
Abstract
The objective of this study was to prepare and evaluate terbutaline sulphate (TBS) bi-layer tablets for once-daily administration. The bi-layer tablets consisted of an immediate-release layer and a sustained-release layer containing 5 and 10 mg TBS, respectively. The sustained-release layer was developed by using Compritol®888 ATO, Precirol® ATO 5, stearic acid, and tristearin, separately, as slowly eroding lipid matrices. A full 4 × 22 factorial design was employed for optimization of the sustained-release layer and to explore the effect of lipid type (X 1), drug–lipid ratio (X 2), and filler type (X 3) on the percentage drug released at 8, 12, and 24 h (Y 1, Y 2, and Y 3) as dependent variables. Sixteen TBS sustained-release matrices (F1–F16) were prepared by melt solid dispersion method. None of the prepared matrices achieved the targeted release profile. However, F2 that showed a relatively promising drug release was subjected to trial and error optimization for the filler composition to develop two optimized matrices (F17 and F18). F18 which consisted of drug–Compritol®888 ATO at ratio (1:6 w/w) and Avicel PH 101/dibasic calcium phosphate mixture of 2:1 (w/w) was selected as sustained-release layer. TBS bi-layer tablets were evaluated for their physical properties, in vitro drug release, effect of storage on drug content, and in vivo performance in rabbits. The bi-layer tablets showed acceptable physical properties and release characteristics. In vivo absorption in rabbits revealed initial high TBS plasma levels followed by sustained levels over 24 h compared to immediate-release tablets.
Similar content being viewed by others

INTRODUCTION
Terbutaline sulphate (TBS) is a β2 adrenergic receptor agonist, used in treatment of bronchial asthma, chronic bronchitis, and emphysema. It is a highly water-soluble compound (250 mg/ml) [1]. It has short biological half-life (3–4 h) [2] and systemic mean residence time of about 3 h [3]. It is given orally in a dose of 5 mg two or three times daily [4]. Such frequent drug administration may reduce patient compliance and therapeutic efficacy [5]. Therefore, a sustained-release TBS formulation is required to improve patient compliance and to maintain a relatively constant plasma level of the drug.
Several sustained-release formulation approaches have been developed for oral TBS administration such as microcapsules [6,7], microspheres [8,9], sustained-release coated pellets [10,11], gastroretentive floating tablets [3], asymmetric membrane osmotic capsules [12], and polymeric matrix tablets [13–15].
Recently, lipids are considered as alternative to polymers in the design of sustained drug delivery systems [16] due to their chemical inertness [17], cost-effectiveness, regulatory acceptance, and above all flexibility to achieve the desired drug release profile [18]. Wide arrays of wax-lipid based hydrophobic materials are available for sustaining drug action as Compritol® 888 ATO (glyceryl behenate) [19–21], Precirol® ATO 5 (glyceryl palmito-stearate) [22–24], stearic acid [23–25], and tristearin [26].
However, it is important to provide faster onset of action of TBS to immediately relieve acute asthmatic attack [27]. Therefore, in the present study, we have prepared bi-layer tablets, in which one layer is formulated to provide immediate drug release to reach a high plasma concentration in a short period of time. The second layer is a sustained release lipid-based matrix designed to maintain an effective drug plasma levels upon once-daily administration to enhance patient compliance.
MATERIALS AND METHODS
Materials
Terbutaline sulphate (TBS) was kindly donated as a gift by Sedico Company (Egypt). Propranolol hydrochloride (internal standard) was kindly donated from Kahira Company (Egypt). Compritol® 888 ATO (glyceryl behenate) and Precirol® ATO 5 (glyceryl palmito-stearate) were kindly donated as a gift by Gattefossé Company (France). Sodium starch glycolate (SSG) was kindly donated as a gift by Epico Company (Egypt). Tristearin and Avicel PH 101 were purchased from Fluka Biochemika Company (Switzerland). Potassium dihydrogen o-phosphate was purchased from Fisher Scientific (UK). Acetonitrile and methanol (HPLC grade) were purchased from Sigma-Aldrich (Saint Louis, MO, USA). Stearic acid, magnesium stearate, and anhydrous dibasic calcium phosphate (DCP) were purchased from El Nasr Pharmaceutical Chemicals Company (Egypt). Aironyl® tablets (manufactured by Sedico Company, Egypt) were purchased from a local pharmacy store. All other chemicals were of analytical grade.
