
Abstract
Long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) deficiency is an inborn error of fatty acid metabolism that affects the degradation of long chain fatty acids and causes insufficient energy production and accumulation of toxic intermediates. The treatment consists of a diet low in fat, with supplementation of medium-chain triglycerides that bypass the metabolic block. In addition, frequent feeds and extra carbohydrates are given during febrile illnesses to reduce lipolysis. Hence, this diet differs from the general dietary recommendations for growing children. Furthermore, the Swedish dietary instructions for fat intake in LCHAD deficiency are given in grams, which differ from most guidelines that recommend fat intake as percentage shares of total caloric intake.
Aims: To assess growth in patients with LCHAD deficiency, in relation to dietary treatment and to evaluate if overweight/obesity is more common than in the normal population.
Results: The growth velocity showed acceleration after diagnosis and the start of treatment, followed by a period of stable or decelerated growth. The majority of the patients developed overweight to a greater extent than children without LCHAD deficiency. Several patients also went through a phase of obesity. Data on final height (FH) showed that three out of five patients had grown according to their genetic potential.
Conclusions: Regular and frequent follow-up and careful monitoring of weight are essential to avoid the development of overweight and obesity. The Swedish dietary instructions defining fat intake in total grams per day may be an alternative approach to achieve a moderate total caloric intake.
Competing interests: None declared
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
LCHAD deficiency (OMIM 609016) was first described in 1989 (Wanders et al. 1989 ; Wanders et al. 1990a; Hagenfeldt et al. 1990) in children who presented with lethargy and hypoketotic hypoglycemia after a short period of fasting often in association with gastroenteritis or other febrile infections. The diagnosis is usually established within the first year of life, often when nightly feedings are weaned (Saudubray et al. 1999; Tyni et al. 1997; Spiekerkoetter et al. 2009a). Some patients present with acute symptoms such as coma, cardiac arrest, and hypoketotic hypoglycemia, while others present with unspecific symptoms such as failure to thrive, poor weight gain, and vomiting. Common clinical findings are liver enlargement, muscular hypotonia, and hypertrophic cardiomyopathy. The mortality rate is high before diagnosis and treatment (den Boer et al. 2002). Measurements of acylcarnitines in plasma or nonesterified dicarboxylic and 3-hydroxy fatty acids in plasma and organic acids in urine and mutation analysis (Hagenfeldt et al. 1995; IJlst et al. 1994) are part of the diagnostic procedure. The most common mutation is G1428C in the α-subunit of the trifunctional protein (IJlst et al. 1996; IJlst et al. 1994; Sims et al. 1995).
Obstetric complications such as acute fatty liver of pregnancy, HELLP syndrome, preeclampsia and growth retardation are common when the fetus is affected with LCHAD deficiency, possibly as a result of placental insufficiency (Tyni et al. 1998a; Wilcken et al. 1993; Yang et al. 2002). The long-term outcome is variable, but a constant feature is retinal pigmentations progressing to impaired retinal function (Fahnehjelm et al. 2007; Tyni et al. 1998b). Peripheral neuropathy and psychomotor delay have been described in some patients (Bennett et al. 2000; den Boer et al. 2002; Saudubray et al. 1999; Spiekerkoetter et al. 2009a, b; Tyni et al. 1997; Wanders et al. 1990b).
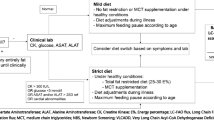
The aim of the treatment is to minimize the necessity of energy production from long-chain fatty acids of both exogenous and endogenous origin, and thereby avoid accumulation of toxic intermediates of the defective β-oxidation. The diet, low in fat content and hence long-chain fatty acids from normal food, is supplemented with medium-chain triglycerides (MCT fat) and thereby bypasses the enzymatic defect. Supplementation with essential fatty acids is necessary to avoid deficiency (Gillingham et al. 1999; Gillingham et al. 2003; Spiekerkoetter et al. 2009a); however, management may vary considerably (Potter et al. 2012). To inhibit lipolysis, frequent feeds are necessary. During catabolic events such as febrile infections, anabolic treatment with an intravenous glucose infusion may be necessary. Even after the diagnosis has been established and treatment started, the patients may have episodes of metabolic decompensation, especially during infections or fasting, with muscle pain, muscular hypotonia, and elevated serum CK and aminotransferases.
