
Abstract
Exenatide is a glucagon-like peptide-1 receptor agonist with both immediate- and extended-release (ER) formulations that are approved for the treatment of type 2 diabetes mellitus. Long-term exposure from the ER formulation is achieved through slow peptide release from a degradable microsphere formulation. The goal of this analysis was to develop a pharmacokinetic model for the ER formulation following single and once-weekly multiple-dose administration. Pharmacokinetic data were collected from two clinical trials—one that evaluated single-dose administration of 2.5, 5, 7, and 10 mg of ER exenatide and a second that included weekly administration of 0.8 and 2 mg for 15 weeks. A population pharmacokinetic model, describing 1586 exenatide concentrations from 64 patients, was developed in the nonlinear mixed-effects modeling software program NONMEM. Pharmacokinetics of the ER formulation was described by a two-compartment model with linear and nonlinear elimination. The complex absorption profile was quantified using three simultaneous processes: a first-order process characterizing a rapid initial release and two series of transit compartments to describe the second (∼3 weeks postinjection) and third phases of drug release (∼7 weeks postinjection). Estimation of the combined single- and multiple-dose data adequately described the rise to steady-state (∼8–10 weeks) and decline to undetectable concentrations that occurred about 10 weeks after treatment cessation. Thus, a population-based pharmacokinetic model was developed that provides a platform for future pharmacodynamic analyses with the ER formulation of exenatide.
Similar content being viewed by others
INTRODUCTION
Exenatide is a glucagon-like peptide-1 (GLP-1) receptor agonist that has been shown to improve glycemic control among patients with type 2 diabetes mellitus (1, 2). Exenatide is available either as a twice-daily, immediate-release (IR) formulation (Byetta®; AstraZeneca, Wilmington, DE), or as a once-weekly, extended-release (ER) formulation (Bydureon®; AstraZeneca). The exenatide peptide elicits its glycemic response through multiple mechanisms of action, including glucose-dependent insulin secretion (first and second phase) (3), slowing of gastric emptying (4), increased satiety, and suppression of glucagon (1, 3–5). The pharmacokinetics (PK), safety, and efficacy of the IR formulation have been extensively documented (4, 6–16). A retrospective population PK analysis describing a wide range of exenatide concentrations associated with multiple routes of administration identified significant covariates that influence systemic exposure of the IR formulation, including renal function on systemic clearance and body weight on the volume of distribution (Cirincione B, Mager DE. Population pharmacokinetics of exenatide. Manuscript accepted). For a typical patient, both the absorption and elimination of the IR formulation following subcutaneous (SC) administration are rapid, with peak concentrations occurring approximately 2 h postdose, followed by a terminal half-life of about 2.4 h (1). Thus, the IR formulation is administered twice daily, within 60 min prior to the morning and evening meals, to achieve effective daily glycemic control (1). In order to improve patient compliance and further improve glycemic benefit, new drug formulations are under development with the potential to extend drug release, resulting in an increased duration of exenatide exposure and effects.
The ER formulation of exenatide was developed to provide patients with long-term exenatide exposure allowing for a once-weekly dosing interval (2). This formulation is based on dispersing exenatide in a structural matrix composed of medical-grade biodegradable poly(D,L-lactide-co-glycolide) (PLGA) polymer (17). After SC administration, the PLGA polymer undergoes hydrolysis in the SC space leading to a continuous release of exenatide (17, 18). The release of exenatide from the PLGA polymers following a single injection results in a total duration of drug exposure of 10–12 weeks (18). The concentration–time profile of the ER formulation after a single SC injection is characterized by three distinct phases. Phase 1 is an initial release of exenatide that is loosely bound to the surface of the microsphere, which is readily released upon hydration of the microsphere and occurs within the first 24 h after SC administration. Following this initial release, the PLGA polymer is slowly hydrolyzed leading to a slow release of exenatide, culminating in a second peak occurring approximately 2 weeks after an SC dose. During the third phase, the microspheres fully hydrolyze and all the remaining drug is slowly released in the SC space to be absorbed into the systemic circulation, producing a third peak that appears in the concentration–time profile approximately 7 weeks postdose. The microspheres themselves are degraded to water and CO2 in the SC space and are not absorbed intact into the circulation (17). Once released from the microspheres and absorbed into the circulation, the exenatide peptide follows the same distribution and elimination properties as the IR formulation (17).