Methods
Differential Scanning Calorimetry
The compatibility of drug–excipients was investigated by differential scanning calorimetry (DSC). The DSC thermograms of pure drug, individual excipients, drug–excipient physical mixtures (1:1 w/w), and drug–lipid melt solid dispersions (1: 1 w/w) were recorded. The analysis was performed using Shimadzu DSC-50 instrument equipped with a computerized data station. Samples (4–5 mg) were placed in an aluminum pan and heated at a rate of 10°C/min with indium in the reference pan in an atmosphere of nitrogen to a temperature of 300°C.
Preparation of Sustained-Release Matrices
A full 4 × 22 factorial design was used to develop TBS sustained-release matrices and to study the effect of three independent variables on the release of the drug using Design-Expert® software (version 7; Stat-Ease Inc., Minneapolis, MN, USA). In this design, lipid type (X 1), drug–lipid ratio (X 2), and filler type (X 3) were selected as independent variables. The X 1 variable was evaluated at four levels while each of the other two variables (X 2) and (X 3) was evaluated at two levels as shown in Table I. The cumulative percentage of drug released at 8, 12, and 24 h (Y 1, Y 2, and Y 3, respectively) were selected as dependent variables. Sixteen sustained-release matrices were prepared by melt solid dispersion method according to the compositions shown in Table II. An accurately weighed amount of lipid (Compritol®888 ATO, Precirol® ATO 5, stearic acid, and tristearin) was melted at a temperature above the melting point of lipid (74.09, 63.59, 56.67, and 70.96°C, respectively) by 10°C. TBS was then added with continuous stirring till homogenous dispersion was obtained. Drug loaded molten dispersion was allowed to cool down and solidify at room temperature. Subsequently, the mass was ground, pulverized, and passed through mesh sieve (less than 300 μm). Then, the resulting solid dispersion was mixed with either Avicel PH 101 or DCP for 10 min in a glass mortar. The obtained powders were mixed with magnesium stearate for 3 min. Finally, 120 mg of the mixture were weighed and fed manually into 8 mm die equipped with concave faced punch and compressed at constant compression force (100 bar) using a hydraulic press (PerkinElmer, Waltham, MA, USA).
In Vitro Drug Release from the Prepared TBS Sustained-Release Matrices
The release studies were performed using USP type II dissolution apparatus (Hanson Research, CA, USA). The paddles rotated at 50 rpm. The temperature was kept at 37 ± 0.5°C. Initial release studies were conducted in 140 ml of 0.1 N HCl (pH = 1.2) for a period of 2 h, then 100 ml 0.2 M tribasic sodium phosphate were added to the dissolution vessels for shifting pH to 6.8 for the following 3 h, and then 50 ml of 0.1 N NaoH were added to the dissolution vessels to change the pH to 7.4 till the end of release experiment. Two-milliliter samples were withdrawn after 0.25, 0.5, 1, 2, 3, 4, 5, 6, 7, 8, 12, 16, 20, and 24 h time intervals and replaced with an equal volume of fresh media. The samples were filtered through 0.45-μm Millipore membrane filters and analyzed spectrophotometrically at 276 nm (PerkinElmer Lambda Ez 201, UK) for TBS content. The release studies were carried out in triplicate.
Optimization of TBS Sustained-Release Matrix Based on In Vitro Release
A numerical optimization technique by the desirability approach was performed for X 1, X 2, and X 3 by utilizing Design-Expert® software followed by trial and error optimization by modifying the filler composition of F2 matrix. Avicel PH 101 was replaced by (Avicel PH 101/DCP) mixture (1:1 or 2:1 w/w) for preparation of the matrices F17 and F18, respectively. The composition of these matrices is detailed in Table II.
Release Kinetics from the Prepared TBS Sustained-Release Matrices
Data obtained from in vitro release were analyzed according to Korsmeyer–Peppas release model [28] given by the equation M t /M ∞ = kt n
where M t is the amount of drug released at time t, M ∞ is the amount of drug released at infinite time, K is the kinetic constant, and n is the release exponent indicative of the release mechanism.