The Swedish dietary instructions for children with an LCHAD deficiency differs from most international guidelines as they are given in grams of long-chain fatty acid intake (Shaw and Lawson 2001) instead of as a percentage of the total energy intake (Gillingham et al. 1999; Gillingham et al. 2003; Spiekerkoetter et al. 2009b, 2010).
The diet in LCHAD is extreme and in disagreement with general nutritional recommendations for growing children. Height may be negatively affected by the low-fat diet or the disease itself, and there is a risk for weight gain with high carbohydrate intake, short fasting periods and continuous night feeds. In addition, a rapid weight gain may result in acceleration in height. This study was designed to examine how the Swedish dietary treatment affects growth, height, and weight development in children with LCHAD deficiency, with particular emphasis on growth patterns, final height, and development of overweight and/or obesity over time.
Materials and Methods
Study Population
Data were retrospectively collected from 10 patients. The age at diagnosis varied between 2 days and 13 months (mean 6.1 months). One patient was diagnosed in the neonatal period before any symptoms had developed because of a family history. The oldest patient has been treated for more than 20 years. The diagnosis was based on increased levels of 3-OH dicarboxylic acids in urine and 3-OH fatty acids in plasma and was confirmed by mutation analysis. Seven patients were homozygous for the common LCHAD deficiency mutation G1528C; the other three were compound heterozygous for G1528C.
The study protocol was approved by the Ethics Committee, and informed consent was given by the patients and families.
Clinical Follow-Up
The patients were followed regularly at our centers. Information on height, weight, and biochemical parameters (plasma 3-OH fatty acids, plasma carnitine, liver enzymes, creatine kinase, and fatty acid profile) was collected and analyzed. Information on clinical presentation and follow-up was collected from the medical records. The number of episodes when the child had fever or muscular pain and had been treated with extra carbohydrate intake, either orally at home or as glucose infusions at home, were noted (Table 1).
Dietary Treatment
Dietary treatment with a low intake of LCT fat was commenced at the time of the diagnosis in all children. The amount of LCT fat in grams was adjusted according to Swedish nutritional minimum requirement recommendations in correlation with age (Shaw and Lawson 2001). Parents were taught how to calculate their child’s LCT intake in grams, to ensure that it did not exceed the maximum prescribed quantity. The carbohydrate and protein intake was not adjusted in detail. Changes in weight or height curves were followed to assess the caloric intake. Observed stagnation or acceleration in weight was compensated for by a moderate increase or reduction of the carbohydrate and protein intake. In addition, the parents were asked to complete food diaries at least once yearly with records of everything their child ate, specified in grams, during 3 consecutive days.
Sufficient amounts of essential fatty acids were given according to Swedish recommendations, predominantly as walnut oil containing a minimum of nonessential fats. The majority of the children (8 out of 10) received docosahexaenoic acid (DHA) supplementation to maintain DHA plasma levels just above or within the upper reference range. In addition, the diet was enriched with vitamins and minerals. All but one of the children had a percutaneous gastrostomy (PEG) with continuous night feeds, predominantly given as a low-fat formula containing whey protein, carbohydrates, MCT fat, vitamins, minerals, and trace elements. The fasting periods were limited to 3–4 h, depending on age. In order to reduce lipolysis, two children were given uncooked corn starch. During febrile infections, a carbohydrate-rich supplement or intravenous glucose infusion was administered. Only patients with a carnitine deficiency were given supplements of 25–50 mg/kg/day, which were omitted during acute decompensations.
Growth
Height and weight were measured and compared to Swedish reference data (Wikland et al. 2002) and plotted as height SDS and BMI SDS. For children born preterm, height and weight SDS were corrected for gestational age up to age 2 years. Overweight was defined as iso-BMI ≥ 25 kg/m2 and obesity as iso-BMI ≥ 30 kg/m2 (Cole et al. 2000; Han et al. 2010; Lindgren et al. 1995). Parental heights were recorded and the target heights (TH) (Tanner et al. 1970) were calculated. In order to assess whether the children achieved the final height (FH) according to their genetic potential, TH SDS was compared to FH SDS. The age at menarche for the girls and their mothers was recorded. The onset of puberty for the boys was assessed by observing start and duration of the pubertal growth spurt in the charts. For patients who had reached FH, the height of the pubertal spurt was calculated and compared with the Tanner reference (28 ± 8 cm for the boys, 25 ± 8 cm for the girls) (Tanner et al. 1976). Birth weight SDS were obtained from the growth charts while the exact SDS of height, weight, BMI, and target height were calculated by the NordiNet® IOS pediatric database and the results used to generate the SDS growth curves.
