
Abstract
Gefitinib (Iressa) is a selective and potent EGFR tyrosine kinase inhibitor. It received an accelerated FDA approval in 2003 for the treatment of patients with nonsmall cell lung cancer (NSCLC) and represents the first-line therapy for NSCLC with EGFR mutations. In the work presented herein, the disposition of gefitinib was investigated extensively in mouse in both plasma and 11 organs (liver, heart, lung, spleen, gut, brain, skin, fat, eye, kidney, and muscle) after a single IV dose of 20 mg/kg. Gefitinib demonstrated extensive distribution in most tissues, except for the brain, and tissue to plasma partition coefficients (K pt) ranged from 0.71 (brain) to 40.5 (liver). A comprehensive whole-body physiologically based pharmacokinetic (PBPK) model of gefitinib in mice was developed, which adequately captured gefitinib concentration-time profiles in plasma and various tissues. Predicted plasma and tissue AUC values agreed well with the values calculated using the noncompartmental analysis (<25% difference). The PBPK model was further extrapolated to humans after taking into account the interspecies differences in physiological parameters. The simulated concentrations in human plasma were in line with the observed concentrations in healthy volunteers and patients with solid malignant tumors after both IV infusion and oral administration. Considering the extensive tissue distribution of gefitinib, plasma concentration may not be an ideal surrogate marker for gefitinib exposure at the target site or organ of toxicity (such as the skin). Since our whole-body PBPK model can predict gefitinib concentrations not only in plasma but also in various organs, our model may have clinical applications in efficacy and safety assessment of gefitinib.
Similar content being viewed by others

INTRODUCTION
Aberrant signaling transduction pathways have been implicated in the development and progression of a number of solid tumors (1). The epidermal growth factor receptor (EGFR) signaling pathway is highly involved in cell proliferation, migration, and differentiation (2). The mutation or overexpression of EGFR can lead to increased cell division and other events involved in cancer development. The EGFR has been found to be highly dysregulated in a variety of human tumors including nonsmall cell lung cancer (NSCLC) and breast, head, and neck cancers along with other tumors (3). In addition, high EGFR expression in cancer has been correlated with advanced tumor stage, resistance to chemotherapy and radiation, and poor patient prognosis (4–6). Thus, inhibition of EGFR has been considered as a promising treatment strategy for novel anticancer therapy, and there are a number of EGFR tyrosine kinase inhibitors under development (7–9).
Gefitinib (Iressa) is an orally active EGFR tyrosine kinase inhibitor. It binds mutated EGFR with high affinity and specificity, produces potent inhibition of EGFR signaling pathway, and thus inhibits a range of EGFR-mediated effects and blocks tumor cell growth (10–12). In several phase 1 and phase 2 clinical trials of nonsmall cell lung cancer, gefitinib offers superior progression-free survival and better tolerability and quality of life when compared to the doublet chemotherapy (13,14). Because of the superior efficacy/safety profile and convenient once-daily oral dosing, gefitinib gained approval in several counties including the USA, Europe, and Japan and now is the first-line treatment of NSCLC with EGFR mutations (15).
The pharmacokinetics of gefitinib in humans has been thoroughly studied. Gefitinib is slowly absorbed into the body with a mean bioavailability around 60% and a peak concentration occurring within 3 to 7 h in healthy volunteers and cancer patients (16,17). Gefitinib is extensively distributed into the tissues and highly bound (∼95%) to human serum albumin (HSA) and alpha 1-acid glycoprotein (AAG), resulting in a long half-life of 48 h (18–20). In vitro and in vivo studies have shown that gefitinib is mainly metabolized in the liver via cytochrome (CYP) 3A4 (21,22). The majority of gefitinib (∼86%) and its metabolites (∼12.1%) are eliminated through the feces over 10 days, while only less than 4% unchanged gefitinib is recovered in the urine (16,18).
Physiologically based pharmacokinetic (PBPK) modeling is a useful tool for evaluating and predicting the disposition of drug of interest into various tissues and organs (23,24). The whole-body PBPK model was constructed using the system of differential equations with known biological and physiological parameters to represent the circulatory system in the body. It usually consists of a number of anatomical compartments interconnected through the body fluid system (24). The use of multicompartmental structure, involving physiological parameters such as organ volume, blood rate, and intrinsic clearance, enables the PBPK model to predict the concentrations of compounds of interest in various tissues and, more importantly, to obtain tissue concentrations in humans through extrapolation. This is very desirable as tissue concentrations are believed to be more relevant than the plasma concentration for the pharmacokinetic/pharmacodynamic activities of many drugs, especially for anticancer agents which have extensive tissue distribution (25,26).
The PBPK models have been developed for many anticancer agents, including mitoxantrone (26), doxorubicin (27), docetaxel (28), topotecan (29), cisplatin (30), and another tyrosine kinase inhibitor lapatinib (31). However, a PBPK model for gefitinib has not been reported. In addition, as almost all gefitinib pharmacokinetic studies only evaluated gefitinib plasma concentrations, the information about the disposition of gefitinib in various tissues and organs is limited in the literature. In the present study, a PK and tissue distribution study for gefitinib was conducted in mice. Based on the experimental data, a whole-body PBPK model was developed in mice to characterize the distribution of gefitinib in plasma and different tissues. The mouse PBPK model was then extrapolated to predict the plasma concentration-time profile in humans by taking into account the interspecies physiology differences.