The n values used for elucidation of drug release mechanism from the prepared sustained-release matrices were determined from the slope of the plot of log cumulative percent of drug release (≤60%) versus log time. If n ≤ 0.45, the release mechanism follows “Fickian diffusion” (case I) and higher values (0.45 < n < 0.89) for mass transfer follow anomalous non-Fickian model (released controlled by combination of diffusion and relaxation). The drug release follows zero-order drug release (case II transport) if the n value is 0.89. For the values of n higher than 0.89, the mechanism of drug release is regarded as super case II transport (relaxation).
Preparation and Evaluation of TBS Immediate-Release Layer
The immediate-release layer, containing 5 mg TBS, 71 mg Avicel PH 101, 3.2 mg sodium starch glycolate, and 0.8 mg magnesium stearate, was prepared by direct compression in 8 mm die using a hydraulic press. The in vitro drug release from the prepared immediate-release layer in 0.1 N HCl was compared to that of a marketed TBS immediate release tablet.
Preparation of TBS Bi-layer Tablets
For the preparation of the bi-layer tablets, the powder mixture of the optimized sustained-release matrix F18 was fed manually into the die and compressed using mild compression force. Then, the powder mixture of the immediate-release layer was fed into the die on the sustained layer and then compressed to the final tablet using a hydraulic press at compression force of 100 bar.
In Vitro Evaluation of the Prepared TBS Bi-layer Tablets
Physical Characterization
The physical properties of the prepared bi-layer tablets were characterized for thickness, diameter, hardness, percent friability, and weight uniformity.
Drug Content Uniformity
Ten tablets from different batches were individually crushed and extracted in 100 ml boiling distilled water. Then, 1 ml was taken and diluted to 2 ml with distilled water. The solution was filtered through a micropore sterile syringe filter 0.45 μm, and the drug content was determined by UV spectroscopy at λ max of 276 nm.
In Vitro Drug Release Studies
The release studies on the prepared TBS bi-layer tablets were performed as described earlier under the in vitro release of TBS sustained-release matrices.
Effect of Storage on TBS Bi-layer Tablets
Effect of Storage on TBS Bi-layer Tablets Was Evaluated in Terms of TBS Content
Tablets were stored in well-stoppered amber glass containers at 25°C for 6 months. Three of the stored tablets were withdrawn and analyzed for TBS content at 0, 3, and 6 months storage. Each tablet was individually crushed and extracted in 100 ml boiling distilled water. Then, 1 ml was taken and diluted to 10 ml with distilled water. The solution was filtered, and the drug content was determined by using a validated stability indicating HPLC method [29] with slight modifications. The HPLC system was Agilent series 1100, Agilent Technologies; Deutschland GmbH. Samples (5 μl) were injected into Inertsil C18 column (250 mm × 4.6 mm, 5 μm). A mobile phase consisting of a mixture of acetonitrile/potassium dihydrogen-o-phosphate buffer (pH 6) at a ratio of 20:80 (v/v %) was used in an analysis that was conducted in an isocratic elution mode. Data acquisition and integration were carried out using software (Chem32). The detection wavelength was 214 nm. All operations were carried out at ambient temperature.
In Vivo Absorption Study
The in vivo absorption study was carried out to explore the potential of the prepared bi-layer tablets as once-daily dosage form of TBS able to maintain adequate drug plasma levels over 24 h compared to a marketed immediate-release tablet.
Study Design
The protocol of the study was approved by the Animal Ethics Committee of Faculty of Pharmacy, Helwan University. Twelve healthy albino rabbits of either sex weighing between 1500 and 1700 g were used in the study. The rabbits were randomly divided into two groups of equal size (six rabbits each). Each rabbit in group 1 received single oral dose of the prepared bi-layer tablets containing 15 mg TBS and rabbits in group 2 received a single oral dose of marketed immediate-release tablet containing 5 mg TBS. The tablets of each formulation were administered orally to the rabbits of each group along with 10 mL of water by using a feeding tube. All the rabbits were fasted over night before drug administration and continued fasting until 4 h postdose, with water allowed.