Results
Clinical Follow-Up
After the confirmed diagnosis and dietary intervention was started, no patients died. Data of birth weight, pregnancy complications, symptoms at diagnosis, and number of episodes with increased carbohydrate intake are listed in Table 1.
Dietary Treatment
The food diaries showed that the reported fat intake adhered quite well to the recommended diet. Table 2 illustrates a characteristic day for a 5-year-old girl with LCHAD deficiency, following the Swedish recommendations. Regrettably we were unable to calculate the dietary intake for the patient group over time, since not enough data was obtained due to noncompliance and/or missing food diaries.
In order to compare the fat intake in grams to other recommendations emphasizing fat restriction as E%, the results from the food diaries of two patients of different age, gender, weight, physical activity levels, and body composition were converted to E%. Calculations were done over several years in the same patients and showed that the reported intake in grams for the two patients was equivalent to 13–24 E% of fat, distributed as 8–19 E% MCT and 4–5 E% LCT. As expected, the children with LCHAD deficiency had a lower total fat intake than recommended for growing children.
Growth
The growth trajectories illustrated that the majority of patients had an initial height and BMI acceleration after the diagnosis, followed by a stabilization or deceleration in height SDS after a couple of years (Figs. 1 and 2). Eight patients (7 girls, 1 boy; patients 1 and 4–10) developed overweight before 6 years of age. Six patients (5 girls, 1 boy; patients 1, 4–6, 8, and 10) had an iso- BMI of slightly more than 30 kg/m2 for a 1–5-year period. Seven patients (5 girls, 2 boys; patients 1, 2, 5, 6, and 8–10) had an iso-BMI ≥ 25 kg/m2, and one of these patients (patient 10) had an iso BMI ≥ 30 kg/m2 at the latest assessment. The other three patients (2 girls, 1 boy; patients 3, 4, and 7) had normal weight for age at the most recent examination (Figure 2). No patients were underweight at any time, apart from patient 2 who showed a different growth pattern than the other patients (Fig. 2), with a fall in BMI below – 2 SDS at 3 years of age, after which the patient showed a markedly increased weight gain. He had poor appetite and was physically very active throughout his younger years, followed by recurrent episodes of muscular pain and elevated CK levels treated with increased energy intake. In addition, this patient had a combination of complex treatment resistant seizures. Following gastrostomy at 5 years of age and commencement with vigabatrin, his weight increased, a side effect known to be associated with vigabatrin therapy (Chadwick 1999). Patient 1 had a rapid weight gain for a short period (+ 4 SDS between 3 and 6 years) when she had recurrent infections.

Height SDS in 10 patients with an LCHAD deficiency. The dotted vertical line illustrates the time at which all patients were diagnosed and dietary treatment was started (13 months). The growth trajectories for height SDS show an initial accelerated growth in childhood after the time of diagnosis

BMI SDS in 10 patients with LCHAD deficiency. The dotted vertical line illustrates the time at which all patients were diagnosed and started on dietary treatment (13 months). Growth trajectories for BMI SDS show early BMI acceleration after the time of diagnosis and development of overweight/obesity in childhood. The majority of patients have a BMI SDS of over ± 1 at the latest assessments
Five patients had reached FH (patients 1–5). FH was within ± 1.0 SDS of TH in three patients (patients 2, 4, and 5) and slightly below target in two patients (patients 1 and 3). Patient 3 had critical hypoglycemia at the time of diagnosis and developed neurological sequele, contractures, and epilepsy with poor seizure control.