The aim of this analysis was to develop a population PK model for the ER formulation of exenatide following both single- and multiple-dose administration. Such a model must account for each phase of drug release, providing a quantitative platform for assessing the factors influencing the time to steady-state and average plasma drug concentrations following multiple-dose administration. The final model may be used to test hypotheses pertaining to the influence of intrinsic factors on components of the concentration–time profile and ultimate pharmacodynamic responses, as well as for simulating alternative dosing regimens.
MATERIALS AND METHODS
Study Design
PK data for model development were obtained from two clinical trials (18). The first study was a phase II, randomized, double-blind, placebo-controlled 12-week trial in patients with type 2 diabetes using a single dose of the ER formulation at doses of 2.5, 5, 7, and 10 mg, which resulted in measurable exenatide concentrations for approximately 9–11 weeks postinjection (18). The study term visit was considered uninformative as it consisted of primarily undetectable exenatide concentrations and thus was not included. Exenatide plasma concentrations were assessed at 23 time points over the duration of the trial. Specific collection times included a rich sampling period (0.25, 0.5, 1, 2, 4, 8, 12, 16, 24, 30, 36, and 48 h postinjection) to capture initial drug release, followed by sampling at 11 additional time points (collected at weekly visits from weeks 1 to 11). A total of 1054 concentration measurements from 47 patients were collected in the single-dose study. Data from six patients were excluded due to one or more of the following reasons: patient was determined not evaluable, more than 90% of the concentrations were assayed as below the limit of quantification (BLQ), or the profile significantly deviated from the anticipated population profile. There were a total of 887 plasma exenatide concentrations from 41 patients collected from this single-dose study. The second study was a phase II, randomized, double-blind, placebo-controlled trial that evaluated two dose levels of ER exenatide (0.8 and 2 mg) administered once weekly for 15 weeks (18). Exenatide plasma concentrations were measured at 22 time points across the 15-week treatment period capturing the initial release and the rise to steady-state conditions, followed by an additional 8 samples during a 12-week washout period. Concentration–time data from 31 patients enrolled in this phase II trial were available, and eight patients were excluded due to incomplete dosing information. There were a total of 699 plasma exenatide concentrations collected from 23 patients with complete dosing in this multiple-dose study. Overall, a total of 1586 exenatide concentrations from 64 patients were available for model development.
Data from 119 patients (408 plasma exenatide concentrations) enrolled in a phase III trial evaluating 2 mg of ER exenatide for 24 weeks were used as an external validation dataset (19). Plasma drug concentrations were obtained at five time points across 24 weeks, with predose samples measured approximately every 4 weeks (weeks 4, 8, 14, 20, and 24 after the start of therapy). All studies were conducted in accordance with the principles described in the Declaration of Helsinki (1946) up to and including the Seoul revision (20). All clinical protocols were approved for each site by an appropriate institutional review board, and all patients provided written informed consent prior to participation.
Exenatide Analytical Method
Plasma samples were analyzed for exenatide concentration using a validated two-site sandwich enzyme-linked immunosorbent assay. The capture monoclonal antibody is specific for exenatide, as it recognizes a C-terminal epitope and does not cross-react with GLP-1(7-36) or glucagon. The detection monoclonal antibody recognizes an N-terminal epitope on exenatide, GLP-1(7-36), and glucagon. The assay is specific for exenatide due to the selectivity of the capture antibody. Since both antibodies need to recognize the peptide in order to generate a signal for this assay, cross-reactivity with other peptides or metabolites is minimized. No cross-reactivity was observed with 1 ng/mL of exendin-4(2-39), exendin-4(3-39), GLP-1(7-36), glucagon, or insulin. The assay lower limit of quantification was 10 pg/mL. Method validation specifications for accuracy, precision, and other performance parameters associated with regulated ligand-binding assays were met at all analytical laboratories (21).