Blood Sampling
Venous blood samples were collected directly from each rabbit into heparinized tubes at 0 (pre-dose), 0.5, 1, 2, 3, 4, 6, 8, 12, and 24 h postdose. Plasma was immediately separated from the blood cells by centrifugation at 4000 rpm for 10 min and stored frozen at −80°C until being analyzed using a reported liquid chromatography-tandem mass spectrometry (LC-MS/MS) assay [30] with slight modifications.
Analysis of TDS Plasma Levels
Internal standard [35 μl of propranolol hydrochloride solution (1 μg/ml)] was mixed with 250 μl plasma samples. Then, 50 μl methanol was added. Then, the volume completed to 1 ml with acetonitrile. Samples were vortexed for 30 s and centrifuged at 4000 rpm for 5 min. The supernatant was filtered and then injected onto the LC-MS/MS system.
LC-MS/MS System
LC-MS/MS system consisted of Agilent 6420 Triple Quadruple with G1311A quaternary pump, G1329A auto-sampler, and G1322A vacuum degasser (Agilent Technologies Deutschland GmbH, Waldbron, Germany). The used column was Inertsil ODS-3 (50 mm × 4.6 mm, 5 μm). A mobile phase consisting of acetonitrile/100 mM ammonium formate (60:40) (% v/v) was conducted in an isocratic elution mode at a flow rate of 0.9 ml/min under ambient temperature. The injection volume for samples was 5 μl. The operation mode of mass spectrometry was electrospray positive mode. Nitrogen gas temperature and flow rate were 350°C and 10 l/min, respectively. Nebulization pressure was 40 psi and capillary voltage was 4000 v. Quantitation of the transitions from m/z 226 to 151.9 for TBS and from m/z 260 to 183 for internal standard was conducted with a scan time of 1.1 min. The retention time of TBS and internal standard was 0.596 and 0.786 min, respectively. Data acquisition was performed using 6400 Series Triple Quadrupole B.07.00 (version B7022.0) software.
Pharmacokinetic and Statistical Analyses
The values of the maximum TBS plasma concentration (C max, ng/ml) and the time to reach C max (T max, h) following oral administration of the two treatments were obtained directly from the plasma data of the individual plasma concentration–time curves. The obtained pharmacokinetic parameters (C max and T max) were analyzed by Student t test using Microsoft Office 2007, Excel package. A statistically significant difference was considered at P < 0.05.
RESULTS AND DISCUSSION
Differential Scanning Calorimetry
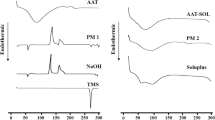
The differential scanning calorimetry (DSC) thermograms of TBS, drug–lipids physical mixtures (1:1 w/w), and their 1:1 (w/w) melt solid dispersions are shown in Fig. 1. The DSC thermogram of TBS showed a characteristic, endothermic melting peak at 263.98°C, indicating its crystalline state. This is in a good agreement with the melting point reported values in literature ranged between 258 to 271°C [2].

DSC Thermograms of A TBS, B TBS/Compritol physical mixture, C TBS/Compritol melt solid dispersion, D TBS/Precirol physical mixture, E TBS/Precirol melt solid dispersion, F TBS/stearic acid physical mixture, G TBS/stearic acid melt solid dispersion, H TBS/tristearin physical mixture, and I TBS/tristearin melt solid dispersion
The thermograms of physical mixtures and melt solid dispersions showed that TBS endothermic peak was within the reported melting point range of the drug, indicating that there was no change in the crystalline state of the drug and absence of drug–lipids interaction.
In Vitro Drug Release from TBS Sustained-Release Matrices
Figure 2a, b shows the in vitro release profiles of TBS from different sustained-release matrices. A burst release of TBS from all the matrices was observed after 15 min. This might be due to the fast dissolution of the drug particles present at the surface, while the embedded drug in the matrices was released at a slower rate [31]. ANOVA test for the selected factorial model was performed to evaluate the level of significance of main effect (one factor effect) of the tested factors (independent variables) on the percentage drug released from different sustained-release matrices after 8, 12, and 24 h as well as the interaction between these factors (two factor interaction).

