosome. The fusion of the autophagosome with lysosomes results in the degradation of the sequestered material and replenishment of the host cell nutrient pool by the degraded products [191, 192]. Depending on the bacteria, the interaction with the autophagic pathway might be either detrimental or beneficial. The overall effect of HIF1α on autophagy still awaits clarification, as there are conflicting reports in the literature. We and others found that HIF1α can activate autophagy and, as a consequence, bacterial degradation [190, 193]. In contrast, during Histoplasma capsulatum infection, HIF1α decreased autolysosome maturation. However, this also led to containment of the pathogen, as H. capsulatum exploited the autophagic pathway for its own survival [194]. Further research is required to understand the different effect of HIF1α and/or hypoxia on autophagy in the different infection models.
In summary, an infected host cell uses several mechanisms to fight invading bacteria under oxygen-limiting conditions (Table 2). While several pathways/factors have been already identified, other still await identification. One of these factors might be itaconate, which is generated from cis-aconitate by the cis-aconitate-decarboxylase (encoded by the immune-responsive gene 1 [IRG1]) and possesses antimicrobial activity [195,196,197]. Our data indicate that infection with Legionella pneumophila or C. burnetii resulted in an increased level of itaconate, supporting previous reports [196]. Its antibacterial activity is at least partially mediated by inhibition of the glyoxylate shunt, which is necessary for bacteria to survive intracellularly [198]. Several pathogens encode genes required for itaconate degradation. These genes are important for the intracellular survival of the corresponding pathogens [199], indicating an important role of itaconate in the containment of infections. However, hypoxia diminished itaconate levels otherwise induced by infection with L. pneumophila or C. burnetii. Importanly, we observed bacterial replication only under normoxia and, thus, in the presence of itaconate [95]. This indicates that the itaconate levels induced by the infection might be insufficient to prevent C. burnetii and L. pneumophila replication. Our data suggest that the itaconate levels correlate with the oxygen concentration during infection. Thus, it is currently unlikely that itaconate contributes to the control of intracellular pathogens under hypoxic conditions.
HIF stabilization as therapeutic strategy for the control of infections in hypoxic tissues
As explained above, HIF1α stabilization is an important regulator of innate immune responses. Therefore, its pharmacological stabilization is a possible treatment strategy to boost the host defense against bacterial infection in an oxygen-independent manner [200]. To stabilize HIF1α, inhibition of prolyl hydroxylases is widely used (reviewed: [201]). These prolyl hydroxylase inhibitors lead to HIF stabilization, mimicking hypoxic effects and have, therefore, been described as potential therapeutic agents [200, 202].
The PHD inhibitor mimosine triggered bactericidal activity in in vitro studies using human phagocytes infected with S. aureus [203]. Mimosine treatment increased HIF1α levels and ameliorated the clinical course of mice infected subcutaneously with S. aureus [203]. AKB-4924, a more potent pharmacological compound, enhanced cutaneous innate defenses against bacterial infections as well [204]. Its stabilizing effect on HIF1α was essential for the enhanced bactericidal activity of phagocytes, which might partially depend on AKB-4924-mediated upregulation of antimicrobial peptides and/or pro-inflammatory cytokines [204]. Inhibition of PHD, and, thus, stabilization of HIF1α by AKB-4924 not only boosted cutaneous defenses, but also improved the host innate immune response against urinary tract infections [205] and protected against colitis induced bacteremia [206].
Another prolyl hydroxylase inhibitor is dimethyloxalylglycine (DMOG). This substance is well tolerated and is able to ameliorate experimental colitis [207]. Its beneficial effects are, however, not limited to gastrointestinal inflammatory disorders. DMOG reduced the cytotoxic effects of P. aeruginosa infection on epithelial cells by decreasing P. aeruginosa internalization [208] as well. Moreover, mice pretreated with DMOG 48 h prior to P. aeruginosa infection showed reduced mortality rates [208]. These results suggest that DMOG might be a possible adjunctive therapeutic option to combat P. aeruginosa infections.
Desferrioxamine, an iron chelator, also leads to HIF1α stabilization by inhibiting PHD enzyme activity [209]. Addition of desferrioxamine to human-derived macrophages alters their cellular metabolism by increasing glycolysis in a HIF1α-dependent manner [210] in uninfected, LPS-stimulated, and M. tuberculosis-infected macrophages. In early stages of M. tuberculosis infection, desferrioxamine increased the immunological function of macrophages by boosting IL-1β through HIF1α [210]. Therefore, desferrioxamine might serve as a possible adjunctive antimicrobial treatment option. However, the possible side effects, like anaphylaxis, anemia, hearing loss and retinopathy have to be taken into account [211]. Inhibition of PHD and HIF stabilization holds potential as an adjunctive therapeutic agent. However, therapeutic stabilization of HIF1α has to be tailored individually. For instance, in a model of progressive pulmonary tuberculosis in BALB/c mice, the blockage of HIF1α worsened the disease during the early phase of infection, while it decreased bacterial load during late tuberculosis [212].
Other bacteria thrive under hypoxia
Although hypoxia damages oxygen-dependent bacteria, induces HIF accumulation and activity, leads to increased release of pro-inflammatory cytokines and antimicrobial peptides, and prolongs the survival of neutrophils, it is not always detrimental to pathogens. First, several antimicrobial pathways such as NOS2 and PHOX require oxygen to produce their toxic compounds. Second, bacterial microorganisms frequently develop strategies to adapt to hypoxic conditions and to benefit from the low O2 level at the site of infection (Table 2).
In the case of Shigella flexneri, the local depletion of oxygen due to the aerobic respiration of the bacteria incapacitated the Ipa (invasion plasmid antigen)-dependent secretion of effector molecules and, thus, the virulence of Shigella flexneri, but at the same time promoted its local proliferation and the establishment of microcolonies within the gut tissue [34].
Furthermore, there is evidence that low oxygen conditions support the replication of Salmonella enterica serovar Typhimurium within macrophages. Hypoxia increased the activity of the Salmonella pathogenicity island 2 (SPI-2)-encoded type III secretion system and simultaneously impaired the activity of the antimicrobial enzymes NOS2 and PHOX ( [33]; Fig. 3; Table 2). At this stage, the role of HIF1α in macrophages during Salmonella pathogenesis is unexplored and requires further investigation. In addition, the mechanisms by which hypoxia increased SPI-2 activity are unclear and warrant further research. Moreover, the metabolic requirements that allow Salmonella survival and replication under low oxygen conditions remain elusive. The ability of Salmonella to generate all its metabolites from simple carbon, nitrogen, and sulfur sources [213] and the expression of high-affinity cytochromes [213] might be the prerequisites that enable Salmonella to thrive within low oxygen environments.
Low oxygen conditions were also reported to impair antimicrobial activity of macrophages directed against E. coli and S. aureus (Fig. 3) [214]. In addition, hypoxia impaired the regular function of mitochondria [214]. It is known that uncoupling of the electron transport chain and mitochondrial ROS production can contribute to the antimicrobial activity of macrophages [215,216,217]. Currently, it is, however, unclear which mitochondrial function is specifically impaired under hypoxic conditions.
Concluding remarks
In this review, we attempted to shed light on the complex and diverse roles of oxygen in host–pathogen interaction. Low levels of oxygen trigger HIF-dependent pathways in host cells, which contribute to containment of bacterial replication and spreading. This is mainly mediated by upregulation of antimicrobial peptides or molecules and the alteration of the host immune response. In recent years, first examples demonstrated that bacterial containment under hypoxia can be accomplished by the depletion of metabolites caused by alteration of the host cell metabolism. A major challenge for future research will be to increase our understanding of the complex interplay between metabolites, immune responses and control of intruding pathogens.
