
Abstract
Background
The microbiota plays a critical role in host homeostasis and has been shown to be a major driving force in host evolution. However, our understanding of these important relationships is hampered by a lack of data for many species, and by significant gaps in sampling of the evolutionary tree. In this investigation we improve our understanding of the host-microbiome relationship by obtaining samples from all seven extant species of sea turtle, and correlate microbial compositions with host evolutionary history.
Results
Our analysis shows that the predominate phyla in the microbiota of nesting sea turtles was Proteobacteria. We also demonstrate a strong relationship between the bacterial phyla SR1 and sea turtle phylogeny, and that sea turtle microbiotas have changed very slowly over time in accordance with their similarly slow phenotypic changes.
Conclusions
This is one of the most comprehensive microbiota studies to have been performed in a single clade of animals and further improves our knowledge of how microbial populations have influenced vertebrate evolution.
Similar content being viewed by others

Background
Residing on and within most metazoan species is a diverse and complex metropolis of microorganisms (viruses, bacteria, fungi and protozoans) known collectively as the microbiota and the occupation of higher organisms by prokaryotic and eukaryotic colonists has been a key factor in driving evolution and radiation of life on Earth [1, 2]. Hosts and their microbiotas form a mutualistic holobiont so intertwined that the amalgamation of these distinct organisms is often referred to as a holobiome, and their combined genetic material collectively defined as the hologenome [3, 4]. This multigenomic microcosm has become so integral to animal homeostasis that it can no longer be considered separate from the individual. Thus, it has been proposed that animals represent a vastly intricate biological ‘super organism’ in which a proportion of the physiological function is derived from microbial activity [5]. The microbiota has been shown to be a heritable trait in a range of vertebrates [6,7,8,9,10,11], and there is evidence to suggest that its composition and function is probably influenced by host phylogeny [12, 13], with co-evolution of the host and microbiota a critical process in shaping metazoan life [3, 4]. Ultimately, it appears that a bidirectional interaction of host physiology and gut microbiota over evolutionary time is responsible for determining host dietary niche and adaptation [12]. Understanding these relationships using a multi-disciplinary approach by combining microbiological techniques with phylogenetic analyses, is fundamental to exploring the origins of complex, multicellular organisms. Investigating closely-related species with clearly defined phylogenies is extremely informative, because analysis is not confounded by vast expanses of evolutionary periods with multiple missing links. Thus, evolutionarily old taxa with relatively simple phylogenetic histories are highly-desirable candidates for such an investigation. Sea turtles represent an excellent group of species to study in this context, because all extant species, including their phylogeny, are well described [14], and they originate from an approximately 200 million year old lineage that has gone through intermittent periods of slow and intermediate evolution and diversification [15], meaning that any described relationships are robust and profound. Exploring the bidirectional interplay of evolutionary forces acting on the host-microbiota relationship is an important step in comprehending the origins of metazoan physiology.
The major site for microbial inhabitation in animals is the gastrointestinal tract, where immense numbers of microorganisms confer myriad beneficial properties to their host. These complex interactions are an exciting and emerging area of evolutionary biology. Historically, the importance of microflora to non-human species has predominately focused on their role during digestion, particularly of complex carbohydrates in herbivorous animals. However, these investigations have begun to broaden with the realisation of the greater role that they play in the health and ecology of all species [2].
Investigations into the microbiota of reptiles, including sea turtles, are limited to studies describing microbial communities [16,17,18,19,20,21,22,23,24,25,26,27,28,29], factors that influence their composition [30,31,32,33,34,35,36,37,38,39,40,41,42,43], and how they affect host physiology [8, 9, 44,45,46], but investigations into the influence of phylogenetic factors affecting microbiota composition in this taxon are rare. Irrespective of the potential host-phylogenetic signal in microbiota composition at a broad taxonomic level, it has been shown that at a more individual level, diet, captivity, geography, and feeding regime all influence the microbiota [9, 17, 32], and that fermenting bacteria are important for digestion in herbivorous species [47].
Sea turtles are among the most imperilled species on Earth and therefore a deeper understanding of their physiology is important to their conservation. The role that microbiota plays in the health, behaviour and physiology of humans and animals is undeniable, and its categorisation in marine turtles is the next important step in understanding how these species adapt to a changing environment. There are seven extant species of sea turtle, the leatherback turtle (Dermochelys coriacea), green turtle (Chelonia mydas), flatback turtle (Natator depressus), hawksbill turtle (Eretmochelys imbricata), loggerhead turtle (Caretta caretta), olive ridley turtle (Lepidochelys olivacea), and the Kemp’s ridley turtle (Lepidochelys kempii). With the exception of the flatback turtle, all are listed as threatened species by the IUCN with various levels of risk, while the flatback turtle is listed as ‘Data Deficient’, but is also likely to be threatened [48]. Marine turtles are highly evolved for a completely aquatic life, but, like almost all reptiles they are still tied to the terrestrial environment for egg-laying [14]. The majority of sea turtle species are scattered unevenly throughout all three tropical oceans, with the exception of the flatback and Kemp’s ridley turtles, which have relatively restricted distributions [14]. Additionally, the leatherback turtle is a more cold adapted species with a more cosmopolitan distribution, and may be found occupying waters at higher latitudes than the other species [14]. Sea turtle diets vary remarkably over life stage, and among species [49]. For example, the leatherback feeds primarily on gelatinous zooplankton for its entire existence, while the green turtle is predominately omnivorous during the oceanic phase of its life, but then undergoes an ontogenic dietary shift to herbivory as it transitions to neritic (nearshore) habitats later in life [49], resulting in a discernible shift in the bacterial communities during this phase of their life [33]. Sea turtles are unique among the Reptilia, in that they are the only members that undergo long-distance migrations, rivalling those of other vertebrate species [50], and during this time they typically do not forage [51], which may have an effect on their microbiota [17, 52,53,54,55,56].