PK Model Development
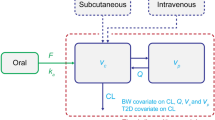
The PK model was developed in two stages, starting with single-dose data to describe the three phases of exenatide release from the microspheres. Subsequently, the single- and multiple-dose data were combined and modeled simultaneously. The final PK model is shown in Fig. 1. The absorption of exenatide from the ER formulation was described with three distinct processes. The initial release was captured with a first-order absorption rate constant (k a), whereas the second and third release processes were each described with a series of transit compartments using an explicit equation derived for such systems (22, 23):

Structural PK model diagram. Exenatide PK was characterized using a two-compartment model with three absorption processes (first-order release and two transit compartment processes) and both linear and nonlinear elimination
The elimination of exenatide (Elim) is described by first-order and saturable elimination processes:
and N i ! is defined using the Sterling approximation:
The estimated parameters associated with the two transit compartment models were the number of transit compartments (N 1 and N 2) and the first-order transit rate constants (k tr1 and k tr2). The sum of the bioavailability of the three parallel absorption processes was parameterized to assure that all of the bioavailable dose relative to the IR formulation (f rel) is absorbed: f 3 = 1 − (f 1 + f 2). For each of the transit series, the absorption rate constant from the final transit compartment into the central compartment is set equal to the k tr of that series. Owing to the complex absorption kinetics, the disposition-related population parameters (i.e., clearance (CL), Michaelis–Menten constants (V max and K m) and volumes of the central and peripheral compartments (V C and V P )) were fixed to the previously developed population model for the IR formulation of exenatide (Cirincione B, Mager DE. Population pharmacokinetics of exenatide. Manuscript accepted).
Data Analysis
All modeling and simulations were conducted using the Laplacian estimation method within the nonlinear mixed-effects modeling software program NONMEM version 7.2 (ICON Development Solutions, Ellicott City, MD). Interindividual variability (IIV) was estimated using a log-normal distribution model on two of the first-order rate constants (k a and k tr2) and the fraction of the bioavailable dose in the initial release (f 1). Alternative distribution models and IIV parameters were evaluated and found to be unsuccessful. The residual variability (RV) was described with a log error structure, and alternative RV structures were evaluated but did not improve model performance.
The single-dose model was adapted to accommodate the weekly dosing regimen in NONMEM using recently published methodology for modeling multiple-dose transit compartment models (23). With the addition of the multiple-dose data, an IIV term was added to the relative bioavailability compared to the IR formulation (f rel). The RV was estimated separately for the single- and multiple-dose trials. Model adequacy was assessed by parameter precision, standard graphical assessments, and evaluation of predictive performance through visual predictive checks (VPCs). The VPC was performed by evaluating the median and 90% prediction interval of 500 simulations from the model in comparison to the observed data (24).
The number of samples obtained following single-dose administration with exenatide concentrations BLQ (10 pg/mL) was largest at the lowest dose level and generally decreased with increasing dose. There were 83 (36.9%), 32 (15.6%), 50 (19.8%), and 26 (14.2%) samples that were BLQ for the 2.5-, 5-, 7-, and 10-mg dose groups, respectively. The BLQ concentrations were primarily in the time period between the first- and second-phase release (within the first 5 days), which is during the lag period for hydration of the microsphere, and at the end of the single-dose profile (i.e., after 60 days following a single dose). Given the number of concentrations that were determined to be BLQ and the time dependency of their occurrence (primarily occurring between the first and second phases), the BLQ data were considered informative and included in the modeling using the Beal M3 method coupled with Laplacian estimation (25). This method allows the BLQ data to be maintained in the dataset but treats them as censored observations (25). VPCs of the frequency of BLQ observations across time were generated (26), in which the simulated median and 90% prediction interval of the fraction of data predicted to be BLQ from 500 simulations were compared with the observed fraction of observed BLQ at each time point. With the weekly dosing regimen, the number of concentrations determined to be BLQ was reduced to 16.3 and 9.1% for the 0.8- and 2-mg weekly dosing regimens, and these occurred primarily at the end of the 12-week washout period. All BLQ samples were also included in the combined analysis.