Low oxygen environments can also be beneficial for the invading pathogen, as several antimicrobial effectors, such as PHOX and NOS2, depend on oxygen for production of their toxic products. Therefore, hypoxia might impede elimination of bacteria and might even result in increased replication under hypoxic condition or trigger bacterial dormancy. In this state, the bacteria can survive for a long period of time and are even protected from antibiotic treatment [218, 219]. As bacterial dormancy is the main cause for recurring and/or chronic infections, the hypoxia-induced bacterial containment might come with a high cost for the patient. It will be of major importance to increase our understanding of how bacterial dormancy and the re-entering into the replication mode are regulated under hypoxic conditions. As bacterial dormancy is a major cause for antibiotic inaccessibility, this knowledge might allow developing novel therapeutics.
The picture is even more complex, as the pathogens have evolved mechanisms to overcome and modulate the hypoxic conditions. Thus, the pathogens rely on oxygen sensors to adapt to hypoxic conditions. These sensors are mainly two component systems and allow the pathogen to transcriptionally react to changes in oxygen availability. Genes important for bacterial metabolism, for the induction of bacterial dormancy, and for virulence factors are transcribed. As different pathogens have distinct metabolic needs and virulence factors, the host–pathogen interaction under hypoxia has to be analyzed for each pathogen individually.
In addition, we are only beginning to understand the role of infection-induced HIF1α stabilization in host–pathogen interaction. This is due to the complex experimental set-up required to differentiate between the role of hypoxia/oxygen deficiency and the infection-induced HIF stabilization. Studies employing HIF stabilization agents may especially interesting to address this issue. From these studies, new targets and therapeutic strategies might emanate, which allow targeting of pathogens in their hypoxic niche. However, therapeutics targeting hypoxia and/or HIF also have to be tailored individually for each pathogen and site of infection and will not be available off the shelf.
Abbreviations
- ATP:
-
Adenosine triphosphate
- DCs:
-
Dendritic cells
- DSS:
-
Dextran sulfate sodium
- DMOG:
-
Dimethyloxalylglycine
- (Dos)R:
-
Dormancy survival regulator
- HIF:
-
Hypoxia-inducible factors
- IDO:
-
Indoleamine 2,3-dioxygenase
- iNOS or NOS2:
-
Inducible or type 2 nitric oxide synthase
- IFNγ:
-
Interferon γ
- IL:
-
Interleukin
- LDH-A:
-
Lactate dehydrogenase-A
- LPS:
-
Lipopolysaccharide
- MtrAB:
-
M. tuberculosis Two-component regulatory system MtrA/MtrB
- NET:
-
Neutrophil extracellular traps
- NO:
-
Nitric oxide
- OXPHOS:
-
Oxidative phosphorylation
- PHOX:
-
Phagocyte NADPH-oxidase
- PMN:
-
Polymorphonuclear neutrophils
- PHDs:
-
Prolyl hydroxylases
- PKM2:
-
Pyruvate kinase M2
- RNS:
-
Reactive nitrogen species
- ROS:
-
Reactive oxygen species
- SPI-2:
-
Salmonella Pathogenicity island 2
- TCA:
-
Tricarboxylic acid cycle
- VEGF:
-
Vascular endothelial growth factor
References
Riera Romo M, Pérez-Martínez D, Castillo Ferrer C (2016) Innate immunity in vertebrates: An overview. Immunology 148:125–139. https://doi.org/10.1111/imm.12597
Taylor CT, Colgan SP (2017) Regulation of immunity and inflammation by hypoxia in immunological niches. Nat Rev Immunol 17:774–785. https://doi.org/10.1038/nri.2017.103
Siegert I, Schödel J, Nairz M et al (2015) Ferritin-mediated iron sequestration stabilizes hypoxia-inducible factor-1α upon LPS activation in the presence of ample oxygen. Cell Rep 13:2048–2055. https://doi.org/10.1016/j.celrep.2015.11.005
Gundra UM, Girgis NM, Gonzalez MA et al (2017) Vitamin A mediates conversion of monocyte-derived macrophages into tissue-resident macrophages during alternative activation. Nat Immunol 18:642–653. https://doi.org/10.1038/ni.3734
Wang A, Huen SC, Luan HH et al (2016) Opposing effects of fasting metabolism on tissue tolerance in bacterial and viral inflammation. Cell 166:1512-1525.e12. https://doi.org/10.1016/j.cell.2016.07.026
Weiss G, Schaible UE (2015) Macrophage defense mechanisms against intracellular bacteria. Immunol Rev 264:182–203. https://doi.org/10.1111/imr.12266
Sitkovsky M, Lukashev D (2005) Regulation of immune cells by local-tissue oxygen tension: HIF1α and adenosine receptors. Nat Rev Immunol 5:712–721
Peyssonnaux C, Boutin AT, Zinkernagel AS et al (2008) Critical role of HIF-1α in keratinocyte defense against bacterial infection. J Invest Dermatol 128:1964–1968. https://doi.org/10.1038/jid.2008.27
Eliasson P, Jönsson JI (2010) The hematopoietic stem cell niche: Low in oxygen but a nice place to be. J Cell Physiol 222:17–22
Scheid A, Wenger RH, Schäffer L et al (2002) Physiologically low oxygen concentrations in fetal skin regulate hypoxia-inducible factor 1 and transforming growth factor-beta3. FASEB J 16:411–413. https://doi.org/10.1096/fj.01-0496fje
Campbell EL, Bruyninckx WJ, Kelly CJ et al (2014) Transmigrating neutrophils shape the mucosal microenvironment through localized oxygen depletion to influence resolution of inflammation. Immunity 40:66–77. https://doi.org/10.1016/j.immuni.2013.11.020
Karhausen J, Furuta GT, Tomaszewski JE et al (2004) Epithelial hypoxia-inducible factor-1 is protective in murine experimental colitis. J Clin Invest 114:1098–1106. https://doi.org/10.1172/jci21086
Marteyn B, West NP, Browning DF et al (2010) Modulation of Shigella virulence in response to available oxygen in vivo. Nature 465:355–358. https://doi.org/10.1038/nature08970
Albenberg L, Esipova TV, Judge CP et al (2014) Correlation between intraluminal oxygen gradient and radial partitioning of intestinal microbiota. Gastroenterology 147:1055-1063.e8. https://doi.org/10.1053/j.gastro.2014.07.020
Kelly CJ, Zheng L, Campbell EL et al (2015) Crosstalk between microbiota-derived short-chain fatty acids and intestinal epithelial HIF augments tissue barrier function. Cell Host Microbe 17:662–671. https://doi.org/10.1016/j.chom.2015.03.005
Jantsch J, Schödel J (2015) Hypoxia and hypoxia-inducible factors in myeloid cell-driven host defense and tissue homeostasis. Immunobiology 220:305–314
Wang XD, Wolfbeis OS (2014) Optical methods for sensing and imaging oxygen: Materials, spectroscopies and applications. Chem Soc Rev 43:3666–3761
Colliez F, Gallez B, Jordan BF (2017) Assessing tumor oxygenation for predicting outcome in radiation oncology: A review of studies correlating tumor hypoxic status and outcome in the preclinical and clinical settings. Front Oncol 7:10
Krohn KA, Link JM, Mason RP (2008) Molecular imaging of hypoxia. J. Nucl. Med. 49:129S–148S