Four out of ten patients (1,4,5,9) (40 %) had a BW between −1.8 and −2.3 SDS when correcting for gestational age (Table 1). Three patients had had menarche (patients 1, 4, 5). One girl (patient 1) had an early puberty, treated with a GnRH agonist between 8 and 10 years of age. Her pubertal growth spurt was normal (24 cm) (Tanner et al. 1976) but she did not reach TH. Patient 4 had menarche at the same age as her mother (at 14 years) while patient 5 had menarche 2 years earlier than her mother (at 11 years). The onset of puberty for the boys was within the normal age. One out of five patients (patient 3) had a subnormal height gain during adolescence, compared to the Tanner reference (Tanner et al. 1976). Mean female FH was 164 cm, constituting – 0.6 SDS, and mean male FH 183 cm, equal to 0.4 SDS. Mean TH SDS for all patients was 0.6 SDS, indicating that this patient population was comparable to the normal population.
Discussion
In this Swedish cohort of ten children with LCHAD deficiency, growth trajectories showed increased growth during a period of months to years after the diagnosis, followed by a period of stable or decelerated growth. The majority of the patients developed overweight and, in addition, obesity during various periods. The growth trajectories were comparable to those of normal children with overweight or obesity (He and Karlberg 2001). Three out of five patients reached FH within their TH; consequently, it seems that neither the disorder itself nor the dietary treatment affected FH negatively.
The Swedish dietary instructions (Shaw and Lawson 2001) for LCT intake were given in grams, in an attempt to achieve metabolic control but avoid overfeeding and obesity. This differs from recent dietary guidelines (Spiekerkoetter et al. 2009b, 2010; Gillingham et al. 1999) that recommend indicating LCT intake as E%. Hence calculating grams of fat instead of recommending fat intake as E% may be an alternative approach to decrease LCT intake since E% will allow a larger amount of LCT the higher the total calorie intake is. In order to compare the intake of LCT fat in patients adhering to the Swedish recommendations with the generally more common instructions in E%, we recalculated the food diaries from two patients. These two examples illustrate that the reported LCT intake is somewhat lower than that recommended by Spiekerkoetter et al. (25–30 E% from fat with 20–25 % as MCT and 5–10 % as LCT) (Spiekerkoetter et al. 2009b) as well as Gillingham (10–20 E% as MCT and 10 E% from LCT) (Gillingham et al. 2003). In this retrospective study, there was no method to really know what the child actually ingested over time. The nutritional diaries give a subjective 3-day snapshot of the patients’ life, often recorded during a period when the child was well (Lichtman et al. 1992). In addition, the available data was not sufficient to allow for more detailed calculations, but the data retrieved indicated good compliance with given dietary recommendations. Previous results indicate that patients with an LCHAD deficiency may have increased lipolysis as soon as after 3–4 hours of fasting (Halldin et al. 2007) underscoring the importance of minimizing the fasting periods and recommending night feeds. This differs from the recommendations by Spiekerkoetter et al., who suggest maximum fasting periods of 10–12 hours at night from 4 years of age (Spiekerkoetter et al. 2009b).
In our cohort, six out of ten patients (60 %) were overweight and one out of ten (10 %) obese at the most recent assessments. However, six patients had a transient period of obesity that regressed to overweight as they became older. In comparison, a Swedish study of healthy schoolchildren aged 8–9 showed that 11 % of the girls and 15 % of boys were overweight, and 3 % of the girls and 4 % of the boys were obese (Rasmussen et al. 2004). This indicates that overweight/obesity was overrepresented in our cohort. Analogous to our findings, Gillingham et al. show an obesity rate of 30 % in nine children with LCHAD deficiency (Gillingham et al. 2007), while Lund et al. do not report the weight development of their patients (Lund et al. 2010).
It is noteworthy that the severe lipid restriction results in a relatively high carbohydrate content which is interesting from an overweight/obesity perspective. There is emerging evidence that the rate of carbohydrate absorption after a meal, the glycaemic index, may affect weight gain (Brand-Miller et al. 2009). Foods with high glycaemic index that result in an initial period of high blood glucose and insulin levels, followed by a rapid fall in blood glucose is often associated with reduced satiety and excessive caloric intake (Ludwig 2002, 2007). On the other hand, low GI foods produce a more consistent blood glucose and insulin release and consequently increased satiety, but may in addition promote fat oxidation and decrease lipogenesis (Brand-Miller and Buyken 2012; Kong et al. 2011; Thomas et al. 2007). This subject warrants future studies since it is not obvious how the glycaemic index could be used to improve the situation for LCHAD patients.