MATERIALS AND METHODS
Chemicals
Gefitinib and dasatinib were purchased from LC Laboratories (Woburn, MA). Ammonium acetate (99%) was obtained from ACROS Organics (NJ, USA). Analytical HPLC grade acetonitrile (ACN), isopropanol, water, ethanol, phosphate-buffered saline (PBS) 10× solution, Triton lysis buffer (pH 8.0), and formic acid were purchased from Fisher Scientific (Pittsburg, PA, USA). Analytical spectrophotometric grade ethyl acetate (99.5%) was obtained from Alfa Aesar (Ward Hill, MA, USA). Hydroxypropyl-β-cyclodextrin (99.5%) was purchased from VWR International LLC (Philadelphia, PA, USA). Heparin-treated mouse plasma was purchased from BioreclamationIVT (East Meadow, NY, USA). Heparin injectable (1000 U/mL) was purchased from Patterson Vet Generics (Devens, MA, USA).
Animals and In Vivo Study
Four- to 6-week-old Harlan ND4 male Swiss Webster mice with an average body weight of 29.6 g were obtained from Harlan Laboratories. Before the experiment, all mice were settled and housed in the University of Florida (UF) Animal Research Facility for a week following a 12-h light/dark cycle. All mice had access to normal/standard diet and water, and this in vivo study was carried out in accordance with the guidelines evaluated and approved by the review board of the UF Animal Care and National Institutes of Health.
In this study, 33 mice were randomly separated into 11 groups based on predetermined time points, with 3 mice at each time point (N = 3). Gefitinib was first dissolved in dimethyl sulfoxide at the concentration of 50 mg/mL, and prior to dosing, it was further diluted as a solution in the vehicle consisting of 10% hydroxypropyl-β-cyclodextrin (HPBCD) at the final concentration of 5 mg/mL. The target dose of 20 mg/kg gefitinib was administered through penile vein injection to the mice. The dose of 20 mg/kg in mice is equivalent to approximately 84 mg in a 70-kg human subject. This dose was selected as it is within the dose range of gefitinib used in humans (50 to 250 mg per day). The mice had free access to food and water before and after IV administration. Blood, heart, lung, gut, brain, liver, kidney, spleen, muscle, eye, skin, and adipose tissues were collected at time points 10 and 30 min and 1, 2, 4, 8, 12, 24, 48, 72, and 96 h after IV dosing. The heparinized blood was immediately centrifuged at 2000g for 8 min with a mini-centrifuge. Plasma was collected and all the plasma and tissue samples were stored at −80°C until analysis.
Sample Preparation and Liquid Chromatography-Tandem Mass Spectroscopy Analysis
The concentrations of gefitinib in the plasma and tissue samples were analyzed based on a liquid chromatography-tandem mass spectroscopy (LC/MS/MS) assay published by Agarwal et al. (32) with some modifications. A 40-μL aliquot of gefitinib samples in plasma samples was deproteinized using a 2-fold volume of ACN. After vortexing, the mixture was centrifuged at 14,000 rpm for 10 min. One hundred microliters of the supernatant was transferred into a 350-μL vial insert for LC/MS/MS analysis. For fat, muscle, heart, and brain, they were weighted and added 3× volume of the PBS buffer (i.e., dilution factor is 4). For eye and skin, 10× volume of the PBS buffer was added (i.e., dilution factor is 11). For liver, kidney, gut, spleen, and lung, 20× volume of the PBS buffer was added for the samples collected between 10 min to 2 h, and 3× volume of the PBS buffer was added for the samples collected after 2 h. Tissue samples were cut into smaller pieces prior to homogenization. Then 4 μL of IS was spiked into 40 μL of tissue homogenate. Other steps were the same as the plasma sample preparation.
LC/MS/MS was performed using an Agilent 6460 triple quadrupole tandem mass spectrometer (Agilent, Santa Clara, CA) linked to an Agilent LC1260 liquid chromatography apparatus. Mass Hunter (version B.06.00) software was used for data acquisition and processing. ZORBAX Eclipse XDB C18 column (2.1 × 50 mm i.d., 1.8 μm; Agilent, Santa Clara, CA) and guard column (2.1 × 50 mm i.d., 1.8 μm; Agilent, Santa Clara, CA) were used. Gefitinib was eluted with a mobile phase of water containing 10 mM ammonium acetate (A) and acetonitrile (B) at a flow rate of 400 μL/min. Mobile phase B was increased from 10 to 90% in the first 5 min and then was kept 90% for 1 min. Another 2 min of postrun was followed to equilibrium for the next injection. The total run time is 8 min. Conditions for MS analysis of gefitinib included a capillary voltage of +4500 V, a nebulizing pressure of 40 psi, and a temperature of 350°C. Sheath gas flow was 11 L/min. The MS was performed in a positive ion mode under multiple reactions monitoring (MRM). The m/z ratios of molecular ion and product ion of gefitinib were 447.1 and 128.1, respectively. The m/z ratios of molecular ion and product ion of dasatinib (internal standard) were 488.1 and 401.1, respectively. The lower limit of quantification (LOQ) of gefitinib was 1 ng/mL in plasma; 3 ng/mL in liver, kidney, spleen, lung, muscle, brain, and fat; 11 ng/mL in eye and skin; and 15 ng/mL in heart and gut. Both standard curves (ranging from 1 to 2500 ng/ml) and QCs (5, 50, and 500 ng/ml) in various tissues were prepared. The calibration curve was linear over the concentration range of 5∼2500 ng/ml in heart and gut and 1∼2500 ng/mL in other tissues.