Mees G, Dierckx R, Vangestel C, Van De Wiele C (2009) Molecular imaging of hypoxia with radiolabelled agents. Eur J Nucl Med Mol Imaging 36:1674–1686
Mirabello V, Cortezon-Tamarit F, Pascu SI (2018) Oxygen sensing, hypoxia tracing and in vivo imaging with functional metalloprobes for the early detection of non-communicable diseases. Front Chem 6:27. https://doi.org/10.3389/fchem.2018.00027
Cortezon-Tamarit F, Sarpaki S, Calatayud DG et al (2016) Applications of “Hot” and “Cold” Bis(thiosemicarbazonato) metal complexes in multimodal imaging. Chem Rec 16:1380–1397. https://doi.org/10.1002/tcr.201500292
Velikyan I (2014) Prospective of 68Ga-Radiopharmaceutical development. Theranostics 4:47–80
Melo T, Duncan J, Ballinger JR, Rauth AM (2000) BRU59-21, a second-generation 99mTc-labeled 2-nitroimidazole for imaging hypoxia in tumors. J Nucl Med 41:169–176
Hoebers FJ, Janssen HL, Olmos RA et al (2002) Phase 1 study to identify tumour hypoxia in patients with head and neck cancer using technetium-99m BRU 59–21. Eur J Nucl Med 29:1206–1211. https://doi.org/10.1007/s00259-002-0888-0
Honess DJ, Hill SA, Collingridge DR, et al (1998) Preclinical evaluation of the novel hypoxic marker 99mTc-HL91 (prognox) in murine and xenograft systems in vivo. In: International Journal of Radiation Oncology Biology Physics. Elsevier, pp 731–735
Long NJ, Wong WT (2014) The chemistry of molecular imaging. Wiley, Hoboken
Melican K, Boekel J, Månsson LE et al (2008) Bacterial infection-mediated mucosal signalling induces local renal ischaemia as a defence against sepsis. Cell Microbiol 10:1987–1998. https://doi.org/10.1111/j.1462-5822.2008.01182.x
Gaertner F, Massberg S (2016) Blood coagulation in immunothrombosis—at the frontline of intravascular immunity. Semin Immunol 28:561–569
Massberg S, Grahl L, Von Bruehl ML et al (2010) Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat Med 16:887–896. https://doi.org/10.1038/nm.2184
Arena ET, Campbell-Valois FX, Tinevez JY et al (2015) Bioimage analysis of Shigella infection reveals targeting of colonic crypts. Proc Natl Acad Sci USA 112:E3282–E3290. https://doi.org/10.1073/pnas.1509091112
Arena ET, Tinevez JY, Nigro G et al (2017) The infectious hypoxia: occurrence and causes during Shigella infection. Microbes Infect 19:157–165. https://doi.org/10.1016/j.micinf.2016.10.011
Jennewein J, Matuszak J, Walter S et al (2015) Low-oxygen tensions found in Salmonella-infected gut tissue boost Salmonella replication in macrophages by impairing antimicrobial activity and augmenting Salmonella virulence. Cell Microbiol 17:1833–1847. https://doi.org/10.1111/cmi.12476
Tinevez JY, Arena ET, Anderson M et al (2019) Shigella-mediated oxygen depletion is essential for intestinal mucosa colonization. Nat Microbiol 4:2001–2009. https://doi.org/10.1038/s41564-019-0525-3
Tu DG, Chang WW, Lin ST et al (2016) Salmonella inhibits tumor angiogenesis by downregulation of vascular endothelial growth factor. Oncotarget 7:37513. https://doi.org/10.18632/oncotarget.7038
Rao P, Suvas S (2019) Development of inflammatory hypoxia and prevalence of glycolytic metabolism in progressing herpes stromal keratitis lesions. J Immunol 202:514–526. https://doi.org/10.4049/jimmunol.1800422
Lopes JP, Stylianou M, Backman E et al (2018) Evasion of immune surveillance in low oxygen environments enhances candida albicans virulence. MBio. https://doi.org/10.1128/mBio.02120-18
Gresnigt MS, Rekiki A, Rasid O et al (2016) Reducing hypoxia and inflammation during invasive pulmonary aspergillosis by targeting the Interleukin-1 receptor. Sci Rep 6:1–12. https://doi.org/10.1038/srep26490
O’Neill LAJ, Kishton RJ, Rathmell J (2016) A guide to immunometabolism for immunologists. Nat Rev Immunol 16:553–565
Davies LC, Rice CM, McVicar DW, Weiss JM (2019) Diversity and environmental adaptation of phagocytic cell metabolism. J Leukoc Biol 105:37–48. https://doi.org/10.1002/JLB.4RI0518-195R
Palsson-Mcdermott EM, O’Neill LAJ (2013) The Warburg effect then and now: From cancer to inflammatory diseases. BioEssays 35:965–973. https://doi.org/10.1002/bies.201300084
Sadiku P, Walmsley SR (2019) Hypoxia and the regulation of myeloid cell metabolic imprinting: consequences for the inflammatory response. EMBO Rep. https://doi.org/10.15252/embr.201847388
Murray PJ, Rathmell J, Pearce E (2015) SnapShot: immunometabolism. Cell Metab 22:190-190.e1
O’Neill LAJ, Pearce EJ (2016) Immunometabolism governs dendritic cell and macrophage function. J Exp Med 213:15–23
Corcoran SE, O’Neill LAJ (2016) HIF1α and metabolic reprogramming in inflammation. J Clin Invest 126:3699–3707
Buck MD, Sowell RT, Kaech SM, Pearce EL (2017) Metabolic Instruction of Immunity. Cell 169:570–586
O’Neill LAJ, Artyomov MN (2019) Itaconate: the poster child of metabolic reprogramming in macrophage function. Nat Rev Immunol 19:273–281
Donnelly RP, Finlay DK (2015) Glucose, glycolysis and lymphocyte responses. Mol Immunol 68:513–519. https://doi.org/10.1016/j.molimm.2015.07.034
Prabhakar NR, Semenza GL (2012) Adaptive and maladaptive cardiorespiratory responses to continuous and intermittent hypoxia mediated by hypoxia-inducible factors 1 and 2. Physiol Rev 92:967–1003
Kaelin WG, Ratcliffe PJ (2008) Oxygen sensing by metazoans: the central role of the hif hydroxylase pathway. Mol Cell 30:393–402
Stothers CL, Luan L, Fensterheim BA, Bohannon JK (2018) Hypoxia-inducible factor-1α regulation of myeloid cells. J Mol Med 96:1293–1306
Fandrey J, Schödel J, Eckardt KU et al (2019) Now a Nobel gas: oxygen. Pflugers Arch Eur J Physiol 471:1343–1358
Serocki M, Bartoszewska S, Janaszak-Jasiecka A et al (2018) miRNAs regulate the HIF switch during hypoxia: a novel therapeutic target. Angiogenesis 21:183–202
Heng TSP, Painter MW, Elpek K et al (2008) The immunological genome project: Networks of gene expression in immune cells. Nat Immunol 9:1091–1094
Tian H, McKnight SL, Russell DW (1997) Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev 11:72–82. https://doi.org/10.1101/gad.11.1.72
Wiesener MS, Jürgensen JS, Rosenberger C et al (2003) Widespread hypoxia-inducible expression of HIF-2alpha in distinct cell populations of different organs. FASEB J 17:271–273. https://doi.org/10.1096/fj.02-0445fje
Imtiyaz HZ, Williams EP, Hickey MM et al (2010) Hypoxia-inducible factor 2α regulates macrophage function in mouse models of acute and tumor inflammation. J Clin Invest 120:2699–2714. https://doi.org/10.1172/JCI39506
Thompson AAR, Elks PM, Marriott HM et al (2014) Hypoxia-inducible factor 2a regulates key neutrophil functions in humans, mice, and zebrafish. Blood 123:366–376. https://doi.org/10.1182/blood-2013-05-500207