Overweight/obesity in childhood is associated with accelerated linear growth, earlier pubertal maturity, and a subnormal height gain in adolescence, and in the end not affecting FH (Dunger et al. 2005; Garn and Haskell 1959; He and Karlberg 2001; Shalitin and Phillip 2003). Two out of three girls in our study had an early puberty, possibly caused by the increased weight gain in childhood. A rapid weight gain in infancy may also cause insulin resistance (Dunger et al. 2005). To the best of our knowledge, insulin sensitivity has not been studied in LCHAD-deficient patients, although one study suggests that children with LCHAD/MTP-deficiency and adiposity have normal insulin sensitivity based on normal insulin secretion (Gillingham et al. 2007). Three out of five patients reached FH within 1 SDS of TH and mean FH was – 0.6 SDS (girls)/0.4 SDS (boys), which indicated adequate growth in the majority of patients. Two patients did not reach TH, likely due to complicating factors other than the fatty acid disorder itself, as one patient developed early puberty and the other had contractures affecting the height measurements.
Earlier reports have demonstrated increased frequencies of IUGR of 47 % (Tyni et al. 1998a) and 43 % (Yang et al. 2002) in patients with LCHAD/MTP defects, not only explained by preeclampsia-related conditions. In this cohort, four out of ten patients (40 %) had some degree of IUGR. Preeclampsia developed in three out of ten pregnancies (30 %) (Table 1), which is consistent with several previous reports (den Boer et al. 2002; Tyni et al. 1998a; Yang et al. 2002). Birth weight is significantly associated with childhood growth (Li et al. 2004). However, this could not be studied in this small patient population.
The causes of obesity and overweight are certainly multifactorial, although the primary determinants of weight gain are excessive caloric intake or insufficient physical activity (Prentice and Jebb 1995) (Han et al. 2010). In LCHAD-deficient patients, there is an obvious risk for overfeeding prompted by the dietary treatment per se, the frequency of meals during the day, the continuous night feeds and short fasting periods as well as altered resting energy expenditure due to the inborn error of metabolism. Parents learning that their child has a disorder with risk for hypoglycemic episodes, treated with frequent feeds, may initially feel insecure and overcompensate resulting in increased weight gain following diagnosis. Children with LCHAD deficiency may be less physically active due to muscular weakness or pain and/or neurological deficits. The increase in BMI seen throughout early childhood may also reflect the increased caloric intake during infections. In the general population, children with parents that have an elevated BMI are at risk of developing obesity (Danielzik et al. 2002; Xu et al. 2011). The children with LCHAD deficiency differ in that they have a specific diet, different from the rest of the family. However, it is likely that hereditary and/or social factors such as attitude to food and physical activity may influence weight development in the patient when other family members have an increased BMI.
Childhood obesity is associated with a higher risk of morbidity in childhood and adolescence (Daniels 2009) with serious disability and premature death in adulthood (Bjorge et al. 2008; Reilly and Kelly 2010). Planned weight reduction is difficult in patients with an LCHAD deficiency, since this is associated with a risk of metabolic decompensation (Gillingham et al. 1999; Hagenfeldt et al. 1990; Halldin et al. 2007; Houten and Wanders 2010). Hence, it is challenging but crucial for the clinician to prevent the development of overweight and obesity. It is necessary to adjust the diet and energy intake and encourage physical activity to achieve normal weight without the development of complications. In conformity with the care of patients with other inborn errors of metabolism, it is essential to evaluate whether the therapeutic effects on the derangements of the disorder justify a strict dietary regimen or whether the patients would have a better quality of life with more liberal dietary recommendations.
Conclusions
Dietary treatment in LCHAD deficiency presents a challenge. The necessity of frequent feeds, high contents of carbohydrates, and periods of high energy intake to maintain good metabolic control increases the risk of overweight and obesity. In this cohort of LCHAD-deficient patients, an accelerated growth during the first years after the diagnosis and start of treatment followed by a period of stable or decelerated growth was seen. The patients developed overweight more often than in the general population, but few were obese despite overnight feeds. To improve metabolic control and weight development, we recommend frequent regular clinical follow-ups and extra vigilance concerning diet and weight. It is possible that giving recommendations for LCT fat intake in grams instead of E% may facilitate weight control and it is important to evaluate the appropriate length of fasting periods to avoid overcompensation.