How the microbiota has contributed to the evolutionary history of sea turtles, and the extraordinary physiological adaptations of these species has been little studied, and as a result, we attempted to address this deficit by examining the microbial populations of the world’s sea turtle species. In this investigation we present the most comprehensive data on the microbiota of sea turtles that has been compiled to date. Furthermore, this is one of the most complete microbiota studies to have been conducted in any taxa, because we obtained samples from an entire clade of the evolutionary tree and we were able to explore some of the phylogenetic relationships that exist between sea turtles and their microbiotas. The aims of this investigation were to categorise the microbiota composition of the world’s sea turtle species, to explore any relationships between sea turtle phylogeny and microbiota composition, and to examine how sea turtle microbiotas have changed over evolutionary time.
Results
Samples were collected from 120 turtles, with sequencing results obtained in all but three, in which failure of DNA extraction meant that results were not obtained for these individuals (Table 1 and Additional file 1).
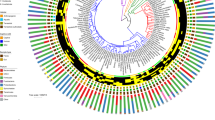
The taxonomic summary of microbial components from all samples yielded a total of 20 bacterial phyla, 36 classes, 63 orders, 122 families, 202 genera, and 362 OTUs (Fig. 1 and Additional file 1). The predominant bacterial phyla were Proteobacteria, Bacteroidetes, Actinobacteria, and Firmicutes (Fig. 1 and Additional file 1), while the phyla Euryarchaeota, Deferribacteres, and Cyanobacteria were only seen in Kemp’s ridley turtles. The prevalence of the phylum SR1 (Asoconditabacteria) was greatest in flatback and green turtles, intermediate in hawksbill and leatherbacks, and lowest in loggerhead and ridley turtles (Fig. 1 and Additional file 1). Analysis of alpha diversity revealed that there were significant differences between Observed OTUs (χ2 = 45.83, df = 6, p < 0.001) (Fig. 2 and Additional file 2), and in species richness between samples as measured by Chao1 (χ2 = 43.28, df = 6, p < 0.001) (Fig. 2 Additional file 2), but not in Shannon diversity (χ2 = 7.48, df = 6, p = 0.28) (Fig. 2 Additional file 2). These observations were consistent when all samples were analysed in entirety, and also for pairwise comparisons (Additional file 3). For beta diversity, we detected clustering patterns for the more ancient species (leatherback and green turtles), as well as the most modern species, Kemp’s ridley (Fig. 3). For the remaining species there was overlap of microbiota compositions (Fig. 3). However, Adonis analysis of the PCoA plot revealed that there was a significant difference between all species when analysed together (df = 6, SST = 15.42, MS = 2.57, f.model = 7.72, R2 = 0.29, p = 0.001), and similarly, when pairwise comparisons were made between all combinations of species, significant differences in microbiota compositions existed for all species combinations (Additional file 4).

Relative abundance of bacterial phyla in each sea turtle species together with their phylogenetic tree

Alpha diversity measures across all species. Individual points and brackets represent the richness estimate and the theoretical standard error range associated with that estimate, respectively. Within each panel, the samples are organized into species, and a boxplot is overlaid on top of this. The mean microbial diversity estimate using Shannon’s diversity index did not differ significantly among all samples (p = 0.28). However, there were significant differences between samples as measured by Observed (p < 0.001) and Chao1 (p < 0.001)

Principle co-ordinate analysis (PCoA) of Bray-Curtis distance showing functional microbial diversity across all species. Significant differences for microbiota composition existed between all species (R2 = 0.29, p = 0.001). For pairwise comparisons between species see Table S4
We did not discover a phylogenetic signal for any alpha diversity measure of microbiota composition for any of the species in this investigation (Table S6 and Additional files 4, 5 and 6). When microbiota composition was analysed in concert with evolutionary history, we found that the bacterial phyla SR1, GN02 (Gracilibacteria) and Actinobacteria had a phylogenetic signal for both Moran’s I and Abouheif’s Cmean calculations (Table S7 and Additional files 7, 8 and 9). However, when these phyla were further examined using Pagels λ and Blomberg’s K-statistic, it was discovered that only SR1 (λ = 1.06, LogL = − 11.67, LogL0 = − 13.20, p = 0.08; K = 1.28, p = 0.02) was correlated to sea turtle phylogeny (GN02: λ = 0.62, LogL = − 9.38, LogL0 = − 9.67, p = 0.44; K = 0.69, p = 0.28; Actinobacteria: λ = 0.69, LogL = − 21.0, LogL0 = − 21.36, p = 0.4; K = 0.9, p = 0.14). The relative abundance of SR1 was greatest in green and flatback turtles, and lowest in olive ridley turtles (Fig. 1). Finally, we reconstructed extinct sea turtle microbiotas (numbered nodes (8–13) on the phylogenetic tree (Fig. 4)), to examine how these have changed over the course of sea turtle evolution. We found that most bacterial phyla have been relatively stable over time, with the exception of Actinobacteria, which decreased in relative abundance as sea turtles evolved, and Spirochaetes, which went through a period of increase between nodes 1–5, but then decreased from nodes 5–6. The phylum Thermi showed a steady increase across nodes as time progressed (Fig. 4).