PBPK Model Development
Global Model
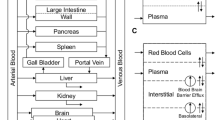
The global (whole-body) PBPK model was developed with gefitinib data from plasma and 11 organs, namely the lung, heart, spleen, gut, liver, kidney, brain, eye, fat, muscle, and skin (Fig. 1). Gefitinib concentrations in other organs such as the bone were not determined and therefore included in the “remainder” compartment. Biotransformation via CYP3A4 in the liver was assumed the only route of elimination according to the results in prior experiments (22). The PBPK model fittings for gefitinib plasma and tissue concentrations in mice were performed in ADAPT 5 (version 5, Biomedical Simulations Resource, Los Angeles, CA) (33). The average weight for the 33 mice in our study was 29.6 g, and the cardiac output (Q) was scaled from the literature data (34) for a 20-g mouse using the following equation:
where BW1 is the body weight of mice (20 g) in the literature, and BW2 is the actual mean body weight of mice used in the current study (29.6 g). The cardiac output for the whole mouse used in the PBPK model was 0.72 L/h.

Schematic representation of a physiologically based pharmacokinetic model for gefitinib in the mouse
Local Model
Two types of local models were used to describe the disposition of gefitinib into different tissues (Fig. 2). A well-stirred model was used for perfusion-limited organs (heart, spleen, gut, liver, kidney, etc.) as it has been shown that gefitinib preferably distributed into these highly perfused tissues (18). A permeability-limited model was used to fit the gefitinib distribution in organs where the permeability across the cell membrane is the limiting process (24). Permeability surface area conduct (PSt) was included in this type of model to describe the movement of drug across the cell membrane into the intracellular tissues.

Model structure for perfusion rate-limited kinetics and permeability rate-limited kinetics
The equation for the perfusion rate-limited model is
The equation for eliminating organ liver is
The equations for the permeability rate-limited model are
where the abbreviations represent gefitinib concentrations in tissue (C t) and plasma (C p), tissue blood flow rate (Q t), tissue volume (V t), tissue to plasma partition coefficient (K pt), permeability surface area product (PSt), unbound fraction of gefitinib in plasma (f u), tissue concentration in the interstitial fluid (C t,ISF) and tissue cells (Ct,c), extracellular volume (V t,ISF), and intracellular volume (V t,c).
Data Analysis
In the data fitting, f u for mice was fixed to 0.051 (19) and all physiological parameters such as blood flow rate and tissue volume in each organ were obtained from Brown et al. and listed in Table I (34). Fractions of intracellular volume in the tissue compartment for the skin, fat, and remainder were fixed to 0.302, 0.135, and 0.333 according to the literature (26,35). All other parameters such as CLint, K pt for all tissues, and PSt for skin, fat, and remainder compartments were estimated in the PBPK model. Initial estimates for the partition coefficient (K pt) for each tissue were calculated by the following equation:
where AUC0 − 24 is the area under the concentration-time curves from 0 to 24 h. AUC values were calculated by the noncompartmental analysis (NCA) based on the trapezoidal rule. The initial estimates for CLint,H were calculated with the assumption of the well-stirred hepatic distribution model:
where Q H is the mouse hepatic blood flow rate, f u is the fraction of unbound gefitinib, and CLH is the hepatic clearance determined by the NCA from the experimental data (76.6 mL/h). The initial estimate for CLint,H was calculated to be 4430 mL/h under this model assumption.
The gefitinib concentration profiles in plasma and 11 tissues are shown in Fig. 3, and because of the sensitivity of the analytical method, only lung and liver samples were detected and quantified by the LC/MS/MS after 24 h. All the plasma and tissue data were fitted simultaneously using the maximum likelihood estimation in ADAPT 5 with the following variance model:
where σ 1 and σ 2 are the additive and proportional variance parameters, Y(t) is the model output at time t, and Var(t) is the variance associated with the output. Goodness-of-fit plots and the Akaike information criterion (AIC) were used to evaluate the adequacy of fitting of the constructed PBPK models.