Model Qualification
Data associated with the 2-mg dose group collected in a phase III multiple-dose study (19) were utilized as an external validation dataset. The prediction error (PE) and absolute prediction error (APE) were calculated to evaluate the bias and precision of the predicted exenatide concentrations:
with PRED ij being the individual predicted concentration for patient i at time point j, and Obs being the measured concentration for the ith patient at the jth time point. In addition, the predictive performance of the model was assessed using VPCs (24).
RESULTS
Patient Demographics
Data were available from a total of 64 patients; 42% were female. The mean (standard deviation) age was 53.5 years (9.8), body weight was 95.7 kg (21), and estimated glomerular filtration rate was 89.6 mL/min/1.73 m2 (23.2). Table I summarizes the patient demographics stratified by the single- and multiple-dose trials. Given the small number of patients relative to the complexities of the model, the influence of patient covariates on the absorption parameters for the PK model was not assessed.
Single-Dose PK
Scatterplots of the observed concentrations, as a function of time and stratified by dose, show the three phases of exenatide release and absorption from the SC microspheres (Fig. 2). The VPC for single-dose PK profiles is shown in Fig. 3 (left panels) and confirms that the model predicts the observed data well, describing all three phases of release from the microspheres across the dose range studied. The right panels in Fig. 3 show the 90% prediction interval for the fraction of BLQ data simulated to occur at each time point (shaded regions), with the solid line reflecting the median simulated fraction of BLQ data at each time point. The use of the M3 method successfully predicted the frequency and the time course of the BLQ data with most of the observed data located within the 90% prediction interval. A comparison of the observed and predicted time-course of exenatide plasma concentrations in representative patients is shown in Fig. 4, further illustrating the range of concentrations and phases of release captured by the model. Together, these figures suggest that the model adequately describes the central tendency and variability of the PK profiles across all doses, as well as provide a robust prediction of the fraction and time-course of the appearance of BLQ samples.

Concentration versus time stratified by single SC doses of ER exenatide. Panel labels (2.5, 5, 7, and 10 mg) indicate the SC doses administered. Circles represent observed concentrations, and solid lines connect the median concentration at each of the scheduled time points

VPCs for the single-dose model (left panels) and the fraction of BLQ concentrations predicted by time (right panels) and stratified by dose group. Row panel labels (2.5, 5, 7, and 10 mg) indicate the administered SC single doses. Circles depict observed concentrations from the single-dose study. Dashed red lines connect the medians of the observed data for time points where >50% of the data is above the BLQ. Black solid lines represent the median of the predicted data. Shaded gray regions are the 90% prediction intervals

Comparison of observed and predicted PK profiles. Panel labels (2.5, 5, 7, and 10 mg) indicate the administered SC doses. Circles depict observed concentrations from the single-dose study, and solid lines represent population mean estimated profiles. Dashed lines represent individual estimated curves following a single SC dose of ER exenatide in representative patients
All model parameters were estimated well (% relative standard error (RSE) <40%) (Table II) with low magnitude of shrinkage (16%) on all IIV and RV terms. IIV was not allowed on the number of transit compartments to maintain the structural model across patients; of the remaining seven estimated parameters, only two did not include IIV. The major model permutations and associated outcomes are listed in Supplementary Table S1. Previously identified covariate relationships were fixed, including renal function on CL and body weight on VC (Table II). The overall relative bioavailability of a single dose of the ER formulation relative to the IR formulation was estimated at 10.2%, with the bioavailable dose distributed across the three absorption processes. Only 1% of the available dose was estimated to be absorbed during the initial-release phase, with approximately 46 and 53% of the remaining available dose being absorbed through the transit compartment pathways (transit series 1 and 2, respectively). The mean (%RSE) absorption rate constant for the initial release (k a) was estimated to be 2.98 days–1 (14.7%). For the first series of transit compartments, the mean transit time was approximately 19 days (∼3 weeks), corresponding well with the observed time of the second drug concentration peak. The second series of transit compartments supported an estimated mean transit time of approximately 46 days (∼7 weeks), corresponding with the third phase of drug release (Fig. 4).