Proposed microbiotas for extinct sea turtle species with phylogenetic tree. Numbered nodes represent extinct ancestors on the sea turtle phylogenetic tree
Discussion
In this investigation we present the most comprehensive data on the microbiota composition of sea turtles to date, and demonstrate a clear link between microbiota composition and sea turtle phylogeny. Furthermore, this is one of the only studies in wild animals to have obtained samples from all extant species of an entire clade of the evolutionary tree. This enabled us to perform a detailed evaluation of the phylogenetic signal that exists between host and microbiota. We showed that microbiota composition differs among all sea turtle species, but in all species, the predominant bacterial phylum in nesting turtles was Proteobacteria. In an investigation into the microbiota of juvenile green turtles from Florida, Proteobacteria was also the major phylum dominating samples [33]. In contrast, the microbiota of juvenile green turtles from coastal areas of Brazil are co-dominated by the phyla Bacteroidetes and Firmicutes, with the investigators speculating that Proteobacteria increased in abundance in three individual turtles in this investigation in response to anthropogenic factors [38]. Similarly, in a study conducted on green turtles from the Great Barrier Reef, Firmicutes was the most common phylum isolated from healthy individuals, but in sick turtles Proteobacteria was the dominate phylum [57]. There are also reports of Firmicutes dominating the microbiota of stranded loggerhead turtles [18, 37], but these investigations are confounded because samples were collected from sick individuals, and in many cases there were delays between when the turtle was rescued and when samples were collected. Given that both captivity [58,59,60,61,62] and health [63] have both been shown to affect the microbiota of individuals, these results should be interpreted with caution as they are unlikely to represent the normal gut microbiota. In our investigation, all turtles sampled were wild and apparently healthy, and some had been sourced from extremely remote locations with no human habitation (e.g. Rosemary Island and Tiwi Islands), so we think that the likelihood of anthropogenic or other factors influencing our results is low. Furthermore, given that Proteobacteria was overwhelmingly the predominate phylum in all sea turtle species, and the strong phylogenetic relationship between microbiota and sea turtle species, we think that in nesting animals (i.e. animals with prolonged periods of inappetence), any microbiota in which Proteobacteria is not the most abundant phylum represents an atypical gut flora.
In comparison to other taxa, there are few studies on the microbiota of wild reptiles, and the relative abundance of specific bacterial phyla in herpetofauna appears to vary greatly. For example, Firmicutes was the dominant phylum identified in anoles (Anolis sp.) [64], Galápagos tortoises (Chelonoidis nigra) [23], gopher tortoises (Gopherus polyphemus) [19], green iguanas (Iguana iguana) [65], Galápagos land iguanas (Conolophus subcristatus) [23], marine iguanas (Amblyrhynchus cristatus) [23], and the montane iguana species Liolaemus parvus, Liolaemus ruibali, and Phymaturus williamsi [9]. Such findings have led some researchers to believe that reptile microbiotas resemble that of mammals [21, 32], but in a wide-ranging investigation into squamate microbiotas, in which individuals representing 22 squamate families were sampled, Proteobacteria was the predominate phylum, and supports a hypothesis that the reptile gut microbiota is similar to that of birds [66]. However, in the single reptile study conducted in the species most closely related to birds, the archosaurian American alligator (Alligator mississippiensis), gut microbiota was overwhelmingly dominated by the phylum Fusobacteria [17]. Furthermore, the results of our investigation are similar to those seen in fish, in which the predominate phyla are Proteobacteria, Bacteroidetes, Actinobacteria, and Fusobacteria [67]. These discrepancies highlight the difficulties associated with making any assumptions on microbial assembly between taxa based solely on compositional data without incorporating any phylogenetic techniques in the analysis.
Unlike their microbiotas, the natural histories of sea turtles differ widely among the seven extant species. Leatherbacks are largely oceanic-pelagic throughout their life-history, and have the most specialised diet, feeding almost entirely on an array of dense gelatinous zooplankton [49]. Their large size means that they are more cold-adapted than other species, allowing them to traverse through the boreal waters that act as a barrier to other warm-water adapted turtles [68]. Green turtles have an oceanic-neritic developmental pattern [69], are found in tropical and temperate waters worldwide [68], and consume a variety of seagrass, marine algae, and invertebrates [49, 70]. Like green turtles, loggerheads also have an oceanic-neritic developmental pattern and prefer temperate to tropical waters [69]. However, unlike green turtles, they are largely carnivorous, feeding on a wide array of prey items including Hydrozoa, Bivalvia, Cephalopodia, Porifera, Scyphoza, Bryozoa, Gastropoda, Polychaeta, Maxillopoda, Malacostraca, Insecta, Holothuroidea, Echinoidea, Anthozoa, Actinopterygii, and occasional plant material [49]. Evidence suggests that hawksbills also have an oceanic-neritic developmental pattern [69], with a preference for tropical waters [69]. Although primarily carnivorous, the composition of prey items varies among populations of hawksbills, with some having a preference for sponges, while others feed predominately on corals [49]. The flatback turtle has a completely neritic life history [69], residing within the tropical waters of the Australian continental shelf [71]. Detailed investigations into their diet are lacking, but it is presumed that they are carnivorous [49]. The olive ridley turtle is predominately oceanic for the duration of its life [69], and is found in both temperate and tropical waters, but most feeding probably occurs in warm water and they are likely omnivorous [49, 68]. Finally, Kemp’s ridleys have an oceanic-neritic developmental pattern [69], have a preference for tropical waters, and as adults are primarily carnivorous ingesting a range of molluscs, fish, jellyfish and gastropods [49]. Given the vast array of niche occupancy and dietary preferences of sea turtles, some with significant overlap between species, it is likely that the similarities seen in relative abundance of the major bacterial phyla are driven by their shared evolutionary history rather than dietary and ecological factors. The similarities seen between gut microbiotas in nesting female sea turtles is remarkable, since there is nearly 100 million years of evolution separating the most ancient species, the leatherback, and the most modern species, the Kemp’s ridley [15]. Such conservation of community similarity may be an indication that specific combinations of bacterial phyla are fundamental to sea turtle normal function, but this remains to be demonstrated in any taxa.