Gefitinib concentrations in plasma and in 11 examined tissues (brain, eye, fat, gut, spleen, heart, kidney, liver, lung, muscle, skin, spleen) following intravenous bolus administration of 20 mg/kg gefitinib in mice. Three mice were used at each time point
Sensitivity Analysis
A normalized sensitivity analysis was conducted as described in Loccisano et al. (36) to evaluate the influence of each PBPK model parameter on the simulated gefitinib plasma AUC for both mouse and human models. Normalized sensitivity coefficients (SC) were calculated as the ratio of percentage change in AUC as a result of 1% increase in a given model parameter:
where A is the simulated AUC resulting from the 1% increase in the given parameter, B is the simulated AUC from the original parameter, C is the parameter value increased by 1%, and D is the original parameter value. Each parameter was analyzed individually, keeping all other parameters fixed at their original levels. The small absolute value of SC indicates that the model output is insensitive to the parameter; the absolute value of a SC greater than 1 suggests that there may be amplified parameter errors (31).
Scale-Up from Mice to Humans
The final gefitinib PBPK model developed for mice was extrapolated to humans to simulate plasma concentrations of gefitinib in humans. The simulation of gefitinib concentration in humans was performed in ADAPT 5 (version 5, Biomedical Simulations Resource, Los Angeles, CA) (33). Human physiological parameters, including organ volume, and blood flow rate, are shown in Table II (34). Hepatic clearance CLH was obtained from the literature (30.8 L/h) (17), and the intrinsic clearance CLint was calculated based on the well-stirred hepatic model assumption. CLint used in human simulations was 1410 L/h. PSt was scaled up to humans by the use of a allometric scaling equation:
where M is the organ weight, A is the model parameter estimated in the mouse PBPK model, and B is the power function coefficient for the allometric equation. A fixed value of 0.75 was used for B. PSt used in the human simulations for skin, fat, and remainder were 62.1, 78.5, and 10.4 L/h, respectively. To simulate the gefitinib PK profile in humans following oral administration, the absorption rate K a and bioavailability F of gefitinib were fixed to the literature reported values obtained in mice (0.88 h−1 and 45%, respectively) (37).
RESULTS
Gefitinib pharmacokinetics was investigated in mice following a single IV dose of 20 mg/kg. As shown in Fig. 3, gefitinib demonstrated extensive distribution in most tissues, except for the brain, with tissue to plasma partition coefficients (K pt) ranging from 0.71 (brain) to 40.5 (liver). Perfusion rate-limited kinetics was used to describe the distribution of gefitinib into highly perfused organs such as the lung, liver, kidney, and gut (K p > 8). It assumes that gefitinib distributes freely and immediately across the cell membrane without diffusion barrier, and blood flow rate acts as the limiting factor of rate of distribution in this model. On the other hand, the gefitinib concentrations in the skin and fat were substantially lower with peak concentrations occurring at around 2 h, which indicates that the permeability across the cell membrane is the limiting process for the distribution of gefitinib in these organs. Therefore, the permeability rate-limited model was applied to fit the gefitinib concentration-time profile in tissue skin, fat, and the “remainder” compartment.
The goodness-of-fit results were shown in Fig. 4. The proposed PBPK model was able to capture the gefitinib concentration-time profiles in plasma and various tissues, with model prediction in close agreement with the experimental observations. The estimated gefitinib PK parameters K pt, CLint,H, and PSt are shown in Table III. In general, the partition coefficients K pt and intrinsic clearance CLint,H are very close to the NCA estimates. Partition coefficient K l in the liver is the only parameter with more than 1-fold difference from the NCA estimate, which is reasonable considering the complexity of the PBPK model.

Observed (dots) and predicted (lines) gefitinib concentrations in plasma and various organs/tissues in mice. Each dot and vertical bar represents the mean and 95% CI of the observations, respectively. Solid lines are the fit predictions based on the proposed PBPK model
High precision was obtained in the estimation of essential PK parameters K pt and CLint,H with coefficient of variation (CV) less than 30%. On the other hand, the variability for the parameters associated with permeability across the cell membrane in the skin, fat, and remainder is relatively high (∼200%), and the possible reason for the high variability associated with PSt and K ISF is because not enough information is available to estimate the interstitial fluid and tissue concentrations simultaneously as well as to fit the equilibrium between these two compartments.
In addition, the model-predicted C max and AUC were in good agreement with the observed values (<25% difference) (Table IV). The half-life calculated from the predicted concentrations in the plasma is 2.9 h, and it is much shorter than the reported value in the literature and does not truly reflect the elimination profile of gefitinib. The real terminal slope in the gefitinib concentration-time profile has not been captured by the plasma data due to the fact that plasma samples after 24 h are below the limit of quantification in the assay. On the other hand, gefitinib concentrations in the lung and liver samples were able to be detected and quantified through the whole study period (i.e., 96 h). As the pharmacokinetic concentration-time profile in the plasma is parallel to that in the tissue, the half-life of gefitinib in plasma was therefore calculated based on the terminal slopes of gefitinib concentration-time profiles in the liver and lung. Based on the terminal slopes of gefitinib kinetics in the liver and lung, the estimated half-life of gefitinib in plasma is 16.4 h, which is much closer to the value reported in the literature (20).