Combined Single- and Multiple-Dose Modeling
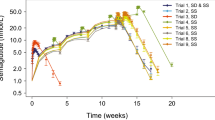
Prediction of the multiple-dose data using the single-dose model adequately described the shape of the concentration–time profiles but showed a slight underprediction of the steady-state concentrations (data not shown). Estimation of the model parameters for the combined dataset did not meaningfully alter the parameter estimates obtained from the single-dose data alone. For the combined analysis, separate relative bioavailability terms were estimated for the single- and multiple-dose studies. The estimated relative bioavailability for the phase II clinical trial was greater than for the phase I clinical trial (15.5% versus 8.86%) (Table II). All parameters were estimated well (Table II, RSE < 40%), and the magnitude of shrinkage was low (<23% on all IIV and RV parameters). The model adequately describes the rise to steady-state concentrations, with steady-state predicted to occur after 6–10 weeks of treatment, and the subsequent decline to undetectable concentrations 10–12 weeks after stopping drug administration (Fig. 5).

VPCs for multiple-dose profiles and the fraction of concentrations that are BLQ. Row panel labels (0.8 and 2.0 mg) indicate the administered SC doses. Circles depict observed concentrations from the multiple-dose study. Dashed red lines connect the medians of the observed data for time points where >50% of the data is above the BLQ. Black solid lines represent the median of the predicted data. Shaded gray regions are the 90% prediction intervals. Arrows indicate weekly administered doses
External Model Qualification
The final PK model was used to predict the concentrations collected in a phase III trial of ER exenatide administered at 2 mg once weekly. The model predicted the exenatide concentration–time profile from the phase III study with high fidelity as assessed by visual assessment (Fig. 6). The observed and predicted profiles show large variability, and the good alignment of the observed and predicted median exposures confirms the predictive performance of the model. Assessment of the PEs further suggests that the model is reasonably accurate in predicting exenatide concentrations with a median PE% of –1.85% across all time points. The median absolute PE was 25%, illustrating that the model shows adequate precision on an individual level.

VPC for phase III external validation. Circles depict observed concentrations from the phase III external validation study. Dashed red line represents the median of the observed data. Dashed black lines represent the 5th and 95th percentiles for the observed data. Black solid line represents the median of the predicted data. Shaded gray regions represent the 90% prediction interval around the median and 5th and 95th percentiles
DISCUSSION
The sustained release of exenatide was achieved by the dispersion of exenatide within PLGA microspheres, such that the active peptide that is released and absorbed into the circulation following SC administration is identical to that found in the IR formulation (17). In terms of the PK properties (absorption, distribution, and elimination) of the ER formulation, only the absorption-related properties differ from the IR formulation. A population PK model of the IR formulation was developed to quantify the influence of weight and renal function, as well as the linear and nonlinear elimination components (Cirincione B, Mager DE. Population pharmacokinetics of exenatide. Manuscript accepted). This model served as the foundation for the development of the PK model of the ER formulation, which was expanded to include a complex absorption system described by three parallel processes.
Previous PK modeling of the ER formulation of exenatide focuses solely on estimating the steady-state concentrations and evaluating the intrinsic factors that influence such values (27). Although useful in describing the exenatide exposure–response relationships, the model is limited to steady-state conditions. Thus, the model developed herein provides a tool that quantifies not only steady state but also the ability to evaluate the time to steady-state and decrease in drug concentrations upon cessation of therapy, which are important clinical considerations when assessing the time to achieve effective clinical response and the time it takes to wash out the drug after halting therapy.