Based on the results of our study, it appears that gut microbial composition and sea turtle phylogeny are intrinsically linked, and it is likely that a process of co-evolution exists between host and microbial community composition. Microbes have been identified as a key driver of vertebrate evolution [2], and more recent research has focused on the role that phylogeny may play in convergence of microbiotas in some species [72]. Investigations in primates suggest that evolutionary trends in host physiology are more important than dietary niche in determining gastrointestinal microbiota [12], which is supported by this research. For example, previous investigations into sea turtle microbiotas have speculated that the high proportion of Firmicutes found in some green turtle samples may be due to this bacterial phylum’s ability to break down plant-derived polysaccharides [57] and thus facilitate digestion of a cellulose-rich food such as the seagrass species that form the primary diet item of green turtles. However, we found that green turtles, along with loggerheads, had the lowest relative abundance of Firmicutes of all the sea turtle species, suggesting that this phylum may not be as important for cellulose digestion in herbivorous reptiles as previously reported. However, it should be remembered that since the animals sampled in our investigation were not feeding, it is possible that there may have been a shift away from phyla important for digestive function and thus they were underrepresented relative to what would be expected in feeding animals. Nevertheless, there are many examples in vertebrates, both terrestrial and aquatic, and from disparate branches of the evolutionary tree, where Firmicutes are lower in abundance than other phyla in herbivorous species [73,74,75,76,77,78,79,80,81]. Therefore, in the absence of specific functional testing, caution should be applied when making assumptions on functionality of the microbiota within a species, especially where phylogeny has not been considered as a component of the analysis. The results of this analysis show that extant sea turtle microbiotas have changed very little over the course of nearly 70 million years of evolution, despite the phenotypic changes that have occurred in their hosts, and this may be an indication that certain combinations of microbes are fundamental to specific aspects of all sea turtle physiology, regardless of differences in natural history between species.
We showed that the bacterial phylum SR1 was strongly linked to sea turtle phylogeny. The candidate phylum SR1 (Absconditabacteria), includes ubiquitous organisms found in marine and terrestrial high-temperature environments, fresh-water lakes, subsurface aquifers, and animals [82, 83]. There are no cultured representatives of SR1, with all current knowledge on their presence and diversity obtained from genomic sequencing [84, 85]. They have a predilection for sulphur-rich and oxygen-limited environments, suggesting a potential microaerophilic, sulphur-based metabolism, and in general, environmental and animal-derived SR1 species appear to cluster differently [83, 86]. SR1 is routinely found in a range of vertebrates, but is most commonly associated with H2S-related malodour and periodontal disease in humans [87, 88]. How SR1 is involved in sea turtle gastrointestinal function remains unknown at this stage. The role of the microbiota in shaping vertebrate phylogeny should be a focus for future investigations, and an effort must be made to sample as diverse an array of species as possible, spanning multiple clades of the evolutionary tree, so that these relationships can be further explored.
Using results obtained from extant turtles, we reconstructed possible microbiotas of extinct sea turtle species. This analysis showed that the composition of sea turtle microbiotas has not changed greatly over time, with only relatively minor fluctuations in the relative abundance of specific phyla responsible for observed differences. In contrast, the human microbiota has diverged rapidly from our closest relatives [89], and among populations of humans, continues to rapidly evolve [90]. Sea turtles are among the most ancient species on Earth, with the leatherback existing in its current form for nearly 70 million years [15], while modern humans first appeared around 350,000 to 260,000 years ago [91]. These seemingly marked differences in rate of evolution of microbiotas, may be reflective of broad differences in rates of evolution between taxa, and warrants further investigation.
A feature of our study was that we were restricted to sampling nesting females. This was because living males are difficult to obtain, and this can only be done in-water. Although this means that our analysis and interpretation is made on a particular subset of animals of the same sex, life stage and reproductive state for each species, it removes potentially confounding variation in the composition of microbiota associated with these traits and thus we think it strengthens our phylogenetic comparisons. It is likely that all of the females sampled in this investigation had not eaten for an extended period, as sea turtles may undergo long periods of fasting, particularly during breeding and migration [51, 92]. Periods of inappetence have been shown to affect the microbiotas of a range of vertebrates including humans [52], fish [53], bears [54], alligators [17], mice [55], penguins [56], and the Burmese python [32], however such changes are not consistent across all taxa, with fasting having negligible effects on the microbiota of geckoes [93]. How this may have influenced our results is unknown, but it may explain the differences in our results and those of other green turtles captured on the Great Barrier Reef [57], and future investigations should focus on obtaining samples from a range of age classes, sexes, and physiological states. Although we were not able to determine if fasting in nesting turtles affected microbial diversity, some authors propose that fasting samples represent the core microbial OTUs, with other OTUs fluctuating in number in response to post-prandial physiological changes [17, 32]. If, as hypothesised, diet has little effect on this core microbiota, then this strengthens the results of our phylogenetic analysis because our results have not been confounded by transient microbial species that might be associated with dietary variation.