Abbreviations
- BW:
-
Birth Weight
- E%:
-
Energy percentage
- EFA:
-
Essential fatty Acid
- FH:
-
Final Height
- FH SDS:
-
Final Height Standard Deviation Score
- IUGR:
-
Intra Uterine Growth Restriction
- LCF:
-
Long Chain Fatty Acid
- LCHAD:
-
Long-chain 3-hydroxyacyl-CoA dehydrogenase
- MTP:
-
Mitochondrial trifunctional protein
- SGA:
-
Small for gestational age
- TH:
-
Target Height
- TH SDS:
-
Target Height Standard Deviation Score
References
Bennett MJ, Rinaldo P, Strauss AW (2000) Inborn errors of mitochondrial fatty acid oxidation. Crit Rev Clin Lab Sci 37:1–44
Bjorge T, Engeland A, Tverdal A, Smith GD (2008) Body mass index in adolescence in relation to cause-specific mortality: a follow-up of 230,000 Norwegian adolescents. Am J Epidemiol 168:30–37
Brand-Miller J , McMillan-Price J , Steinbeck K , Caterson I (2009) Dietary glycemic index: health implications. J Am Coll Nutr 28 Suppl:446S–449S
Brand-Miller J, Buyken AE (2012) The glycemic index issue. Curr Opin Lipidol 23:62–67
Chadwick D (1999) Safety and efficacy of vigabatrin and carbamazepine in newly diagnosed epilepsy: a multicentre randomised double-blind study. Vigabatrin European Monotherapy Study Group. Lancet 354:13–19
Cole TJ, Bellizzi MC, Flegal KM, Dietz WH (2000) Establishing a standard definition for child overweight and obesity worldwide: international survey. Brit Med J 320:1240–1243
Daniels SR (2009) Complications of obesity in children and adolescents. Int J Obes (Lond) 33(Suppl 1):S60–65
Danielzik S, Langnase K, Mast M, Spethmann C, Muller MJ (2002) Impact of parental BMI on the manifestation of overweight 5–7 year old children. Eur J Nutr 41:132–138
den Boer ME, Wanders RJ, Morris AA, IJlst L, Heymans HS, Wijburg FA (2002) Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: clinical presentation and follow-up of 50 patients. Pediatrics 109:99–104
Dunger DB, Ahmed ML, Ong KK (2005) Effects of obesity on growth and puberty. Best Pract Res Clin Endocrinol Metab 19:375–390
Fahnehjelm KT, Holmstrom G, Ying L et al (2007) Ocular characteristics in 10 children with long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: a cross-sectional study with long-term follow-up. Acta Ophthalmol Scand 86(3):329–337
Garn SM, Haskell JA (1959) Fat and growth during childhood. Science 130:1711–1712
Gillingham M, Van Calcar S, Ney D, Wolff J, Harding C (1999) Dietary management of long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency (LCHADD). A case report and survey. J Inherit Metab Dis 22:123–131
Gillingham MB, Connor WE, Matern D et al (2003) Optimal dietary therapy of long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. Mol Genet Metab 79:114–123
Gillingham MB, Purnell JQ, Jordan J, Stadler D, Haqq AM, Harding CO (2007) Effects of higher dietary protein intake on energy balance and metabolic control in children with long-chain 3-hydroxy acyl-CoA dehydrogenase (LCHAD) or trifunctional protein (TFP) deficiency. Mol Genet Metab 90:64–69
Hagenfeldt L, von Dobeln U, Holme E et al (1990) 3-Hydroxydicarboxylic aciduria–a fatty acid oxidation defect with severe prognosis. J Pediatrics 116:387–392
Hagenfeldt L, Venizelos N, von Dobeln U (1995) Clinical and biochemical presentation of long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. J Inherit Metab Dis 18:245–248
Halldin MU, Forslund A, von Dobeln U, Eklund C, Gustafsson J (2007) Increased lipolysis in LCHAD deficiency. J Inherit Metab Dis 30:39–46
Han JC, Lawlor DA, Kimm SY (2010) Childhood obesity. Lancet 375:1737–1748
He Q, Karlberg J (2001) BMI in childhood and its association with height gain, timing of puberty, and final height. Pediatric research 49:244–251