The final PK model incorporates BLQ measurements using the M3 method (25). During the initial stages of model development, the BLQ concentrations were excluded and resulted in minimization difficulties. The dissolution of the microsphere is known to have periods of time with minimal release, thus resulting in two main sections of the typical concentration–time profile where BLQ concentrations occurred. The first appearance is between Day 1 and 1 week following drug administration. This phase reflects the lag time that occurs between the initial release and the second-phase release (i.e., microsphere hydration) (17), with the other occurrence within the terminal elimination phase. Thus, the incorporation of the BLQ measurements was critical for adequately capturing each phase of drug release.
The microsphere technology has been used with other injectable ER products. Absorption kinetics for these products can also be complex and are commonly comprised of an initial release followed by a longer sustained-release profile. The specific release characteristics can be customized to maximize unique therapeutic goals. Several PK modeling approaches have been reported for these products, including initial deconvolution techniques to identify specific absorption rate constants for subsequent use in a PK model (28), as well as the estimation of combinations of sequential dual and triple first-order absorption rate constants and associated lag times (29, 30). The use of transit compartments to capture the delay in absorption processes has been reported (22, 23), and we utilized two series of transit compartments to describe the delays in the system associated with the second and third phases of drug release. The absorption of a long-acting formulation of risperidone from a PLGA implant was also characterized using a similar series of transit compartments, and a first-order process was included for the initial drug release (31). In contrast to modeling the absorption profile with estimated absorption rate constants and lag times, this approach provides a more gradual input into the central compartment, resulting in a profile that is more representative of the observed exposure profiles for ER formulations. Although the use of transit compartments is empirical and simplifies the release and absorption processes that occur from the microspheres, it was found to capture the three distinct peaks of plasma exenatide concentrations well, thus allowing for simulations that will accurately describe the rise to steady-state and better predict plasma exenatide concentrations under various conditions, such as missed doses and incomplete dosing.
The appearance of antidrug antibodies to exenatide has been reported (32), and a retrospective analysis of the immunogenic response to exenatide showed that the peak immune response occurs at 6–22 weeks and subsequently decreases (32). The binding of the antibody to the peptide prevents the renal clearance of exenatide; however, the influence of antibodies on the elimination of exenatide has not been evaluated in the IR model. Additional data are needed to evaluate the effect of antibody titer on total systemic clearance. The incidence of antidrug antibodies has been reported to be greater with the ER formulation than with the IR formulation, and further analysis of the potential effect on both formulations is warranted.
CONCLUSIONS
In summary, a population-based PK model was successfully developed for an ER formulation of exenatide. Since the prolonged exenatide release is achieved by drug dispersion in PLGA microspheres, an ER profile is accomplished without alteration of the exenatide peptide. This allows the model to reconcile the PK properties of the ER and IR formulations and should also serve as a platform for studying subsequent pharmacodynamic properties. The enhanced features of this model facilitate addressing critical clinical questions, including the effects of intrinsic factors on the time to steady-state, steady-state exposure, and the time at which concentrations become undetectable after cessation of therapy.
References
Byetta (exenatide) injection. US prescribing information. Wilmington: AstraZeneca LP; 2014.
Bydureon (exenatide extended-release for injectable suspension). US prescribing information. Wilmington: AstraZeneca LP; 2014.
Fehse F, Trautmann M, Holst JJ, Halseth AE, Nanayakkara N, Nielsen LL, et al. Exenatide augments first- and second-phase insulin secretion in response to intravenous glucose in subjects with type 2 diabetes. J Clin Endocrinol Metab. 2005;90(11):5991–7.
Linnebjerg H, Park S, Kothare PA, Trautmann ME, Mace K, Fineman M, et al. Effect of exenatide on gastric emptying and relationship to postprandial glycemia in type 2 diabetes. Regul Pept. 2008;151(1-3):123–9.
Nielsen LL, Young AA, Parkes DG. Pharmacology of exenatide (synthetic exendin-4): a potential therapeutic for improved glycemic control of type 2 diabetes. Regul Pept. 2004;117(2):77–88.
Buse JB, Henry RR, Han J, Kim DD, Fineman MS, Baron AD, et al. Effects of exenatide (exendin-4) on glycemic control over 30 weeks in sulfonylurea-treated patients with type 2 diabetes. Diabetes Care. 2004;27(11):2628–35.