Duan C (2016) Hypoxia-inducible factor 3 biology: complexities and emerging themes. Am J Physiol - Cell Physiol 310:C260–C269. https://doi.org/10.1152/ajpcell.00315.2015
Cockman ME, Lippl K, Tian YM et al (2019) Lack of activity of recombinant HIF prolyl hydroxylases (PHDs) on reported non-HIF substrates. Elife. https://doi.org/10.7554/eLife.46490
Greer SN, Metcalf JL, Wang Y, Ohh M (2012) The updated biology of hypoxia-inducible factor. EMBO J 31:2448–2460
Mole DR, Blancher C, Copley RR et al (2009) Genome-wide association of hypoxia-inducible factor (HIF)-1α and HIF-2α DNA binding with expression profiling of hypoxia-inducible transcripts. J Biol Chem 284:16767–16775. https://doi.org/10.1074/jbc.M901790200
Schödel J, Mole DR, Ratcliffe PJ (2013) Pan-genomic binding of hypoxia-inducible transcription factors. Biol Chem 394:507–517
Palazon A, Goldrath AW, Nizet V, Johnson RS (2014) HIF transcription factors, inflammation, and immunity. Immunity 41:518–528
Devraj G, Beerlage C, Brüne B, Kempf VAJ (2017) Hypoxia and HIF-1 activation in bacterial infections. Microbes Infect 19:144–156. https://doi.org/10.1016/j.micinf.2016.11.003
Dehne N, Brüne B (2009) HIF-1 in the inflammatory microenvironment. Exp Cell Res 315:1791–1797
Werth N, Beerlage C, Rosenberger C et al (2010) Activation of hypoxia inducible factor 1 is a general phenomenon in infections with human pathogens. PLoS ONE 5:1–12. https://doi.org/10.1371/journal.pone.0011576
Blouin CC, Pagé EL, Soucy GM, Richard DE (2004) Hypoxic gene activation by lipopolysaccharide in macrophages: Implication of hypoxia-inducible factor 1α. Blood 103:1124–1130. https://doi.org/10.1182/blood-2003-07-2427
Frede S, Stockmann C, Freitag P, Fandrey J (2006) Bacterial lipopolysaccharide induces HIF-1 activation in human monocytes via p44/42 MAPK and NF-κB. Biochem J 396:517–527. https://doi.org/10.1042/BJ20051839
Rius J, Guma M, Schachtrup C et al (2008) NF-κB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1α. Nature 453:807–811. https://doi.org/10.1038/nature06905
Nicholas SA, Sumbayev VV (2010) The role of redox-dependent mechanisms in the downregulation of ligand-induced Toll-like receptors 7, 8 and 4-mediated HIF-1α prolyl hydroxylation. Immunol Cell Biol 88:180–186. https://doi.org/10.1038/icb.2009.76
Bailey JD, Diotallevi M, Nicol T et al (2019) Nitric oxide modulates metabolic remodeling in inflammatory macrophages through TCA cycle regulation and itaconate accumulation. Cell Rep 28:218-230.e7. https://doi.org/10.1016/j.celrep.2019.06.018
Lawless SJ, Kedia-Mehta N, Walls JF et al (2017) Glucose represses dendritic cell-induced T cell responses. Nat Commun 8:1–14. https://doi.org/10.1038/ncomms15620
Mills EL, Kelly B, Logan A et al (2016) Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell 167:457-470.e13. https://doi.org/10.1016/J.CELL.2016.08.064
Tannahill GM, Curtis AM, Adamik J et al (2013) Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 496:238–242. https://doi.org/10.1038/nature11986
Jantsch J, Wiese M, Schödel J et al (2011) Toll-like receptor activation and hypoxia use distinct signaling pathways to stabilize hypoxia-inducible factor 1α (HIF1A) and result in differential HIF1A-dependent gene expression. J Leukoc Biol 90:551–562. https://doi.org/10.1189/jlb.1210683
Solis AG, Bielecki P, Steach HR et al (2019) Mechanosensation of cyclical force by PIEZO1 is essential for innate immunity. Nature 573:69–74. https://doi.org/10.1038/s41586-019-1485-8
Kurbel S, Kurbel B, Belovari T et al (2001) Model of interstitial pressure as a result of cyclical changes in the capillary wall fluid transport. Med Hypotheses 57:161–166. https://doi.org/10.1054/mehy.2001.1288
Hartmann H, Eltzschig HK, Wurz H et al (2008) Hypoxia-independent activation of HIF-1 by enterobacteriaceae and their siderophores. Gastroenterology. https://doi.org/10.1053/j.gastro.2007.12.008
Riess T, Andersson SGE, Lupas A et al (2004) Bartonella adhesin A mediates a proangiogenic host cell response. J Exp Med 200:1267–1278. https://doi.org/10.1084/jem.20040500
Kempf VAJ, Lebiedziejewski M, Alitalo K et al (2005) Activation of hypoxia-inducible factor-1 in bacillary angiomatosis: evidence for a role of hypoxia-inducible factor-1 in bacterial infections. Circulation 111:1054–1062. https://doi.org/10.1161/01.CIR.0000155608.07691.B7
Legendre C, Reen FJ, Mooij MJ et al (2012) Pseudomonas aeruginosa Alkyl quinolones repress hypoxia-inducible factor 1 (HIF-1) signaling through HIF-1α degradation. Infect Immun 80:3985–3992. https://doi.org/10.1128/IAI.00554-12
Rupp J, Gieffers J, Klinger M et al (2007) Chlamydia pneumoniae directly interferes with HIF-1α stabilization in human host cells. Cell Microbiol 9:2181–2191. https://doi.org/10.1111/j.1462-5822.2007.00948.x
Walmsley SR, Cadwallader KA, Chilvers ER (2005) The role of HIF-1α in myeloid cell inflammation. Trends Immunol 26:434–439
Maianski NA, Geissler J, Srinivasula SM et al (2004) Functional characterization of mitochondria in neutrophils: A role restricted to apoptosis. Cell Death Differ 11:143–153. https://doi.org/10.1038/sj.cdd.4401320
Cramer T, Yamanishi Y, Clausen BE et al (2003) HIF-1α is essential for myeloid cell-mediated inflammation. Cell 112:645–657. https://doi.org/10.1016/S0092-8674(03)00154-5
Walmsley SR, Print C, Farahi N et al (2005) Hypoxia-induced neutrophil survival is mediated by HIF-1α-dependent NF-κB activity. J Exp Med 201:105–115. https://doi.org/10.1084/jem.20040624
Schuster DP, Brody SL, Zhou Z et al (2007) Regulation of lipopolysaccharide-induced increases in neutrophil glucose uptake. Am J Physiol Lung Cell Mol Physiol. https://doi.org/10.1152/ajplung.00350.2006
Sadiku P, Willson JA, Dickinson RS et al (2017) Prolyl hydroxylase 2 inactivation enhances glycogen storage and promotes excessive neutrophilic responses. J Clin Invest 127:3407–3420. https://doi.org/10.1172/JCI90848
Peyssonnaux C, Datta V, Cramer T et al (2005) HIF-1alpha expression regulates the bactericidal capacity of phagocytes. J Clin Invest 115:1806–1815. https://doi.org/10.1172/JCI23865
McInturff AM, Cody MJ, Elliott EA et al (2012) Mammalian target of rapamycin regulates neutrophil extracellular trap formation via induction of hypoxia-inducible factor 1 α. Blood 120:3118–3125. https://doi.org/10.1182/blood-2012-01-405993
Monceaux V, Chiche-Lapierre C, Chaput C et al (2016) Anoxia and glucose supplementation preserve neutrophil viability and function. Blood 128:993–1002. https://doi.org/10.1182/blood-2015-11-680918
Palsson-Mcdermott EM, Curtis AM, Goel G et al (2015) Pyruvate kinase M2 regulates hif-1α activity and il-1β induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab 21:65–80. https://doi.org/10.1016/j.cmet.2014.12.005