Gefitinib Model Simulations in Humans
A human PBPK model was generated using human physiological parameters, such as organ volume and blood flow rate from a healthy subject weighing 70 kg. As gefitinib undergoes minimal renal elimination, hepatic metabolism was considered as the only elimination pathway in our model. Human plasma gefitinib observations were obtained from three previously published studies (17,38). In the first study (17), 12 healthy volunteers were randomized to receive IV infusion administered over 1 h of 50 mg, 100 mg gefitinib or a placebo. In the second study (17), 19 patients with advanced solid malignant tumor of the colon/rectum (n = 8), lung (n = 5), stomach (n = 2), ovary (n = 1), liver (n = 1), pancreas (n = 1) and kidney (n = 1) refractory to standard therapy received IV infusion over 5 min of 50 mg gefitinib. In the third study (38), 30 Japanese patients with solid malignant tumors of nonsmall cell lung (n = 23), colorectal (n = 5), head (n = 2), and breast (n = 1) cancers received a single oral dose of gefitinib, followed by 10–14 days of observation. Each of the three observed human datasets was compared with the simulation results after the same gefitinib dosing regimen, respectively. As shown in Fig. 5, the simulated gefitinib plasma concentrations in general were in good agreement with the observed values in both healthy volunteers and patients following IV or oral dose regimens.

Observed and PBPK model-simulated plasma concentrations of gefitinib following different dosing regimens in both healthy subjects and patients with solid tumors. Measured gefitinib data (black dots) was digitalized from following published articles: a–c Swaisland et al. (17); d Nakagawa et al. (38). Solid lines and dotted lines represent the mean and 90% CI of the simulations from the proposed PBPK model, respectively
Sensitivity Analysis
The normalized sensitivity coefficients for the mouse (20 mg/kg dose) and human (50 mg infusion in 5 min) PBPK model with regard to plasma AUC were calculated. Most parameters had sensitivity coefficients smaller than 0.01, indicating that the model output is very insensitive to the variation of the given parameter. The only parameter that stood out among others was the intrinsic clearance in the human PBPK model, but its absolute SC value is still relatively small (0.51). We also tested the change of plasma AUC when the parameter was randomly increased by 1-fold. Results showed that AUC did not change (<1%) even if most parameters were as twice high as their original values. We only observed a noticeable decrease in AUC (18 and 27% in mouse and human models, respectively) when CLint was increased by 1-fold, which further confirms that the model output is insensitive to the variation of model parameters.
DISCUSSION
Gaining insight into the pharmacokinetic profiles of drug in various organs is very desirable in the evaluation of anticancer drugs which have extensive tissue distribution. With the PBPK model, the dynamics of drug distribution in various tissues can be evaluated and predicted, thus leading to a better understanding of the relationship between target tissue exposure and both drug safety and efficacy. The present analysis features a PBPK model that is able to characterize the concentration-time profiles of gefitinib in plasma and in various organs after IV bolus of 20 mg/kg in mice. Our results showed that gefitinib preferably distributed to the highly perfused organs such as the lung, liver, and kidney. The partition coefficients (K pt) in these tissues were many times higher than for plasma. A classical perfusion-limited kinetic model well described the pharmacokinetic profile of gefitinib in these organs. In contrast, the K p values in the skin and fat are smaller, and the perfusion-limited kinetic model is not able to capture the peak concentration around 2 h, suggesting that the permeability across the cell membrane is the limiting process for the distribution in these organs. Among the different approaches attempted, the permeability-limited kinetics model, which separates the tissue compartment into interstitial space and intracellular space, provided the best fitting and successfully captured both the peak and terminal slope of gefitinib concentration-time profiles in the skin and fat. The addition of a deep binding compartment was also explored; however, it did not improve the model fitting and suffered from instability and imprecision of parameter estimates. McKillop et al. have found that gefitinib persisted in melanin-containing tissues, including the eye and skin in male pigmented rats in one study (18). Such observations did not recur in our study. One possible explanation could be that mice in our experiment are different from the pigmented rats in terms of melanin concentrations in the skin and eyes.
One of the most attractive features of the PBPK model is its scalability from one species to another species. The proposed PBPK model was successfully extrapolated from mice to humans to predict the human plasma concentrations after taking into account the interspecies differences in physiology. As shown in Fig. 5, in general, the model predictions are very close to the human plasma observations in three clinical studies, and almost all observations are within the 90% confidence interval of the simulated concentrations. It seems that the distribution phase human observations distribute faster than our simulation results (Fig. 5). Possible reasons for this minor discrepancy include the inability to differentiate venous blood from artery blood in the PBPK model or the uncharacterized differences in the drug transporters and metabolic enzymes between mice and humans. We also noticed that simulations in the healthy subjects were more accurate than those in patients with solid tumors. Simulations in the patient populations tend to underestimate the gefitinib concentrations after 96 h. This error may be caused by the inherent differences of hepatic clearance between two populations: the hepatic clearance was reported to be 2-fold smaller in patients than that in healthy subjects (17). It should also be pointed out that the sample size of healthy volunteers (n = 12) and cancer patients (n = 19) in these two studies is relatively small and may cause a small bias in evaluating the difference of model performance between healthy subjects and cancer patients. Another noticeable discrepancy in the human simulation occurred in the peak concentration following oral administration. C max of the human simulation is approximately 2-fold higher than the value observed in the Japanese patients administered with 100 mg oral dose. Since the fixed model parameters K a and F used in the simulation were obtained from the PK/PD model developed in the mice, the 2-fold difference observed in the C max is quite understandable and within the acceptable range for the prediction from in vivo to humans.