Cirincione B, Edwards J, Aisporna M, MacConell L. Gastrointestinal tolerability with twice-daily or once-weekly exenatide formulations was not predicted by concentration with long-term treatment. Diabetologia. 2013;56 Suppl 1:S363.
DeFronzo RA, Okerson T, Viswanathan P, Guan X, Holcombe JH, MacConell L. Effects of exenatide versus sitagliptin on postprandial glucose, insulin and glucagon secretion, gastric emptying, and caloric intake: a randomized, cross-over study. Curr Med Res Opin. 2008;24(10):2943–52.
DeFronzo RA, Ratner RE, Han J, Kim DD, Fineman MS, Baron AD. Effects of exenatide (exendin-4) on glycemic control and weight over 30 weeks in metformin-treated patients with type 2 diabetes. Diabetes Care. 2005;28(5):1092–100.
Fineman MS, Bicsak TA, Shen LZ, Taylor K, Gaines E, Varns A, et al. Effect on glycemic control of exenatide (synthetic exendin-4) additive to existing metformin and/or sulfonylurea treatment in patients with type 2 diabetes. Diabetes Care. 2003;26(8):2370–7.
Kendall DM, Riddle MC, Rosenstock J, Zhuang D, Kim DD, Fineman MS, et al. Effects of exenatide (exendin-4) on glycemic control over 30 weeks in patients with type 2 diabetes treated with metformin and a sulfonylurea. Diabetes Care. 2005;28(5):1083–91.
Kolterman OG, Buse JB, Fineman MS, Gaines E, Heintz S, Bicsak TA, et al. Synthetic exendin-4 (exenatide) significantly reduces postprandial and fasting plasma glucose in subjects with type 2 diabetes. J Clin Endocrinol Metab. 2003;88(7):3082–9.
Kolterman OG, Kim DD, Shen L, Ruggles JA, Nielsen LL, Fineman MS, et al. Pharmacokinetics, pharmacodynamics, and safety of exenatide in patients with type 2 diabetes mellitus. Am J Health Syst Pharm. 2005;62(2):173–81.
Linnebjerg H, Kothare PA, Park S, Mace K, Reddy S, Mitchell M, et al. Effect of renal impairment on the pharmacokinetics of exenatide. Br J Clin Pharmacol. 2007;64(3):317–27.
Linnebjerg H, Kothare PA, Seger M, Wolka AM, Mitchell MI. Exenatide - pharmacokinetics, pharmacodynamics, safety and tolerability in patients ≥75 years of age with type 2 diabetes. Int J Clin Pharmacol Ther. 2011;49(2):99–108.
Linnebjerg H, Seger M, Kothare PA, Hunt T, Wolka AM, Mitchell MI. A thorough QT study to evaluate the effects of single-dose exenatide 10 μg on cardiac repolarization in healthy subjects. Int J Clin Pharmacol Ther. 2011;49(10):594–604.
DeYoung MB, MacConell L, Sarin V, Trautmann M, Herbert P. Encapsulation of exenatide in poly-(D, L-lactide-co-glycolide) microspheres produced an investigational long-acting once-weekly formulation for type 2 diabetes. Diabetes Technol Ther. 2011;13(11):1145–54.
Fineman M, Flanagan S, Taylor K, Aisporna M, Shen LZ, Mace KF, et al. Pharmacokinetics and pharmacodynamics of exenatide extended-release after single and multiple dosing. Clin Pharmacokinet. 2011;50(1):65–74.
Blevins T, Pullman J, Malloy J, Yan P, Taylor K, Schulteis C, et al. DURATION-5: exenatide once weekly resulted in greater improvements in glycemic control compared with exenatide twice daily in patients with type 2 diabetes. J Clin Endocrinol Metab. 2011;96(5):1301–10.
World Medical Association Declaration of Helsinki. Recommendations guiding physicians in biomedical research involving human subjects. JAMA. 1997;277(11):925–6.