Kim SY, Choi YJ, Joung SM et al (2010) Hypoxic stress up-regulates the expression of Toll-like receptor 4 in macrophages via hypoxia-inducible factor. Immunology 129:516–524. https://doi.org/10.1111/j.1365-2567.2009.03203.x
Hayek I, Fischer F, Schulze-Luehrmann J et al (2019) Limitation of TCA cycle intermediates represents an oxygen-independent nutritional antibacterial effector mechanism of macrophages. Cell Rep 26:3502-3510.e6. https://doi.org/10.1016/j.celrep.2019.02.103
Elks PM, Brizee S, van der Vaart M et al (2013) Hypoxia inducible factor signaling modulates susceptibility to mycobacterial infection via a nitric oxide dependent mechanism. PLoS Pathog 9:1–16. https://doi.org/10.1371/journal.ppat.1003789
Bayele HK, Peyssonnaux C, Giatromanolaki A et al (2007) HIF-1 regulates heritable variation and allele expression phenotypes of the macrophage immune response gene SLC11A1 from a Z-DNA-forming microsatellite. Blood 110:3039–3048. https://doi.org/10.1182/blood-2006-12-063289
Schatz V, Strüssmann Y, Mahnke A et al (2016) Myeloid cell-derived HIF-1α promotes control of leishmania major. J Immunol 197:4034–4041. https://doi.org/10.4049/jimmunol.1601080
Takeda N, O’Dea EL, Doedens A et al (2010) Differential activation and antagonistic function of HIF-α isoforms in macrophages are essential for NO homeostasis. Genes Dev 24:491–501. https://doi.org/10.1101/gad.1881410
Kelly B, O’Neill LAJ (2015) Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res 25:771–784
Everts B, Amiel E, Van Der Windt GJW et al (2012) Commitment to glycolysis sustains survival of NO-producing inflammatory dendritic cells. Blood 120:1422–1431. https://doi.org/10.1182/blood-2012-03-419747
Winning S, Fandrey J (2016) Dendritic cells under hypoxia: how oxygen shortage affects the linkage between innate and adaptive immunity. J Immunol Res 2016:5134329–5134329. https://doi.org/10.1155/2016/5134329
Perrin-Cocon L, Aublin-Gex A, Diaz O et al (2018) Toll-like receptor 4–induced glycolytic burst in human monocyte-derived dendritic cells results from p38-dependent stabilization of HIF-1α and increased hexokinase II expression. J Immunol 201:1510–1521. https://doi.org/10.4049/jimmunol.1701522
Guak H, Al Habyan S, Ma EH et al (2018) Glycolytic metabolism is essential for CCR7 oligomerization and dendritic cell migration. Nat Commun. https://doi.org/10.1038/s41467-018-04804-6
Köhler T, Reizis B, Johnson RS et al (2012) Influence of hypoxia-inducible factor 1α on dendritic cell differentiation and migration. Eur J Immunol 42:1226–1236. https://doi.org/10.1002/eji.201142053
Liu J, Zhang X, Chen K et al (2019) CCR7 chemokine receptor-inducible lnc-Dpf3 restrains dendritic cell migration by inhibiting HIF-1α-mediated glycolysis. Immunity 50:600-615.e15. https://doi.org/10.1016/j.immuni.2019.01.021
Bhandari T, Olson J, Johnson RS, Nizet V (2013) HIF-1α influences myeloid cell antigen presentation and response to subcutaneous OVA vaccination. J Mol Med 91:1199–1205. https://doi.org/10.1007/s00109-013-1052-y
Jantsch J, Chakravortty D, Turza N et al (2008) Hypoxia and hypoxia-inducible factor-1α modulate lipopolysaccharide-induced dendritic cell activation and function. J Immunol 180:4697–4705. https://doi.org/10.4049/jimmunol.180.7.4697
Pantel A, Teixeira A, Haddad E et al (2014) Direct type I IFN but Not MDA5/TLR3 activation of dendritic cells is required for maturation and metabolic shift to glycolysis after poly IC stimulation. PLoS Biol 12:e1001759. https://doi.org/10.1371/journal.pbio.1001759
Spirig R, Djafarzadeh S, Regueira T et al (2010) Effects of TLR agonists on the hypoxia-regulated transcription factor HIF-1α and dendritic cell maturation under normoxic conditions. PLoS ONE 5:e10983. https://doi.org/10.1371/journal.pone.0010983
Everts B, Amiel E, Huang SCC et al (2014) TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKε supports the anabolic demands of dendritic cell activation. Nat Immunol 15:323–332. https://doi.org/10.1038/ni.2833
Netea MG, Quintin J, Van Der Meer JWM (2011) Trained immunity: a memory for innate host defense. Cell Host Microbe 9:355–361
Penkov S, Mitroulis I, Hajishengallis G, Chavakis T (2019) Immunometabolic crosstalk: an ancestral principle of trained immunity? Trends Immunol 40:1–11
Cheng SC, Quintin J, Cramer RA et al (2014) MTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science (80-). https://doi.org/10.1126/science.1250684
de Laval B, Maurizio J, Kandalla PK et al (2020) Erratum: C/EBPβ-dependent epigenetic memory induces trained immunity in hematopoietic stem cells. Cell Stem Cell 26:793. https://doi.org/10.1016/j.stem.2020.01.017 ((26(5) (657–674.e8))
Fensterheim BA, Young JD, Luan L et al (2018) The TLR4 agonist monophosphoryl lipid A drives broad resistance to infection via dynamic reprogramming of macrophage metabolism. J Immunol 200:3777–3789. https://doi.org/10.4049/jimmunol.1800085
Thompson AAR, Dickinson RS, Murphy F et al (2017) Hypoxia determines survival outcomes of bacterial infection through HIF-1α–dependent reprogramming of leukocyte metabolism. Sci Immunol. https://doi.org/10.1126/sciimmunol.aal2861
Wallace N, Zani A, Abrams E, Sun Y (2016) The impact of oxygen on bacterial enteric pathogens. Adv Appl Microbiol 95:179–204. https://doi.org/10.1016/bs.aambs.2016.04.002
Kamath KS, Krisp C, Chick J et al (2017) Pseudomonas aeruginosa proteome under hypoxic stress conditions mimicking the cystic fibrosis lung. J Proteome Res 16:3917–3928. https://doi.org/10.1021/acs.jproteome.7b00561
Scotti JS, Leung IKH, Ge W et al (2014) Human oxygen sensing may have origins in prokaryotic elongation factor Tu prolyl-hydroxylation. Proc Natl Acad Sci U S A 111:13331–13336. https://doi.org/10.1073/pnas.1409916111
Schaible B, Rodriguez J, Garcia A et al (2017) Hypoxia reduces the pathogenicity of Pseudomonas aeruginosa by decreasing the expression of multiple virulence factors. J Infect Dis 215:1459–1467. https://doi.org/10.1093/infdis/jix139
Schaible B, Crifo B, Schaffer K, Taylor CT (2019) Pseudomonas Prolyl Hydroxylase (PPHD), a putative bacterial oxygen sensor suppresses antibiotic resistance and pathogenicity in Pseudomonas aeruginosa. J Biol Chem. https://doi.org/10.1074/jbc.ra119.010033
Kaihami GH, Breda LCD, de Almeida JRF et al (2017) The atypical response regulator AtvR is a new player in Pseudomonas aeruginosa response to hypoxia and virulence. Infect Immun. https://doi.org/10.1128/IAI.00207-17
Eichner A, Günther N, Arnold M et al (2014) Marker genes for the metabolic adaptation of Pseudomonas aeruginosa to the hypoxic cystic fibrosis lung environment. Int J Med Microbiol 304:1050–1061. https://doi.org/10.1016/j.ijmm.2014.07.014