Overall, this proposed whole-body PBPK model is able to accurately predict the gefitinib concentrations in both healthy volunteers and cancer patients. The predictions of human PK profile using the PBPK model is believed to be more accurate than the allometric prediction (39,40). It is not surprising as the PBPK model is able to account for the differences between humans and animals with regard to the activity and specificity of metabolic enzymes and transport processes, and all the parameters such as blood flow rate, tissue volume, and intrinsic clearance have physiological meaning and can be scaled from one species to another species, whereas the allometric prediction of human PK assumes that the only difference between humans and other mammals is the size.
Almost all human gefitinib PK studies are based on the plasma/blood data as tissue samples are usually not feasible in humans. However, plasma concentration may not be a good indicator for the gefitinib exposure considering its extensive distribution into highly perfused organs like the liver and limited distribution across the blood-brain barrier into the central nervous system (CNS). Intuitively, gefitinib concentration in the target organ is believed to be a better surrogate marker of its safety and efficacy.
The constructed PBPK model was able to fit the gefitinib concentrations in mice up to 96 h in the lung and liver even though most observations in the dataset were obtained in the first 24 h. The simulated gefitinib concentrations in human plasma were in line with the observed data reported in the literature in healthy volunteers or patients up to 150 days. Despite that gefitinib has a high response rate of 80% in the NSCLC with EGFR mutation (41), it is common to observe acquired resistance and disease progression in patients after initial positive response (13,14). A secondary T970M mutation has been found to be highly associated with acquired resistance to gefitinib in patients with NSCLC in several studies and case reports (41–43). The ability to predict the plasma and potentially lung concentrations beyond clinically convenient sampling times is clinically meaningful as it provides a tool to monitor the antitumor response and possible resistance of gefitinib in the long-term treatment of lung cancer.
Genetic mutations in the EGFR pathway have been found to be associated with malignant gliomas (44,45). However, in a phase II clinical trial evaluating the efficacy of gefitinib in the treatment of glioblastomas, patients showed no objective response and no improvement in overall survival (46,47). One of the possible reasons for this lack of efficacy is that gefitinib fails to achieve sufficient intratumoral concentrations in the central nervous system due to active efflux by P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP) at the blood-brain barrier (BBB). Agarwal et al. have demonstrated in a mice model that gefitinib steady-state brain to plasma concentration ratios were improved by 70-fold through the inhibition of P-gp and BCRP receptors with coadministration of the dual inhibitor elacridar (32). Our in vivo animal studies are underway to evaluate the gefitinib concentration with and without administration of potent efflux transporter inhibitor. The proposed PBPK model could be further modified to include P-gp- and BCRP-mediated gefitinib efflux.
In summary, we reported for the first time a whole-body PBPK model of gefitinib which is able to capture gefitinib concentration-time profiles in plasma and various target tissues in mice. The PBPK model was successfully extrapolated to humans after taking into account the interspecies differences in physiological parameters. The simulated concentrations in human plasma agreed well with the observed concentrations in healthy volunteers and patients with solid malignant tumors after both IV infusion and oral administration, with minor discrepancy in the distribution phase and C max of the oral dosage possibly due to uncharacterized species differences. Since our whole-body PBPK model can predict gefitinib concentrations not only in plasma but also in various organs, our model may have clinical applications in efficacy and safety assessment of gefitinib.
References
Ciardiello F, Tortora G. Anti-epidermal growth factor receptor drugs in cancer therapy. Expert Opin Invest Drugs. 2002;11(6):755–68.
Salomon DS, Brandt R, Ciardiello F, Normanno N. Epidermal growth factor-related peptides and their receptors in human malignancies. Crit Rev Oncol Hematol. 1995;19(3):183–232.
Huang SM, Harari PM. Epidermal growth factor receptor inhibition in cancer therapy: biology, rationale and preliminary clinical results. Investig New Drugs. 1999;17(3):259–69.
Nicholson RI, Gee JM, Harper ME. EGFR and cancer prognosis. Eur J Cancer. 2001;37 Suppl 4:S9–S15.
Sainsbury JR, Malcolm AJ, Appleton DR, Farndon JR, Harris AL. Presence of epidermal growth factor receptor as an indicator of poor prognosis in patients with breast cancer. J Clin Pathol. 1985;38(11):1225–8.
Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350(21):2129–39.