Houten SM, Wanders RJ (2010) A general introduction to the biochemistry of mitochondrial fatty acid beta-oxidation. J Inherit Metab Dis 33:469–477
IJlst L, Wanders RJ, Ushikubo S, Kamijo T, Hashimoto T (1994) Molecular basis of long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: identification of the major disease-causing mutation in the alpha-subunit of the mitochondrial trifunctional protein. Biochim Biophys Acta. Dec 8;1215(3):347–350
IJlst L, Ruiter JPN, Hoovers JMN, Jakobs ME, Wanders RJ (1996) Common missense mutation G1528C in long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency - Characterization and expression of the mutant protein, mutation analysis on genomic DNA and chromosomal localization of the mitochondrial trifunctional protein alpha subunit gene. J Clin Invest 98:1028–1033
Kong APS, Chan RSM, Nelson EAS, Chan JCN (2011) Role of low-glycemic index diet in management of childhood obesity. Obesity Reviews 12:492–498
Li L, Manor O, Power C (2004) Early environment and child-to-adult growth trajectories in the 1958 British birth cohort. Am J Clin Nutr 80:185–192
Lichtman SW, Pisarska K, Berman ER et al (1992) Discrepancy between self-reported and actual caloric-intake and exercise in obese subjects. New Engl J Med 327:1893–1898
Lindgren G, Strandell A, Cole T, Healy M, Tanner J (1995) Swedish population reference-standards for height, weight and body-mass index attained at 6 to 16 years (girls) or 19 years (boys). Acta Paediatrica 84:1019–1028
Ludwig DS (2002) The glycemic index: physiological mechanisms relating to obesity, diabetes, and cardiovascular disease. JAMA 287:2414–2423
Ludwig DS (2007) Clinical update: the low-glycaemic-index diet. Lancet 369:890–892
Lund AM, Skovby F, Vestergaard H, Christensen M, Christensen E (2010) Clinical and biochemical monitoring of patients with fatty acid oxidation disorders. J Inher Metabolic Dis 33:495–500
Potter BK, Little J, Chakraborty P et al (2012) Variability in the clinical management of fatty acid oxidation disorders: results of a survey of Canadian metabolic physicians. J Inherit Metab Dis 35:115–123
Prentice AM, Jebb SA (1995) Obesity in Britain: gluttony or sloth? BMJ 311:437–439
Rasmussen F, Eriksson M, Bokedal C, Elinder L (2004) [Fysisk aktivitet, matvanor, övervikt och självkänsla bland ungdomar. In: Rapport/Epidemiologiska enheten Samhällsmedicin, Stockholmsläns landsting (eds) Epidemiologiska enheten, Samhällsmedicin Statens folklhälsoinstitut, Stockholm]
Reilly JJ, Kelly J (2010) Long-term impact of overweight and obesity in childhood and adolescence on morbidity and premature mortality in adulthood: systematic review. Int J Obes (Lond) 35(7):891–898
Saudubray JM, Martin D, de Lonlay P et al (1999) Recognition and management of fatty acid oxidation defects: a series of 107 patients. J Inherit Metab Dis 22:488–502
Shalitin S, Phillip M (2003) Role of obesity and leptin in the pubertal process and pubertal growth–a review. Int J Obes Relat Metab Disord 27:869–874
Shaw V, Lawson M (2001) Clinical paediatric dietetics. Blackwell Science, Oxford
Sims HF, Brackett JC, Powell CK et al (1995) The molecular basis of pediatric long chain 3-hydroxyacyl-CoA dehydrogenase deficiency associated with maternal acute fatty liver of pregnancy. Proc Natl Acad Sci U S A 92:841–845
Spiekerkoetter U, Lindner M, Santer R et al (2009a) Management and outcome in 75 individuals with long-chain fatty acid oxidation defects: results from a workshop. J Inherit Metab Dis 32:488–497
Spiekerkoetter U, Lindner M, Santer R et al (2009b) Treatment recommendations in long-chain fatty acid oxidation defects: consensus from a workshop. J Inherit Metab Dis 32:498–505
Spiekerkoetter U, Bastin J, Gillingham M, Morris A, Wijburg F, Wilcken B (2010) Current issues regarding treatment of mitochondrial fatty acid oxidation disorders. J Inherit Metab Dis 33:555–561