Pfyffer GE (2015) Mycobacterium: general characteristics, laboratory detection, and staining procedures. In: Manual of clinical microbiology. ASM Press, Washington, DC, pp 536–569
King AN, de Mets F, Brinsmade SR (2020) Who’s in control? Regulation of metabolism and pathogenesis in space and time. Curr Opin Microbiol 55:88–96
Jacob-Dubuisson F, Mechaly A, Betton JM, Antoine R (2018) Structural insights into the signalling mechanisms of two-component systems. Nat Rev Microbiol 16:585–593
Parish T (2014) Two-component regulatory systems of mycobacteria. Microbiol Spectr. https://doi.org/10.1128/microbiolspec.mgm2-0010-2013
Sivaramakrishnan S, De Montellano PRO (2013) The DosS-DosT/DosR mycobacterial sensor system. Biosensors 3:259–282
Banerjee SK, Lata S, Sharma AK et al (2019) The sensor kinase MtrB of Mycobacterium tuberculosis regulates hypoxic survival and establishment of infection. J Biol Chem 294:19862–19876. https://doi.org/10.1074/jbc.RA119.009449
Yang H, Sha W, Liu Z et al (2018) Lysine acetylation of DosR regulates the hypoxia response of Mycobacterium tuberculosis article. Emerg Microbes Infect 7:1–14. https://doi.org/10.1038/s41426-018-0032-2
Kumar V, Aneesh Kumar A, Sanawar R et al (2019) Chronic pressure overload results in deficiency of mitochondrial membrane transporter ABCB7 which contributes to iron overload, mitochondrial dysfunction, metabolic shift and worsens cardiac function. Sci Rep. https://doi.org/10.1038/s41598-019-49666-0
Sun X, Wang Y, Sui N (2018) Transcriptional regulation of bHLH during plant response to stress. Biochem Biophys Res Commun 503:397–401
Galagan JE, Minch K, Peterson M et al (2013) The Mycobacterium tuberculosis regulatory network and hypoxia. Nature 499:178–183. https://doi.org/10.1038/nature12337
Sun C, Yang G, Yuan J et al (2017) Mycobacterium tuberculosis hypoxic response protein 1 (Hrp1) augments the pro-inflammatory response and enhances the survival of Mycobacterium smegmatis in murine macrophages. J Med Microbiol 66:1033–1044. https://doi.org/10.1099/jmm.0.000511
Zheng H, Colvin CJ, Johnson BK et al (2017) Inhibitors of Mycobacterium tuberculosis DosRST signaling and persistence. Nat Chem Biol 13:218–225. https://doi.org/10.1038/nchembio.2259
Zheng H, Williams JT, Aleiwi B et al (2019) Inhibiting Mycobacterium tuberculosis DosRST signaling by targeting response regulator DNA binding and sensor kinase heme. ACS Chem Biol 15:52–62. https://doi.org/10.1021/acschembio.8b00849
Oehlers SH, Cronan MR, Scott NR et al (2015) Interception of host angiogenic signalling limits mycobacterial growth. Nature 517:612–615. https://doi.org/10.1038/nature13967
Ramakrishnan L (2020) Mycobacterium tuberculosis pathogenicity viewed through the lens of molecular Koch’s postulates. Curr Opin Microbiol 54:103–110
Hayek I, Berens C, Lührmann A (2019) Modulation of host cell metabolism by T4SS-encoding intracellular pathogens. Curr Opin Microbiol 47:59–65
Yuan J, Chen C, Cui J et al (2019) Fatty liver disease caused by high-alcohol-producing Klebsiella pneumoniae. Cell Metab 30:675-688.e7. https://doi.org/10.1016/j.cmet.2019.08.018
Elshaghabee FMF, Bockelmann W, Meske D et al (2016) Ethanol production by selected intestinal microorganisms and lactic acid bacteria growing under different nutritional conditions. Front Microbiol 7:47. https://doi.org/10.3389/fmicb.2016.00047
Crocker AW, Harty CE, Hammond JH et al (2019) Pseudomonas aeruginosa ethanol oxidation by AdhA in low-oxygen environments. J Bacteriol. https://doi.org/10.1128/JB.00393-19
Wauven CV, Pierard A, Kley-Raymann M, Haas D (1984) Pseudomonas aeruginosa mutants affected in anaerobic growth on arginine: evidence for a four-gene cluster encoding the arginine deiminase pathway. J Bacteriol 160:918–934. https://doi.org/10.1128/jb.160.3.928-934.1984
Eschbach M, Schreiber K, Trunk K et al (2004) Long-term anaerobic survival of the opportunistic pathogen Pseudomonas aeruginosa via pyruvate fermentation. J Bacteriol 186:4596–4604. https://doi.org/10.1128/JB.186.14.4596-4604.2004
Borrero-de Acuña JM, Timmis KN, Jahn M, Jahn D (2017) Protein complex formation during denitrification by Pseudomonas aeruginosa. Microb Biotechnol 10:1523–1534
Sershen CL, Plimpton SJ, May EE (2016) Oxygen modulates the effectiveness of granuloma mediated host response to Mycobacterium tuberculosis: a multiscale computational biology approach. Front Cell Infect Microbiol. https://doi.org/10.3389/fcimb.2016.00006
Honaker RW, Leistikow RL, Bartek IL, Voskui MI (2009) Unique roles of DosT and DosS in DosR regulon induction and Mycobacterium tuberculosis dormancy. Infect Immun 77:3258–3263. https://doi.org/10.1128/IAI.01449-08
Jakkala K, Ajitkumar P (2019) Hypoxic non-replicating persistent Mycobacterium tuberculosis develops thickened outer layer that helps in restricting rifampicin entry. Front Microbiol. https://doi.org/10.3389/fmicb.2019.02339
Iacobino A, Giannoni F, Pardini M et al (2019) The combination rifampin-nitazoxanide, but not rifampin-isoniazid-pyrazinamide-ethambutol, kills dormant Mycobacterium tuberculosis in hypoxia at neutral pH. Antimicrob Agents Chemother. https://doi.org/10.1128/AAC.00273-19
Schaible B, Taylor CT, Schaffer K (2012) Hypoxia increases antibiotic resistance in Pseudomonas aeruginosa through altering the composition of multidrug efflux pumps. Antimicrob Agents Chemother 56:2114–2118. https://doi.org/10.1128/AAC.05574-11
Du P, Sohaskey CD, Shi L (2016) Transcriptional and physiological changes during Mycobacterium tuberculosis reactivation from non-replicating persistence. Front Microbiol. https://doi.org/10.3389/fmicb.2016.01346
McGillivray A, Golden NA, Kaushal D (2015) The Mycobacterium tuberculosis Clp gene regulator is required for in vitro reactivation from hypoxia-induced dormancy. J Biol Chem 290:2351–2367. https://doi.org/10.1074/jbc.M114.615534
Eoh H, Wang Z, Layre E et al (2017) Metabolic anticipation in Mycobacterium tuberculosis. Nat Microbiol. https://doi.org/10.1038/nmicrobiol.2017.84
Bogdan C (2015) Nitric oxide synthase in innate and adaptive immunity: an update. Trends Immunol 36:161–178
Nathan C, Shiloh MU (2000) Reactive oxygen and nitrogen intermediates in the relationship between mammalian hosts and microbial pathogens. Proc Natl Acad Sci USA 97:8841–8848. https://doi.org/10.1073/pnas.97.16.8841
McCormick CC, Li WP, Calero M (2000) Oxygen tension limits nitric oxide synthesis by activated macrophages. Biochem J 350:709–716. https://doi.org/10.1042/0264-6021:3500709
Cunningham-Bussel A, Zhang T, Nathan CF (2013) Nitrite produced by Mycobacterium tuberculosis in human macrophages in physiologic oxygen impacts bacterial ATP consumption and gene expression. Proc Natl Acad Sci USA. https://doi.org/10.1073/pnas.1316894110