Conclusions
Our investigation represents the most comprehensive microbiota study to have been conducted in sea turtles to date, but most importantly, we were able to obtain samples from an entire clade of the evolutionary tree, which allowed us to perform a comprehensive phylogenetic analysis. While other investigations have sampled multiple species in the single study [22, 78, 81, 94, 95], the breadth of taxonomic coverage in these analyses was less comprehensive from a phylogenetic perspective. We were able to show that in nesting turtles, the microbiota is predominated by the phylum Proteobacteria and that the phylum SR1 is strongly linked to sea turtle phylogeny. Understanding the structure of microbial populations, and the complexities of the host-microbiome relationship, is the next critical step in managing threatened species populations. Furthermore, only by unravelling these mysteries can we truly understand the origins of life, and the forces that have shaped the diversity in form and function we see today.
Methods
Study populations
All samples were collected from adult female turtles as they nested during their respective breeding seasons. Flatback turtles (n = 17) were sampled from Port Hedland, Western Australia (20.3107° S, 118.5878° E) in November 2016. Green sea turtles (n = 18), were sampled from Heron Island, Queensland, Australia (23.4423° S, 151.9148° E) in January 2017. Loggerhead turtles (n = 20) were sampled from Mon Repos, Queensland, Australia (24.8059° S, 152.4416° E) in January 2017. Hawksbill turtles (n = 20) were sampled from Rosemary Island, Western Australia (20.2846° S, 116.3540° E) in October 2017. Olive ridley turtles (n = 10) were sampled from Tiwi Islands, Northern Territory, Australia (11.6969° S, 130.8779° E) in April 2018. Leatherback turtles (n = 18) were sample from Juno Beach, Florida, USA (26.5224° N, 80.315° E) in May 2018. Kemp’s ridley turtles (n = 20) were sampled from Playa Rancho Nuevo, Tamaulipas, Mexico (23.11° N, 97.46° W) in June 2019.
Sample collection
For leatherback turtles we waited until the female was covering the nest and then we dug a channel in the sand behind the turtle so that a swab could be inserted into the cloaca while the animal was still in a ventral position. For all other species, we waited until they had finished laying and were returning to the ocean and the turtle was then flipped into a dorsal position for sample collection. In all turtles an equine uterine swab (Minitube, Smythesdale, Victoria, Australia) was inserted into the cloaca so that it entered the distal colon. We were confident of correct placement of the swab because they are 90 cm long and were inserted to a depth of at least 60 cm, which is much longer than the length of the cloaca based on the primary author’s experience with endoscopic examination of chelonian cloacas, [96]. These swabs were housed in a sterile sheath, the entire apparatus was inserted into the cloaca and the swab tip was extruded to collect the colonic sample when correct placement of the sheath had been achieved. The swab tip was then retracted back into the sheath prior to extraction to shield it from any environmental contamination. By using these swabs in this manner, we did not collect any negative control swabs as we were confident that there was no risk for contamination and samples represented true colonic samples. Turtles were then permitted to return to the ocean and then the tip of the swab was cut using a sterile wire cutter, placed into a sterile Eppendorf tube and sealed. Total sample collection time was approximately 10 min. The Eppendorf tube containing the swab was then immediately placed into a portable cool box filled with ice, and once back at the field station they were frozen at − 20 °C for approximately 3–4 days. Swabs were transported back to the laboratory using dry ice (− 78.5 °C), where they were stored at − 80 °C until extraction could take place approximately 1 week later.
DNA extraction
DNA was extracted manually. In each Eppendorf tube, 500 μL of extraction buffer (20 mM EDTA, 0.1 M Tris, 1% CTAB, 56 mM NaCl, pH 8) was added so that swabs were completely covered. We then added 20 μL of Proteinase K (QIAGEN Proteinase K (10 ml(> 600 mAU/ml)) to each vial, along with 60 μL of 10% SDS. The mixture was then incubated at 55 °C overnight. The next day, 50 μL of 5 M NaCl and 500 μL of Phenol was added, and the tubes shaken until an emulsion was formed. They were then incubated at room temperature for 10 min, with intermittent mixing. The tubes were then centrifuged at 11,200 RCF for 10 min and the supernatant removed and added to a new tube containing 250 μL Phenol and 250 μL Chlorophorm:Isoamyl-Alcohol (24:1). The tubes were again centrifuged at 11,200 RCF for 10 min and the resultant supernatant added to a new tube containing 500 μL of Chlorophorm:Isoamyl-Alcohol. Once again, the tubes were centrifuged at 11,200 RCF for 10 min. The supernatant was then added to a new tube containing 3 M Sodium Acetate at a volume equal to 10% of the extraction solution. We then added 1 ml of ice-cold 99% ethanol to each test tube and then placed them into a freezer at − 20 °C for 1 h. The tubes were then centrifuged at 4 °C at 16,128 RCF for 10 min. The fluid in the test tube was then removed with a glass pipette and 1 ml of ice-cold 70% alcohol was added. The tubes were centrifuged a final time at 4 °C at 16,128 RCF for 5 min. After centrifugation the alcohol was removed and the lids left off the tubes to allow the DNA pellet to dry. Once dried, 25 μL of 1 x TE was added to each tube and the extracted DNA was stored at − 20 °C until amplicon sequencing could take place.
16S rRNA gene amplicon sequencing
The V3-V4 region of 16S rRNA genes were amplified with forward primer 5′ ACTCCTACGGGAGGCAGCAG 3′ and reverse primer 5′ GGACTACHVGGGTWTCTAAT 3′ using Q5 high fidelity polymerase (New England Biolabs). Sequencing was performed on an Illumina MiSeq system (2 × 300 bp) by the method of Fadrosh DW, Ma B, Gajer P, Sengamalay N, Ott S, Brotman RM and Ravel J [97].