Release profiles of TBS from different sustained-release matrices formulae a F1–F8 and b F9–F16
One-Factor Effect on the Drug Release
Effect of Lipid Type
A significant impact of the lipid type (X 1) on drug release appeared only after 8 h (Y 1) (P < 0.05), while after 12 (Y 2) and 24 h (Y 3), there was no significant effect of lipid type on the drug release (P > 0.1). However, the lipid type variable had four levels, so recall from ANOVA was the overall difference between responses after 8 h. Moreover, Tukey–Kramer multicomparison post test, for drug release after 8 h from the matrices having similar composition but different type of lipid, revealed that there was no significant (P > 0.05) difference between each pair of the matrices (F1, F5, F9, and F13). However, there was a significant difference between each pair of matrices (F2, F6, F10, and F14) except the pair (F10–F14). These results indicate that when Avicel PH 101 was used as filler, the lipid type had no effect on percent drug released at 1:3 drug–lipid ratio. However, at 1:6 drug–lipid ratio, there was no significant (P > 0.05) difference between stearic acid and tristearin and both exhibited a significantly (P < 0.05) higher drug release than Compritol and Precirol.
On replacement of Avicel PH 101 by DCP as filler, the results showed that the percent drug released from the lipids followed the following order: tristearin < Compritol = Precirol < stearic acid, at 1:3 drug–lipid ratio, while at 1:6 drug–lipid ratio, Compritol significantly (P < 0.05) provided the highest percent drug release compared to other used lipids.
Effect of Drug–Lipid Ratio
A significant impact of the drug–lipid ratio on the drug release was demonstrated. Changing the drug–lipid ratio from 1:3 to 1:6 resulted in a significant decrease in %TBS released after 8, 12, and 24 h (P < 0.0001). This agrees with previously reported results [32,33]. The authors reported that increasing the hydrophobic content of the matrix significantly reduced the rate and extent of drug release, as a result of increased drug diffusion path length, the decrease in the total porosity of the matrices, and the negative effect of hydrophobic matrix against the penetration of the aqueous dissolution medium.
Effect of Filler Type
Avicel PH 101 significantly increased the %TBS released compared to DCP (P < 0.0001) after 8, 12, and 24 h. This may be attributed to difference in solubility and swelling nature of the two fillers [34–36].
Two-Factor Interaction
Figure 3 shows X 1 X 2, X 1 X 3, and X 2 X 3 interaction after 8 h (Y 1). It can be shown from this figure that there is a highly significant (P < 0.0001) interaction between lipid type and lipid–drug ratio (X 1 X 2) and between lipid type and filler type (X 1 X 3). However, the interaction between drug–lipid ratio and filler type (X 2 X 3) is not significant (P < 0.05). Moreover, the two-factor interactions between all the independent variables were found to be highly significant (P < 0.0001) at 12 (Y 2) and 24 h (Y 3) (data not shown).

Two-factor interaction charts of Y 1 response between a lipid type and drug–lipid ratio (X 1 X 2), b lipid type and filler type (X 1 X 3), and c drug–lipid ratio and filler type (X 2 X 3)
Optimization of TBS Sustained-Release Matrix
In order to find the level of each independent variable for the optimized sustained-release matrix, a numerical optimization technique by the desirability approach was performed for X 1, X 2, and X 3 using the following target ranges: 50% ≤ Y 1 ≤ 60%, 65% ≤ Y 2 ≤ 75%, and 90% ≤ Y 3 ≤ 100%. The target ranges of these responses were based on the standard values of percent drug released at different time points for the 24-h sustained-release profiles reported by Dhameliy et al. [16].
Design-Expert® software revealed that there is no possibility to get an optimized sustained-release matrix from the analyzed formulae. However, F2 matrix achieved a relatively promising release profile compared to the targeted release percent after 8, 12, and 24 h. Trial and error optimization was carried out for the filler composition in formula (F2) to reach the targeted release profile. The optimized formulae (F17 and F18) which consisted of drug–Compritol®888 ATO at ratio (1:6 w/w) and Avicel PH 101/DCP (1:1 and 2:1 w/w), respectively, were prepared. Figure 4 shows the in vitro release from the matrices F17 and F18. The matrix F17 released 39.78 ± 4.09, 57.95 ± 3.77, and 82.85 ± 3.56% after 8, 12, and 24 h, respectively. The matrix F18 released 48.97 ± 0.61, 67.25 ± 1.14, and 94.84 ± 0.62% after 8, 12, and 24 h, respectively. Accordingly, F18 was selected as a sustained-release layer based on its closely matched release profile with the targeted set of values of drug release at different time points.