McGovern NN, Cowburn AS, Porter L et al (2011) Hypoxia selectively inhibits respiratory burst activity and killing of staphylococcus aureus in human neutrophils. J Immunol 186:453–463. https://doi.org/10.4049/jimmunol.1002213
Branitzki-Heinemann K, Möllerherm H, Völlger L et al (2016) Formation of neutrophil extracellular traps under low oxygen level. Front Immunol 7:518. https://doi.org/10.3389/fimmu.2016.00518
Möllerherm H, Branitzki-Heinemann K, Brogden G et al (2017) Hypoxia modulates the response of mast cells to Staphylococcus aureus infection. Front Immunol. https://doi.org/10.3389/fimmu.2017.00541
Kinkel TL, Roux CM, Dunman PM, Fang FC (2013) The Staphylococcus aureus SrrAB two-component system promotes resistance to nitrosative stress and hypoxia. MBio. https://doi.org/10.1128/mBio.00696-13
Harris GD, Johanson WG, Pierce AK (1977) Determinants of lung bacterial clearance in mice after acute hypoxia. Am Rev Respir Dis 116:671–677. https://doi.org/10.1164/arrd.1977.116.4.671
Nickel D, Busch M, Mayer D et al (2012) Hypoxia triggers the expression of human β Defensin 2 and antimicrobial activity against Mycobacterium tuberculosis in human macrophages. J Immunol 188:4001–4007. https://doi.org/10.4049/jimmunol.1100976
Zenk SF, Vollmer M, Schercher E et al (2016) Hypoxia promotes Mycobacterium tuberculosis-specific up-regulation of granulysin in human T cells. Med Microbiol Immunol 205:219–229. https://doi.org/10.1007/s00430-015-0442-x
Schaffer K, Taylor CT (2015) The impact of hypoxia on bacterial infection. FEBS J 282:2260–2266
Gil-Marqués ML, Pachón-Ibáñez ME, Pachón J, Smani Y (2018) Effect of hypoxia on the pathogenesis of Acinetobacter baumannii and Pseudomonas aeruginosa in vitro and in murine experimental models of infection. Infect Immun 86:e00543-e618. https://doi.org/10.1128/IAI.00543-18
Berger EA, McClellan SA, Vistisen KS, Hazlett LD (2013) HIF-1α is essential for effective PMN bacterial killing, antimicrobial peptide production and apoptosis in Pseudomonas aeruginosa keratitis. PLoS Pathog 9:e1003457. https://doi.org/10.1371/journal.ppat.1003457
Kirienko NV, Kirienko DR, Larkins-Ford J et al (2013) Pseudomonas aeruginosa disrupts Caenorhabditis elegans iron homeostasis, causing a hypoxic response and death. Cell Host Microbe 13:406–416. https://doi.org/10.1016/j.chom.2013.03.003
Meyer JM, Neely A, Stintzi A et al (1996) Pyoverdin is essential for virulence of Pseudomonas aeruginosa. Infect Immun 64:518–523. https://doi.org/10.1128/iai.64.2.518-523.1996
Osada-Oka M, Goda N, Saiga H et al (2019) Metabolic adaptation to glycolysis is a basic defense mechanism of macrophages for Mycobacterium tuberculosis infection. Int Immunol 31:781–793. https://doi.org/10.1093/intimm/dxz048
Infantino V, Convertini P, Cucci L et al (2011) The mitochondrial citrate carrier: a new player in inflammation. Biochem J 438:433–436. https://doi.org/10.1042/BJ20111275
Infantino V, Iacobazzi V, Menga A et al (2014) A key role of the mitochondrial citrate carrier (SLC25A1) in TNFα- and IFNγ-triggered inflammation. Biochim Biophys Acta Gene Regul Mech 1839:1217–1225. https://doi.org/10.1016/j.bbagrm.2014.07.013
Moon JS, Hisata S, Park MA et al (2015) MTORC1-induced HK1-dependent glycolysis regulates NLRP3 inflammasome activation. Cell Rep 12:102–115. https://doi.org/10.1016/j.celrep.2015.05.046
Rother M, Gonzalez E, Teixeira da Costa AR et al (2018) Combined human genome-wide RNAi and metabolite analyses identify IMPDH as a host-directed target against chlamydia infection. Cell Host Microbe 23:661-671.e8. https://doi.org/10.1016/j.chom.2018.04.002
Resende M, Ferreira CM, Barbosa AM et al (2020) Myeloid HIF-1α regulates pulmonary inflammation during experimental Mycobacterium tuberculosis infection. Immunology 159:121–129. https://doi.org/10.1111/imm.13131
Ogryzko NV, Lewis A, Wilson HL et al (2019) Hif-1α–induced expression of Il-1β protects against Mycobacterial infection in zebrafish. J Immunol 202:494–502. https://doi.org/10.4049/jimmunol.1801139
Green AM, DiFazio R, Flynn JL (2013) IFN-γ from CD4 T cells is essential for host survival and enhances CD8 T cell function during Mycobacterium tuberculosis infection. J Immunol 190:270–277. https://doi.org/10.4049/jimmunol.1200061
Flesch I, Kaufmann SH (1987) Mycobacterial growth inhibition by interferon-gamma-activated bone marrow macrophages and differential susceptibility among strains of Mycobacterium tuberculosis. J Immunol 138:4408–4413
Shenoy AR, Kim BH, Choi HP et al (2008) Emerging themes in IFN-γ-induced macrophage immunity by the p47 and p65 GTPase families. Immunobiology 212:771–784. https://doi.org/10.1016/j.imbio.2007.09.018
Braverman J, Sogi KM, Benjamin D et al (2016) HIF-1α is an essential mediator of IFN-γ–dependent immunity to Mycobacterium tuberculosis. J Immunol 197:1287–1297. https://doi.org/10.4049/jimmunol.1600266
Song X, Zhang Y, Zhang L et al (2018) Hypoxia enhances indoleamine 2,3-dioxygenase production in dendritic cells. Oncotarget 9:11572–11580. https://doi.org/10.18632/oncotarget.24098
Mellor AL, Chandler P, Baban B et al (2004) Specific subsets of murine dendritic cells acquire potent T cell regulatory functions following CTLA4-mediated induction of indoleamine 2,3 dioxygenase. Int Immunol 16:1391–1401. https://doi.org/10.1093/intimm/dxh140
Pfefferkorn ER (1984) Interferon γ blocks the growth of Toxoplasma gondii in human fibroblasts by inducing the host cells to degrade tryptophan. Proc Natl Acad Sci USA 81:908–912. https://doi.org/10.1073/pnas.81.3.908
Byrne GI, Lehmann LK, Landry GJ (1986) Induction of tryptophan catabolism is the mechanism for gamma-interferon-mediated inhibition of intracellular Chlamydia psittaci replication in T24 cells. Infect Immun 53:347–351. https://doi.org/10.1128/iai.53.2.347-351.1986
Ganesan S, Roy CR (2019) Host cell depletion of tryptophan by IFNγ-induced Indoleamine 2,3-dioxygenase 1 (IDO1) inhibits lysosomal replication of Coxiella burnetii. PLoS Pathog 15:e1007955. https://doi.org/10.1371/journal.ppat.1007955
Roth A, König P, Zandbergen VG et al (2010) Hypoxia abrogates antichlamydial properties of IFN-γ in human fallopian tube cells in vitro and ex vivo. Proc Natl Acad Sci U S A 107:19502–19507. https://doi.org/10.1073/pnas.1008178107
Anand RJ, Gribar SC, Li J et al (2007) Hypoxia causes an increase in phagocytosis by macrophages in a HIF-1α-dependent manner. J Leukoc Biol 82:1257–1265. https://doi.org/10.1189/jlb.0307195
Bellot G, Garcia-Medina R, Gounon P et al (2009) Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol 29:2570–2581. https://doi.org/10.1128/mcb.00166-09
Mimouna S, Bazin M, Mograbi B et al (2014) HIF1A regulates xenophagic degradation of adherent and invasive Escherichia coli (AIEC). Autophagy 10:2333–2345. https://doi.org/10.4161/15548627.2014.984275