Data processing
Sequence data was analysed using QIIME version 1.9.1 [98] using default parameters and a Phred quality threshold of > 20. The UCLUST algorithm [99] was used to pick OTUs at 97% sequence identity and a Biome table was produced. Potentially chimeric sequences were identified using Pintail [100]. Blast was used to assign taxonomy against the Greengenes database [101] and QIIME version 1.9.1 defaults. Additional assignment of taxonomy was performed using a command line version of BLASTN [102] against the NCBI 16S Microbial database.
Statistics and data analysis
Initial exploration of the Biome table data was performed using the online Calypso software [103]. Data was further analysed in R, utilising the package ‘phyloseq’ [104]. Alpha diversity was explored using Observed OTUs, Shannon index and Chao1 estimates. Alpha diversity was tested for normality using the Shapiro-Wilks test, with all metrics being non-normally distributed (Observed: W = 0.95, p < 0.001; Chao1: W = 0.96, p = 0.001; Shannon: W = 0.9, p < 0.001), and so comparisons between groups were first made using the Kruskal-Wallis test, and then paired comparisons between groups were made using the pairwise Wilcoxon rank sum test with (Holm) p-values that were adjusted for multiple comparisons. Beta diversity was investigated using principle co-ordinate analysis (PCoA) (Bray-Curtis) and Adonis tests to compare all species, and then pairwise comparisons were made between all combinations of species with Holm correction of p-values for multiple comparisons.
To test phylogenetic signal related to microbial composition in sea turtles, we first obtained genetic sequence data for the sea turtle species from NCBI (www.ncbi.nlm.nih.gov), and used these to construct a phylogenetic tree, with branch lengths, using the online database “Interactive Tree of Life” [105]. We then compiled the microbial data for all species into a Microsoft Excel spread sheet, and imported both the tree and microbiota data into R, and used the package ‘phytools’ to explore the effects of phylogeny on microbiota composition [106]. We used Moran’s I, and Abouheif’s Cmean to determine if specific bacterial phyla had a phylogenetic signal and whether or not there was a phylogenetic signal in alpha diversity. Where a significant result was found, this was further confirmed using Blomberg’s K-statistics, and Pagel’s λ for the individual bacterial phylum [107]. Finally, we reconstructed probable microbiota compositions of extinct sea turtle species at each of the nodes in our phylogenetic tree. For all statistical analyses significance was accepted if p > 0.05.
Availability of data and materials
All data not presented here or in the supplementary material have been submitted to The National Center for Biotechnology Information www.ncbi.nlm.nih.gov.
Abbreviations
- CTAB:
-
Cetrimonium bromide
- DNA:
-
Deoxyribonucleic acid
- EDTA:
-
Ethylenediaminetetraacetic acid
- GN02:
-
Gracilibacteria
- IUCN:
-
International Union for Conservation of Nature
- NCBI:
-
National Centre for Biotechnology Information
- OTU:
-
Operational taxonomic unit
- PCoA:
-
Principal coordinate analysis
- RCF:
-
Relative centrifugal force
- rRNA:
-
Ribosomal ribonucleic acid
- SDS:
-
Sodium dodecyl sulfate
- SR1:
-
Asoconditabacteria
- USA:
-
United States of America
References
Lee YK, Mazmanian SK. Has the microbiota played a critical role in the evolution of the adaptive immune system? Science. 2010;330:1768–73.
McFall-Ngai M, Hadfield MG, Bosch TC, Carey HV, Domazet-Loso T, Douglas AE, et al. Animals in a bacterial world, a new imperative for the life sciences. PNAS. 2013;110:3229–36.
Theis KR, Dheilly NM, Klassen JL, Brucker RM, Baines JF, Bosch TC, et al. Getting the hologenome concept right: an eco-evolutionary framework for hosts and their microbiomes. mSystems. 2016;1:e00028–16.
Shapira M. Gut microbiotas and host evolution: scaling up symbiosis. Trends Ecol Evol. 2016;31:539–49.
Dethlefsen L, McFall-Ngai M, Relman DA. An ecological and evolutionary perspective on human-microbe mutualism and disease. Nature. 2007;449:811–8.
Sylvain FE, Derome N. Vertically and horizontally transmitted microbial symbionts shape the gut microbiota ontogenesis of a skin-mucus feeding discus fish progeny. Sci Rep. 2017;7:5263.
Ding J, Dai R, Yang L, He C, Xu K, Liu S, et al. Inheritance and establishment of gut microbiota in chickens. Front Microbiol. 2017;8:1967.
Trevelline BK, MacLeod KJ, Knutie SA, Langkilde T, Kohl KD. In ovo microbial communities: a potential mechanism for the initial acquisition of gut microbiota among oviparous birds and lizards. Biol Lett. 2018;14:20180225.
Kohl KD, Brun A, Magallanes M, Brinkerhoff J, Laspiur A, Acosta JC, et al. Gut microbial ecology of lizards: insights into diversity in the wild, effects of captivity, variation across gut regions and transmission. Mol Ecol. 2017;26:1175–89.
Dominguez-Bello MG, Costello EK, Contreras M, Magris M, Hidalgo G, Fierer N, et al. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. PNAS. 2010;107:11971–5.
Scheelings TF. The microbiota of sea turtlesPhD. Monash University; 2019.
Amato KR, Sanders JG, Song SJ, Nute M, Metcalf JL, Thompson LR, et al. Evolutionary trends in host physiology outweigh dietary niche in structuring primate gut microbiomes. ISME J. 2018;13:576–87.