Ciardiello F, Tortora G. EGFR antagonists in cancer treatment. N Engl J Med. 2008;358(11):1160–74.
Ciardiello F, Tortora G. A novel approach in the treatment of cancer: targeting the epidermal growth factor receptor. Clin Cancer Res. 2001;7(10):2958–70.
Raymond E, Faivre S, Armand JP. Epidermal growth factor receptor tyrosine kinase as a target for anticancer therapy. Drugs. 2000;60 Suppl 1:15–23.
Wakeling AE, Guy SP, Woodburn JR, Ashton SE, Curry BJ, Barker AJ, et al. ZD1839 (Iressa): an orally active inhibitor of epidermal growth factor signaling with potential for cancer therapy. Cancer Res. 2002;62(20):5749–54.
Moulder SL, Yakes FM, Muthuswamy SK, Bianco R, Simpson JF, Arteaga CL. Epidermal growth factor receptor (HER1) tyrosine kinase inhibitor ZD1839 (Iressa) inhibits HER2/neu (erbB2)-overexpressing breast cancer cells in vitro and in vivo. Cancer Res. 2001;61(24):8887–95.
Ciardiello F, Caputo R, Bianco R, Damiano V, Pomatico G, De Placido S, et al. Antitumor effect and potentiation of cytotoxic drugs activity in human cancer cells by ZD-1839 (Iressa), an epidermal growth factor receptor-selective tyrosine kinase inhibitor. Clin Cancer Res. 2000;6(5):2053–63.
Kris MG, Natale RB, Herbst RS, Lynch Jr TJ, Prager D, Belani CP, et al. Efficacy of gefitinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in symptomatic patients with non-small cell lung cancer: a randomized trial. JAMA. 2003;290(16):2149–58.
Fukuoka M, Yano S, Giaccone G, Tamura T, Nakagawa K, Douillard JY, et al. Multi-institutional randomized phase II trial of gefitinib for previously treated patients with advanced non-small-cell lung cancer (the IDEAL 1 Trial) [corrected]. J Clin Oncol. 2003;21(12):2237–46.
Cohen MH, Williams GA, Sridhara R, Chen G, Pazdur R. FDA drug approval summary: gefitinib (ZD1839) (Iressa) tablets. Oncologist. 2003;8(4):303–6.
Swaisland H, Laight A, Stafford L, Jones H, Morris C, Dane A, et al. Pharmacokinetics and tolerability of the orally active selective epidermal growth factor receptor tyrosine kinase inhibitor ZD1839 in healthy volunteers. Clin Pharmacokinet. 2001;40(4):297–306.
Swaisland HC, Smith RP, Laight A, Kerr DJ, Ranson M, Wilder-Smith CH, et al. Single-dose clinical pharmacokinetic studies of gefitinib. Clin Pharmacokinet. 2005;44(11):1165–77.
McKillop D, Hutchison M, Partridge EA, Bushby N, Cooper CM, Clarkson-Jones JA, et al. Metabolic disposition of gefitinib, an epidermal growth factor receptor tyrosine kinase inhibitor, in rat, dog and man. Xenobiotica. 2004;34(10):917–34.
Li J, Brahmer J, Messersmith W, Hidalgo M, Baker SD. Binding of gefitinib, an inhibitor of epidermal growth factor receptor-tyrosine kinase, to plasma proteins and blood cells: in vitro and in cancer patients. Invest New Drugs. 2006;24(4):291–7.
Cohen MH, Williams GA, Sridhara R, Chen G, McGuinn Jr WD, Morse D, et al. United States Food and Drug Administration Drug Approval summary: gefitinib (ZD1839; Iressa) tablets. Clin Cancer Res. 2004;10(4):1212–8.
Li J, Zhao M, He P, Hidalgo M, Baker SD. Differential metabolism of gefitinib and erlotinib by human cytochrome P450 enzymes. Clin Cancer Res. 2007;13(12):3731–7.
Swaisland HC, Cantarini MV, Fuhr R, Holt A. Exploring the relationship between expression of cytochrome P450 enzymes and gefitinib pharmacokinetics. Clin Pharmacokinet. 2006;45(6):633–44.
Gerlowski LE, Jain RK. Physiologically based pharmacokinetic modeling: principles and applications. J Pharm Sci. 1983;72(10):1103–27.
Jones H, Rowland-Yeo K. Basic concepts in physiologically based pharmacokinetic modeling in drug discovery and development. CPT Pharmacometrics Syst Pharmacol. 2013;2:e63.
Jones HM, Chen Y, Gibson C, Heimbach T, Parrott N, Peters SA, et al. Physiologically based pharmacokinetic modeling in drug discovery and development: a pharmaceutical industry perspective. Clin Pharmacol Ther. 2015;97(3):247–62.
An G, Morris ME. A physiologically based pharmacokinetic model of mitoxantrone in mice and scale-up to humans: a semi-mechanistic model incorporating DNA and protein binding. AAPS J. 2012;14(2):352–64.