Cong Y, Kumar ND, Mauthe M et al (2020) Manipulation of selective macroautophagy by pathogens at a glance. J Cell Sci. https://doi.org/10.1242/jcs.240440
Keller MD, Torres VJ, Cadwell K (2020) Autophagy and microbial pathogenesis. Cell Death Differ 27:872–886
Neubert P, Weichselbaum A, Reitinger C et al (2019) HIF1A and NFAT5 coordinate Na+-boosted antibacterial defense via enhanced autophagy and autolysosomal targeting. Autophagy 15:1899–1916. https://doi.org/10.1080/15548627.2019.1596483
Friedrich D, Zapf D, Lohse B et al (2019) The HIF-1/LC3-II axis impacts fungal immunity in human macrophages. Infect Immun. https://doi.org/10.1128/IAI.00125-19
Patil NK, Bohannon JK, Hernandez A et al (2019) Regulation of leukocyte function by citric acid cycle intermediates. J Leukoc Biol 106:105–117
Naujoks J, Tabeling C, Dill BD et al (2016) IFNs modify the proteome of legionella-containing vacuoles and restrict infection via IRG1-derived itaconic acid. PLoS Pathog 12:e1005408. https://doi.org/10.1371/journal.ppat.1005408
Chen M, Sun H, Boot M et al (2020) Itaconate is an effector of a Rab GTPase cell-autonomous host defense pathway against Salmonella. Science (80-) 369:450–455. https://doi.org/10.1126/science.aaz1333
Cordes T, Michelucci A, Hiller K (2015) Itaconic acid: the surprising role of an industrial compound as a mammalian antimicrobial metabolite. Annu Rev Nutr 35:451–473. https://doi.org/10.1146/annurev-nutr-071714-034243
Sasikaran J, Ziemski M, Zadora PK et al (2014) Bacterial itaconate degradation promotes pathogenicity. Nat Chem Biol 10:371–377. https://doi.org/10.1038/nchembio.1482
Bhandari T, Nizet V (2014) Hypoxia-inducible factor (HIF) as a pharmacological target for prevention and treatment of infectious diseases. Infect Dis Ther 3:159–174
Maxwell PH, Eckardt KU (2016) HIF prolyl hydroxylase inhibitors for the treatment of renal anaemia and beyond. Nat Rev Nephrol 12:157–168. https://doi.org/10.1038/nrneph.2015.193
Colgan SP, Furuta GT, Taylor CT (2020) Hypoxia and innate immunity: keeping up with the HIFsters. Annu Rev Immunol. https://doi.org/10.1146/annurev-immunol-100819-121537
Zinkernagel AS, Peyssonnaux C, Johnson RS, Nizet V (2008) Pharmacologic augmentation of hypoxia-inducible factor–1α with mimosine boosts the bactericidal capacity of phagocytes. J Infect Dis 197:214–217. https://doi.org/10.1086/524843
Okumura CYM, Hollands A, Tran DN et al (2012) A new pharmacological agent (AKB-4924) stabilizes hypoxia inducible factor-1 (HIF-1) and increases skin innate defenses against bacterial infection. J Mol Med 90:1079–1089. https://doi.org/10.1007/s00109-012-0882-3
Lin AE, Beasley FC, Olson J et al (2015) Role of hypoxia inducible factor-1α (HIF-1α) in innate defense against uropathogenic Escherichia coli infection. PLoS Pathog. https://doi.org/10.1371/journal.ppat.1004818
Keely S, Campbell EL, Baird AW et al (2014) Contribution of epithelial innate immunity to systemic protection afforded by prolyl hydroxylase inhibition in murine colitis. Mucosal Immunol 7:114–123. https://doi.org/10.1038/mi.2013.29
Cummins EP, Seeballuck F, Keely SJ et al (2008) The hydroxylase inhibitor dimethyloxalylglycine is protective in a murine model of colitis. Gastroenterology 134:156-165.e1. https://doi.org/10.1053/j.gastro.2007.10.012
Schaible B, McClean S, Selfridge A et al (2013) Hypoxia modulates infection of epithelial cells by Pseudomonas aeruginosa. PLoS ONE 8:e56491. https://doi.org/10.1371/journal.pone.0056491
Jaakkola P, Mole DR, Tian YM et al (2001) Targeting of HIF-α to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science (80-) 292:468–472. https://doi.org/10.1126/science.1059796
Phelan JJ, McQuaid K, Kenny C et al (2020) Desferrioxamine supports metabolic function in primary human macrophages infected with Mycobacterium tuberculosis. Front Immunol 11:836. https://doi.org/10.3389/fimmu.2020.00836
Velasquez J, Wray AA (2020) Deferoxamine. StatPearls [Internet]. StatPearls Publishing, Treasure Island, FL
Baay-Guzman GJ, Duran-Padilla MA, Rangel-Santiago J et al (2018) Dual role of hypoxia-inducible factor 1 α in experimental pulmonary tuberculosis: Its implication as a new therapeutic target. Future Microbiol 13:785–798. https://doi.org/10.2217/fmb-2017-0168
Eisenreich W, Rudel T, Heesemann J, Goebel W (2019) How viral and intracellular bacterial pathogens reprogram the metabolism of host cells to allow their intracellular replication. Front Cell Infect Microbiol 9:42
Wiese M, Gerlach RG, Popp I et al (2012) Hypoxia-mediated impairment of the mitochondrial respiratory chain inhibits the bactericidal activity of macrophages. Infect Immun 80:1455–1466. https://doi.org/10.1128/IAI.05972-11
Garaude J, Acín-Pérez R, Martínez-Cano S et al (2016) Mitochondrial respiratory-chain adaptations in macrophages contribute to antibacterial host defense. Nat Immunol 17:1037–1045. https://doi.org/10.1038/ni.3509
West AP, Brodsky IE, Rahner C et al (2011) TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 472:476–480. https://doi.org/10.1038/nature09973
Davies LC, Rice CM, Palmieri EM et al (2017) Peritoneal tissue-resident macrophages are metabolically poised to engage microbes using tissue-niche fuels. Nat Commun. https://doi.org/10.1038/s41467-017-02092-0
Wood TK, Knabel SJ, Kwan BW (2013) Bacterial persister cell formation and dormancy. Appl Environ Microbiol 79:7116–7121
Harms A, Maisonneuve E, Gerdes K (2016) Mechanisms of bacterial persistence during stress and antibiotic exposure. Science (80-). https://doi.org/10.1126/science.aaf4268
Szaszák M, Shima K, Käding N et al (2013) Host metabolism promotes growth of Chlamydia pneumoniae in a low oxygen environment. Int J Med Microbiol 303:239–246. https://doi.org/10.1016/j.ijmm.2013.03.005
Acknowledgements
The preparation of this article and the performance of some of the studies reviewed were supported by the Deutsche Forschungsgemeinschaft (DFG; consortium CRC1181, project fund A06 to AL and project fund C04 to CB) and by the Bundesministerium für Bildung und Forschung (BMBF; consortium Q-GAPS as part of the research network zoonotic infection diseases [01KI1726A] to AL). JJ received funding from the Bavarian Ministry of Science and the Arts in the framework of the Bavarian Research Network ‘New Strategies Against Multi-Resistant Pathogens by Means of Digital Networking–bayresq.net’.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hayek, I., Schatz, V., Bogdan, C. et al. Mechanisms controlling bacterial infection in myeloid cells under hypoxic conditions. Cell. Mol. Life Sci. 78, 1887–1907 (2021). https://doi.org/10.1007/s00018-020-03684-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-020-03684-8