Tanner JM, Goldstein H, Whitehouse RH (1970) Standards for children’s height at ages 2–9 years allowing for height of parents. Arch Disease Childhood 45:755–762
Tanner JM, Whitehouse RH, Marubini E, Resele LF (1976) The adolescent growth spurt of boys and girls of the Harpenden growth study. Ann Hum Biol 3:109–126
Thomas DE, Elliott EJ, Baur L (2007) Low glycaemic index or low glycaemic load diets for overweight and obesity. Cochrane Database Syst Rev CD005105
Tyni T, Palotie A, Viinikka L et al (1997) Long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency with the G1528C mutation: clinical presentation of thirteen patients. J Pediatrics 130:67–76
Tyni T, Ekholm E, Pihko H (1998a) Pregnancy complications are frequent in long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency. Am J Obst Gynecol 178:603–608
Tyni T, Kivela T, Lappi M, Summanen P, Nikoskelainen E, Pihko H (1998b) Ophthalmologic findings in long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency caused by the G1528C mutation: a new type of hereditary metabolic chorioretinopathy. Ophthalmology 105:810–824
Wanders RJ, Duran M, IJlst L et al (1989) Sudden infant death and long-chain 3-hydroxyacyl-CoA dehydrogenase. Lancet 2:52–53
Wanders RJ, IJlst L, van Gennip AH et al (1990a) Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: identification of a new inborn error of mitochondrial fatty acid beta-oxidation. J Inherit Metab Dis 13:311–314
Wanders RJ, van Roermund CW, Schutgens RB et al (1990b) The inborn errors of peroxisomal beta-oxidation: a review. J Inherit Metab Dis 13:4–36
Wikland KA, Luo ZC, Niklasson A, Karlberg J (2002) Swedish population-based longitudinal reference values from birth to 18 years of age for height, weight and head circumference. Acta Paediatrica 91:739–754
Wilcken B, Leung K, Hammond J, Kamath R, Leonard JV (1993) Pregnancy and fetal long-chain 3-hydroxyacyl coenzyme A dehydrogenase deficiency. Lancet 341:407–408
Xu L, Dubois L, Burnier D, Girard M, Prud'homme D (2011) Parental overweight/obesity, social factors, and child overweight/obesity at 7 years of age. Pediatr Int 53:826–831
Yang Z, Zhao Y, Bennett MJ, Strauss AW, Ibdah JA (2002) Fetal genotypes and pregnancy outcomes in 35 families with mitochondrial trifunctional protein mutations. Am J Obst Gynecol 187:715–720
Acknowledgments
We thank the patients and their families for taking part in the study. We are indebted to nurses Jenny Gårdman and dieticians Gunn Nyberg, Agnes Pal, and Christina Eklund for their skillful know-how and clinical assessments. This research has been financially supported by the regional agreement on medical training and clinical research (ALF) between Stockholm County Council and the Karolinska Institute, the Sven Jerring Foundation, the Samariten Foundation, the Child Care Society, and the Vera Ekström Foundation.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Additional information
Communicated by: Carlo Dionisi-Vici
Rights and permissions
This chapter is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
Copyright information
© 2012 SSIEM and Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Haglind, C.B. et al. (2012). Growth in Long-Chain 3-Hydroxyacyl-CoA Dehydrogenase Deficiency. In: Zschocke, J., Gibson, K., Brown, G., Morava, E., Peters, V. (eds) JIMD Reports - Case and Research Reports, 2012/5. JIMD Reports, vol 8. Springer, Berlin, Heidelberg. https://doi.org/10.1007/8904_2012_164
Download citation
DOI: https://doi.org/10.1007/8904_2012_164
Received:
Revised:
Accepted:
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-33432-0
Online ISBN: 978-3-642-33433-7
eBook Packages: MedicineMedicine (R0)