Gustafson DL, Rastatter JC, Colombo T, Long ME. Doxorubicin pharmacokinetics: macromolecule binding, metabolism, and excretion in the context of a physiologic model. J Pharm Sci. 2002;91(6):1488–501.
Bradshaw-Pierce EL, Eckhardt SG, Gustafson DL. A physiologically based pharmacokinetic model of docetaxel disposition: from mouse to man. Clin Cancer Res. 2007;13(9):2768–76.
Shah DK, Balthasar JP. Physiologically based pharmacokinetic model for topotecan in mice. J Pharmacokinet Pharmacodyn. 2011;38(1):121–42.
Farris FF, Dedrick RL, King FG. Cisplatin pharmacokinetics: applications of a physiological model. Toxicol Lett. 1988;43(1–3):117–37.
Hudachek SF, Gustafson DL. Physiologically based pharmacokinetic model of lapatinib developed in mice and scaled to humans. J Pharmacokinet Pharmacodyn. 2013;40(2):157–76.
Agarwal S, Sane R, Gallardo JL, Ohlfest JR, Elmquist WF. Distribution of gefitinib to the brain is limited by P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2)-mediated active efflux. J Pharmacol Exp Ther. 2010;334(1):147–55.
D’Argenio DZ, Schumitzky A, Wang X. ADAPT 5 user’s guide: pharmacokinetic/pharmacodynamic systems analysis software. Biomedical Simulations Resource, Los Angeles, 2009.
Davies B, Morris T. Physiological parameters in laboratory animals and humans. Pharm Res. 1993;10(7):1093–5.
Kawai R, Mathew D, Tanaka C, Rowland M. Physiologically based pharmacokinetics of cyclosporine A: extension to tissue distribution kinetics in rats and scale-up to human. J Pharmacol Exp Ther. 1998;287(2):457–68.
Loccisano AE, Campbell Jr JL, Butenhoff JL, Andersen ME, Clewell 3rd HJ. Comparison and evaluation of pharmacokinetics of PFOA and PFOS in the adult rat using a physiologically based pharmacokinetic model. Reprod Toxicol. 2012;33(4):452–67.
Wang S, Zhou Q, Gallo JM. Demonstration of the equivalent pharmacokinetic/pharmacodynamic dosing strategy in a multiple-dose study of gefitinib. Mol Cancer Ther. 2009;8(6):1438–47.
Nakagawa K, Tamura T, Negoro S, Kudoh S, Yamamoto N, Yamamoto N, et al. Phase I pharmacokinetic trial of the selective oral epidermal growth factor receptor tyrosine kinase inhibitor gefitinib (‘Iressa’, ZD1839) in Japanese patients with solid malignant tumors. Ann Oncol. 2003;14(6):922–30.
Tang H, Mayersohn M. A global examination of allometric scaling for predicting human drug clearance and the prediction of large vertical allometry. J Pharm Sci. 2006;95(8):1783–99.
Rowland M, Peck C, Tucker G. Physiologically-based pharmacokinetics in drug development and regulatory science. Annu Rev Pharmacol Toxicol. 2011;51:45–73.
Kosaka T, Yatabe Y, Endoh H, Yoshida K, Hida T, Tsuboi M, et al. Analysis of epidermal growth factor receptor gene mutation in patients with non-small cell lung cancer and acquired resistance to gefitinib. Clin Cancer Res. 2006;12(19):5764–9.
Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352(8):786–92.
Balak MN, Gong Y, Riely GJ, Somwar R, Li AR, Zakowski MF, et al. Novel D761Y and common secondary T790M mutations in epidermal growth factor receptor-mutant lung adenocarcinomas with acquired resistance to kinase inhibitors. Clin Cancer Res. 2006;12(21):6494–501.
Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321(5897):1807–12.
Cancer Genome Atlas Research N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(7216):1061–8.
Brandes AA, Franceschi E, Tosoni A, Hegi ME, Stupp R. Epidermal growth factor receptor inhibitors in neuro-oncology: hopes and disappointments. Clin Cancer Res. 2008;14(4):957–60.
Rich JN, Reardon DA, Peery T, Dowell JM, Quinn JA, Penne KL, et al. Phase II trial of gefitinib in recurrent glioblastoma. J Clin Oncol. 2004;22(1):133–42.
Acknowledgments
This work was supported by PhRMA Foundation (Research Starter Grant awarded to Dr. Guohua An) and by Dr. Guohua An’s Start-up fund (provided by the University of Florida).
Authorship Contributions
Guohua An was responsible for the research design and animal experiment; Jiexin Deng for gefitinib analysis using LC/MS/MS; Youwei Bi for data analysis and PBPK model construction using ADAPT 5; and Youwei Bi, Guohua An, and Daryl J Murry for the writing or contributed to the writing of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Bi, Y., Deng, J., Murry, D.J. et al. A Whole-Body Physiologically Based Pharmacokinetic Model of Gefitinib in Mice and Scale-Up to Humans. AAPS J 18, 228–238 (2016). https://doi.org/10.1208/s12248-015-9836-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1208/s12248-015-9836-3