Carrillo-Araujo M, Tas N, Alcantara-Hernandez RJ, Gaona O, Schondube JE, Medellin RA, et al. Phyllostomid bat microbiome composition is associated to host phylogeny and feeding strategies. Front Microbiol. 2015;6:447.
Pritchard PCH. Evolution, phylogeny, and current status. In: Lutz PL, Musick JA, editors. The biology of sea turtles. Boca Raton: CRC Press; 1997. p. 1–28.
Duchene S, Frey A, Alfaro-Nunez A, Dutton PH, Thomas PGM, Morin PA. Marine turtle mitogenome phylogenetics and evolution. Mol Phylogenet Evol. 2012;65:241–50.
Zhang B, Ren J, Yang D, Liu S, Gong X. Comparative analysis and characterization of the gut microbiota of four farmed snakes from southern China. PeerJ. 2019;7:e6658.
Keenan SW, Engel AS, Elsey RM. The alligator gut microbiome and implications for archosaur symbioses. Sci Rep. 2013;3:2877.
Arizza V, Vecchioni L, Caracappa S, Sciurba G, Berlinghieri F, Gentile A, et al. New insights into the gut microbiome in loggerhead sea turtles Caretta caretta stranded on the Mediterranean coast. PLoS One. 2019;14:e0220329.
Yuan ML, Dean SH, Longo AV, Rothermel BB, Tuberville TD, Zamudio KR. Kinship, inbreeding and fine-scale spatial structure influence gut microbiota in a hindgut-fermenting tortoise. Mol Ecol. 2015;24:2521–36.
McLaughlin RW, Cochran PA, Dowd SE. Metagenomic analysis of the gut microbiota of the timber rattlesnake, Crotalus horridus. Mol Biol Rep. 2015;42:1187–95.
Hong PY, Wheeler E, Cann IK, Mackie RI. Phylogenetic analysis of the fecal microbial community in herbivorous land and marine iguanas of the Galapagos Islands using 16S rRNA-based pyrosequencing. ISME J. 2011;5:1461–70.
Ren T, Kahrl AF, Wu M, Cox RM. Does adaptive radiation of a host lineage promote ecological diversity of its bacterial communities? A test using gut microbiota of Anolis lizards. Mol Ecol. 2016;25:4793–804.
Hong P-Y, Mao Y, Ortiz-Kofoed S, Shah R, Cann I, Mackie RI. Metagenomic-based study of the phylogenetic and functional gene diversity in Galápagos land and marine iguanas. Microb Ecol. 2015;69:444–56.
Holmes IA, Monagan IV Jr, Rabosky DL, Davis Rabosky AR. Metabolically similar cohorts of bacteria exhibit strong cooccurrence patterns with diet items and eukaryotic microbes in lizard guts. Ecol Evol. 2019;9:12471–81.
Willson NL, Van TTH, Lever J, Moore RJ, Stanley D. Characterisation of the intestinal microbiota of commercially farmed saltwater crocodiles, Crocodylus porosus. Appl Microbiol Biotechnol. 2019;103:8977–85.
Lin M, Zeng C, Li Z, Ma Y, Jia X. Comparative analysis of the composition and function of fecal-gut bacteria in captive juvenile Crocodylus siamensis between healthy and anorexic individuals. MicrobiologyOpen. 2019;8:e929.
Tang W, Zhu G, Shi Q, Yang S, Ma T, Mishra SK, et al. Characterizing the microbiota in gastrointestinal tract segments of Rhabdophis subminiatus: dynamic changes and functional predictions. MicrobiologyOpen. 2019;8:e789.
Fugate HM, Kapfer JM, McLaughlin RW. Analysis of the microbiota in the fecal material of painted turtles (Chrysemys picta). Curr Microbiol. 2020;77:11–4.
Gaillard DL. Population genetics and microbial communities of the gopher tortoise (Gopherus polyphemus)Doctor of Philosophy. Hattiesburg: The University of Southern Mississippi; 2014.
Jiang HY, Ma JE, Li J, Zhang XJ, Li LM, He N, et al. Diets alter the gut microbiome of crocodile lizards. Front Microbiol. 2017;8:2073.
Colston TJ, Noonan BP, Jackson CR. Phylogenetic analysis of bacterial communities in different regions of the gastrointestinal tract of Agkistrodon piscivorus, the cottonmouth snake. PLoS One. 2015;10:e0128793.
Costello EK, Gordon JI, Secor SM, Knight R. Postprandial remodeling of the gut microbiota in Burmese pythons. ISME J. 2010;4:1375–85.
Price JT, Paladino FV, Lamont MM, Witherington BE, Bates ST, Soule T. Characterization of the juvenile green turtle (Chelonia mydas) microbiome throughout an ontogenetic shift from pelagic to neritic habitats. PLoS One. 2017;12:e0177642.
Indest KJ, Everman SJ, Lindsay JH, Jung CM, Smith JC, Newell SB. Effects of acute exposures of 2,4,6-trinitrotoluene and inorganic lead on the fecal microbiome of the green anole (Anolis carolinensis). PLoS One. 2018;13:e0208281.
Littleford-Colquhoun BL, Weyrich LS, Kent N, Frere CH. City life alters the gut microbiome and stable isotope profiling of the eastern water dragon (Intellagama lesueurii). Mol Ecol. 2019;28:4592–607.
Bloodgood JCG, Hernandez SM, Isaiah A, Suchodolski JS, Hoopes LA, Thompson PM, et al. The effect of diet on the gastrointestinal microbiome of juvenile rehabilitating green turtles (Chelonia mydas). PLoS One. 2020;15:e0227060.