Release profiles of TBS from optimized sustained-release matrices
Release Kinetics of TBS from Sustained-Release Matrices
The release kinetics of TBS from the prepared sustained-release matrices (F1–F18) were studied by applying the Korsmeyer–Peppas model. All the matrices exhibited Fickian release except F10 and F14 which had (n) values more than 0.45 and less than 0.89 indicating non-Fickian anomalous transport. In addition, matrices (F1, F5, F9, and F13) showed (n) values more than 0.89, indicating super case II (relaxation).
TBS Immediate-Release Layer
The immediate-release layer was prepared and evaluated for in vitro release of TBS compared to a marketed immediate-release tablet. The drug release profile of the prepared formula was similar to that of the marketed immediate-release tablet (data not shown).
In Vitro Evaluation of the Prepared TBS Bi-layer Tablets
The prepared tablets showed accepted limits for thickness, diameter uniformity, and %friability. Hardness was 12.2 ± 1.13 kg which is within the accepted limits for sustained release tablets [37]. The tablets met the requirements concerning the uniformity of weight where the percent deviation from the average did not exceed ±7.5% [38]. The amount of drug in each of the ten tablets lies within the range of 85–115% and RSD% was (<6%) [39], which reflects a high degree of distribution homogeneity of the drug among the prepared tablets.
The release profile of TBS bi-layer tablet (Fig. 5) showed that the bi-layer tablets released 35.33 ± 1.36% of TBS after 15 min and 68.98 ± 3.66, 81.17 ± 1.35, and 95.56 ± 1.33% of TBS after 8, 12, and 24 h, respectively. This result indicated that the bi-layer tablets are able to provide a sustained release of TBS over 24 h with immediate release of (~5 mg) of the drug after 15 min as loading dose.

Release profile of TBS from bi-layer tablet
Effect of Storage on TBS Bi-layer Tablets
After storage of the bi-layer tablet at 25°C, the percent drug content after 3 and 6 months was 99 ± 1.68 and 98.38±1.65% from the initial content that was measured at zero time. This result indicates the stability of TBS in the bi-layer tablets when stored at room temperature.
In Vivo Absorption Study
Figure 6 shows the mean TBS plasma concentration–time curves after oral administration of single dose of bi-layer tablet (15 mg) and immediate-release marketed tablet (5 mg) in rabbits. The mean C max values for the bi-layer tablets and the marketed immediate-release tablets were 354.12 ± 106.64 and 172.07 ± 40.70 ng/ml, respectively. The significantly (P < 0.05) higher C max produced by the bi-layer tablet could be attributed to concomitant drug release from the immediate-release layer and the burst release of TBS from the sustained layer as previously observed in the in vitro release studies. The mean T max values for the bi-layer tablets and the marketed immediate-release tablets were 2.6 ± 0.55 and 2.2 ± 0.45 h, respectively. The difference between the mean T max values of both formulations was found to be statistically insignificant (P < 0.05). It could be observed from Fig. 6 that the plasma TBS level after administration of the bi-layer tablets was sustained over 24 h, while the drug level of the marketed immediate-release tablets dropped below the detection limit after 12 h. This indicates that the prepared TBS bi-layer tablets have the potential to be used as once-daily dosage regimen that could enhance patient compliance in treatment of chronic asthmatic patients using TBS. However, further in vivo absorption studies in humans and clinical investigations in asthmatic patients should be conducted to support the obtained results.

Mean (±SD, n = 6) plasma concentration–time curves of TBS in rabbits after single oral administration of bi-layer tablet (15 mg) and marketed immediate-release tablet (5 mg)
CONCLUSION
TBS bi-layer tablets comprising immediate-release layer and lipid-based sustained-release matrix [melt solid dispersion of (1:6 w/w) drug–Compritol®888 ATO ratio and Avicel PH 101/DCP mixture (2:1 w/w)] were developed and showed a promising in vitro drug release. In vivo absorption in rabbits indicated that the bi-layer tablets could be a potential dosage form for once-daily administration of TBS and improving patient compliance. However, further in vivo absorption studies in humans and clinical investigations in asthmatic patients should be conducted to support the obtained results.