US Food and Drug Administration. Draft guidance for industry: bioanalytical method validation. Rockville: US Food and Drug Administration; 2001.
Savic RM, Jonker DM, Kerbusch T, Karlsson MO. Implementation of a transit compartment model for describing drug absorption in pharmacokinetic studies. J Pharmacokinet Pharmacodyn. 2007;34(5):711–26.
Shen J, Boeckmann A, Vick A. Implementation of dose superimposition to introduce multiple doses for a mathematical absorption model (transit compartment model). J Pharmacokinet Pharmacodyn. 2012;39(3):251–62.
Holford N. The visual predictive check—superiority to standard diagnostic (Rorschach) plots. PAGE. 2005;14: Abstract 738.
Beal SL. Ways to fit a PK model with some data below the quantification limit. J Pharmacokinet Pharmacodyn. 2001;28(5):481–504.
Bergstrand M, Hooker AC, Karlsson MO. Visual predictive checks for censored and categorical data. PAGE. 2009;18:Abstract 1604.
Cirincione B, Passarell J, Kothare P, Cummings M, Grasela T, Fineman M. Development of a steady-state exposure-response model for exenatide once weekly. Presented at American Society for Clinical Pharmacology and Therapeutics (ASCPT) Annual Meeting; Washington, DC, March 18–21, 2009.
Sun YN, Lee HJ, Almon RR, Jusko WJ. A pharmacokinetic/pharmacodynamic model for recombinant human growth hormone effects on induction of insulin-like growth factor I in monkeys. J Pharmacol Exp Ther. 1999;289(3):1523–32.
Dunbar JL, Turncliff RZ, Hayes SC, Farrell CB. Population pharmacokinetics of extended-release injectable naltrexone (XR-NTX) in patients with alcohol dependence. J Stud Alcohol Drugs. 2007;68(6):862–70.
Rhee YS, Sohn M, Woo BH, Thanoo BC, DeLuca PP, Mansour HM. Sustained-release delivery of octreotide from biodegradable polymeric microspheres. AAPS PharmSciTech. 2011;12(4):1293–301.
Laffont CM, Gomeni R, Zheng B, Heidbreder C, Fudala PJ, Nasser AF. Population pharmacokinetics and prediction of dopamine D2 receptor occupancy after multiple doses of RBP-7000, a new sustained-release formulation of risperidone, in schizophrenia patients on stable oral risperidone treatment. Clin Pharmacokinet. 2014;53(6):533–43.
Fineman MS, Mace KF, Diamant M, Darsow T, Cirincione BB, Booker Porter TK, et al. Clinical relevance of anti-exenatide antibodies: safety, efficacy and cross-reactivity with long-term treatment. Diabetes Obes Metab. 2012;14(6):546–54.
Acknowledgments
The authors thank the exenatide development team formerly at Amylin for their scientific consultation and support of the development of this manuscript. The authors thank David Clawson and Carl LaCerte for their SAS programming, consultation, and quality control support. The authors also acknowledge Dr. Frank LaCreta for his review and clinical perspectives in support of this manuscript. This analysis was supported by Amylin Pharmaceuticals Inc. and Bristol-Myers Squibb. In addition, we thank inScience Communications, Springer Healthcare (Philadelphia, PA, USA), for providing copyright editorial support for submission and AstraZeneca for funding this support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
All studies were conducted in accordance with the principles described in the Declaration of Helsinki (1946) up to and including the Seoul revision. All clinical protocols were approved for each site by an appropriate institutional review board, and all patients provided written informed consent prior to participation.
Conflict of Interest
B.C. is an employee of Bristol-Myers Squibb. J.E. was an employee of Amylin at the time of the initial analyses. D.E.M. declares no conflicts of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOCX 12 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Cirincione, B., Edwards, J. & Mager, D.E. Population Pharmacokinetics of an Extended-Release Formulation of Exenatide Following Single- and Multiple-Dose Administration. AAPS J 19, 487–496 (2017). https://doi.org/10.1208/s12248-016-9975-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1208/s12248-016-9975-1