References
Borgström L, Bondesson E, Morén F, Trofast E, Newman SP. Lung deposition of budesonide inhaled via Turbuhaler®: a comparison with terbutaline sulphate in normal subjects. Eur Respir J. 1994;7:69–73.
Ahuja S, Ashman J. Terbutaline sulfate. In: Florey K, editor. Analytical profiles of drug substances. New York: Academic; 1990. p. 601–25.
Kumar L, Singh V, Meel RK. Formulation and evaluation of gastro retentive floating tablets of terbutaline sulphate. Int J Pharma Sci Res. 2014;5(10):639–45.
Reynolds JEF. Martindale: the extra pharmacopoeia. 29th ed. London: The Pharmaceutical Press; 1989.
Hirtz J. The GIT, absorption of drugs in man a review of current concepts and methods of investigation. Br J Clin Pharmacol. 1985;19:77S–83S.
Manekar NC, Puranik PK, Joshi SB. Prolonged released terbutaline sulphate microcapsules. J Microencapsul. 1991;8(4):521–3.
Manekar NC, Puranik PK, Joshi SB. Microencapsulation of terbutaline sulphate by solvent evaporation technique. J Microencapsul. 1992;9(4):481–7.
Kim CK, Kim MJ, Oh KH. Preparation and evaluation of sustained release microspheres of terbutaline sulfate. Int J Pharm. 1994;106:213–9.
Kotadiya R, Patel V, Patel H, Salaniya B. Evaluation of terbutaline sulphate encapsulated ethylcellulose microspheres: a factorial approach. Int J PharmTech Res. 2009;1(4):1271–8.
Umprayn K, Chitropas P, Amarekajorn S. Development of terbutaline sulfate sustained release coated pellets. Drug Dev Ind Pharm. 1999;25(4):477–91.
Abdul Hadi M, Rao AS, Abhinetri V, Rao AHP. Design and in-vitro evaluation of multiparticulate drug delivery system of terbutaline sulphate for the treatment of nocturnal asthma. Der Pharmacia Lett. 2012;4(6):1777–85.
Gobade NG, Koland M, Harish KH. Asymmetric membrane osmotic capsules for terbutaline sulphate. Indian J Pharm Sci. 2012;74(1):69–72.
Varshosaz J, Emami J, Jafari E. Comparison of hydrophilic natural gums and cellulosic polymers in formulation of sustained-release matrix tablets of terbutalin sulfate. Res Pharmaceut Sci. 2006;1:30–9.
Harish NM, Narayana CR, Shenoy KRP, Prabhu P, Narendra C, Nisha GS. Formulation design and optimization of sustained release tablets of terbutaline sulphate. Ind J Pharm Edu Res. 2011;45(3):259–66.
Kola R, Deepa RN, Pragati KB. Formulation and in-vitro evaluation of terbutaline sulphate sustained release tablets. Ind J Res Pharma Biotechnol. 2013;1(5):621–4.
Dhameliya PB, Vyas JR, Narola M, Patel K, Upadhyay U. Preparation and evaluation of sustained release tablet of eperisone hydrochloride by Compritol ATO888 as matrix forming agent. J Drug Del Therapeut. 2014;4(3):132–7.
Miyagawa Y, Okabe T, Yamaguchi Y, Miyajima M, Sato H, Sunada H. Controlled release of diclofenac sodium from wax matrix granules. Int J Pharm. 1996;138(2):215–24.
Goodhart FW, McCoy RH, Ninger FC. Release of a water-soluble drug from a wax matrix timed-release tablet. J Pharm Sci. 1974;63(11):1748–51.
Li FQ, Hu JH, Deng JX, Su H, Xu S, Liu JY. In vitro controlled release of sodium ferulate from compritol 888 ATO-based matrix tablets. Int J Pharm. 2006;324:152–7.
Barakat NS, Elbagory IM, Almurshedi AS. Controlled-release carbamazepine granules and tablets comprising lipophilic and hydrophilic matrix components. AAPS Pharm Sci Tech. 2008;9(4):1054–62.
Roberts M, Daniele Vellucci D, Mostafa S, Miolane C, Marchaud D. Development and evaluation of sustained-release Compritol®888 ATO matrix mini-tablets. Drug Dev Ind Pharm. 2012;38(9):1068–76.
Hamdani J, Moës AJ, Amighi K. Physical and thermal characterization of Precirol® and Compritol® as lipophilic glycerides used for the preparation of controlled-release matrix pellets. Int J Pharm. 2003;260(1):47–57.
O¨zyazıcı M, Go¨kce EH, Ertan G. Release and diffusional modeling of metronidazole lipid matrices. Eur J Pharm Biopharm. 2006;63(3):331–9.
Abd-Elbary A, Tadros MI, Alaa-Eldin AA. Sucrose stearate-enriched lipid matrix tablets of etodolac: modulation of drug release, diffusional modeling and structure elucidation studies. AAPS Pharm Sci Tech. 2013;14(2):656–68.
Abdul Quadir M, Rahman MS, Karim MZ, Akter S, Bin Awkat MT, Reza MDS. Evaluation of hydrophobic materials as matrices for controlled-release drug delivery. Pak J Pharm Sci. 2003;16(2):17–28.
Nagarani B, Ashwinkumar K, Srikanth P, Julapally D. A review on controlled drug delivery and brief information on matrix system. Int J Innov Pharmaceutl Sci Res. 2014;2(7):1555–86.
Suresh S, Pandit N, Joshi HP. Preparation and evaluation of mouth dissolving tablets of salbutamol sulphate. Indian J Pharm Sci. 2007;69(3):467–9.
Korsmeyer RW, Gurny R, Doelker E, Buri P, Peppas NA. Mechanism of solute release from porous hydrophilic polymers. Int J Pharm. 1983;15:25–35.
Bapatu HR, Kumar MR, Mallu UR, Ganthi HKR, Kota CMR, Pyreddy VR. Stability indicating RP-HPLC method for the determination of terbutaline sulphate, guaifenesin, ambroxol hydrochloride and preservatives content in liquid formulations. J Pharm Res. 2011;4(11):4117–22.
Faiyazuddin M, Ahmad N, Iqbal Z, Talegaonkar S, Bhatnagar A, Khar RK, et al. Development and validation of UHPLC/ESI-Q-TOF-MS method for terbutaline estimations in experimental rodents: stability effects and plasma pharmacokinetics. Curr Pharma Anal. 2012;8(2):1–7.
Obaidat AA, Obaidat RM. Controlled release of tramadol hydrochloride from matrices prepared using glyceryl behenate. Eur J Pharm Biopharm. 2001;52:231–5.
Quadir MA, Rahman MS, Karim MZ, Akter S, Awkat MT, Reza MS. Evaluation of hydrophobic materials as matrices for controlled-release drug delivery. Pak J Pharm Sci. 2003;16:17–28.
Aboelwafa AA, Basalious EB. Optimization and in vivo pharmacokinetic study of a novel controlled release venalafaxine hydrochloride three layer tablet. AAPS Pharm Sci Tech. 2010;2(3):1026–37.
Abd El-Halim SM, Amin MM, El-Gazayerly ON, Abd El-Gawad NA. Comparative study on the different techniques for the preparation of sustained-release hydrophobic matrices of a highly water-soluble drug. Drug Discov Ther. 2010;4(6):484–92.
Liu J, Zhang F, McGinity JW. Properties of lipophilic matrix tablets containing phenylpropanolamine hydrochloride prepared by hot-melt extrusion. Eur J Pharm Biopharm. 2001;52(2):181–90.
El-Shanawany S. Sustained release of nitrofurantoin from inert wax matrices. J Control Release. 1993;26:11–9.
Pandey S, Niraj, Gupta MM, Chauhan BS. Bilayer tablets: recent trends in oral drug delivery with present and future prospects. Int J UnivPharma Bio Sci. 2013;2(3):360–78.
The British pharmacopoeia, London: HMSO;1993. p.753.
US pharmacopeia, 22nd ed. USA: Mack printing Company, Easton, PA; 1990.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Hashem, F.M., Nasr, M., Fathy, G. et al. Formulation and In Vitro and In Vivo Evaluation of Lipid-Based Terbutaline Sulphate Bi-layer Tablets for Once-Daily Administration. AAPS PharmSciTech 17, 727–734 (2016). https://doi.org/10.1208/s12249-015-0404-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1208/s12249-015-0404-z