Abdelrhman KF, Bacci G, Mancusi C, Mengoni A, Serena F, Ugolini A. A first insight into the gut microbiota of the sea turtle Caretta caretta. Front Microbiol. 2016;7:1060.
Campos P, Guivernau M, Prenafeta-Boldu FX, Cardona L. Fast acquisition of a polysaccharide fermenting gut microbiome by juvenile green turtles Chelonia mydas after settlement in coastal habitats. Microbiome. 2018;6:69.
Martin MO, Gilman FR, Weiss SL. Sex-specific asymmetry within the cloacal microbiota of the striped plateau lizard, Sceloporus virgatus. Symbiosis. 2010;51:97–105.
Bestion E, Jacob S, Zinger L, Di Gesu L, Richard M, White J, et al. Climate warming reduces gut microbiota diversity in a vertebrate ectotherm. Nat Ecol Evol. 2017;1:161.
Zhang W, Li N, Tang X, Liu N, Zhao W. Changes in intestinal microbiota across an altitudinal gradient in the lizard Phrynocephalus vlangalii. Ecol Evol. 2018;8:4695–703.
Montoya-Ciriaco N, Gomez-Acata S, Munoz-Arenas LC, Dendooven L, Estrada-Torres A, de la Vega-Perez AH D, et al. Dietary effects on gut microbiota of the mesquite lizard Sceloporus grammicus (Wiegmann, 1828) across different altitudes. Microbiome. 2020;8:6.
García-De La Peña C, Garduño-Niño E, Vaca-Paniagua F, Díaz-Velásquez C, Barrows CW, Gomez-Gil B, et al. Comparison of the fecal bacterial microbiota composition between wild and captive bolson tortoises (Gopherus flavomarginatus). Herpetol Conserv Bio. 2019;14:587–600.
Kohl KD, Brun A, Magallanes M, Brinkerhoff J, Laspiur A, Acosta JC, et al. Physiological and microbial adjustments to diet quality permit facultative herbivory in an omnivorous lizard. J Exp Biol. 2016;219:1903–12.
Rawski M, Kieronczyk B, Dlugosz J, Swiatkiewicz S, Jozefiak D. Dietary probiotics affect gastrointestinal microbiota, histological structure and shell mineralization in turtles. PLoS One. 2016;11:e0147859.
Trevelline BK, MacLeod KJ, Langkilde T, Kohl KD. Gestation alters the gut microbiota of an oviparous lizard. FEMS Microbiol Ecol. 2019;95:fiz086.
Mackie RI, Rycyk M, Ruemmler RL, Aminov RI, Wikelski M. Biochemical and microbiological evidence for fermentative digestion in free-living land iguanas (Conolophus pallidus) and marine iguanas (Amblyrhynchus cristatus) on the Galapagos archipelago. Physiol Biochem Zool. 2004;77:127–38.
IUCN. The IUCN red list of threatened species. Version 2019–1; 2019. http://www.iucnredlist.org/details/4615/0. Accessed 1 June 2019.
Jones TT, Seminoff JA. Feeding biology: advances from field-based observations, physiological studies, and molecular techniques. In: Wyneken J, Lohmann KJ, Musick JA, editors. The biology of sea turtles. Boca Raton: CRC Press; 2013. p. 211–48.
Plotkin P. Adult migrations and habitat use. In: Lutz PL, Musick JA, Wyneken J, editors. The biology of sea turtles. Boca Raton, Florida: CRC Press; 2003. p. 225–41.
Hays GC, Broderick AC, Glen F, Godley BJ. Change in body mass associated with long-term fasting in a marine reptile: the case of green turtles (Chelonia mydas) at Ascension Island. Can J Zool. 2002;80:1299–302.
Remely M, Hippe B, Geretschlaeger I, Stegmayer S, Hoefinger I, Haslberger A. Increased gut microbiota diversity and abundance of Faecalibacterium prausnitzii and Akkermansia after fasting: a pilot study. Wien Klin Wochenschr. 2015;127:394–8.
Xia JH, Lin G, Fu GH, Wan ZY, Lee M, Wang L, et al. The intestinal microbiome of fish under starvation. BMC Genomics. 2014;15:266.
Sommer F, Stahlman M, Ilkayeva O, Arnemo JM, Kindberg J, Josefsson J, et al. The gut microbiota modulates energy metabolism in the hibernating brown bear Ursus arctos. Cell Rep. 2016;14:1655–61.
Beli E, Yan Y, Moldovan L, Vieira CP, Gao R, Duan Y, et al. Restructuring of the gut microbiome by intermittent fasting prevents retinopathy and prolongs survival in db/db mice. Diabetes. 2018;67:1867–79.
Dewar ML, Arnould JP, Krause L, Trathan P, Dann P, Smith SC. Influence of fasting during moult on the faecal microbiota of penguins. PLoS One. 2014;9:e99996.
Ahasan MS, Waltzek TB, Huerlimann R, Ariel E. Fecal bacterial communities of wild-captured and stranded green turtles (Chelonia mydas) on the great barrier reef. FEMS Microbiol Ecol. 2017;93:fix13.
Kohl KD, Skopec MM, Dearing MD. Captivity results in disparate loss of gut microbial diversity in closely related hosts. Conserv Physiol. 2014;2:cou009.
Clayton JB, Vangay P, Huang H, Ward T, Hillmann BM, Al-Ghalith GA, et al. Captivity humanizes the primate microbiome. PNAS. 2016;113:10376–81.
Delport TC, Power ML, Harcourt RG, Webster KN, Tetu SG. Colony location and captivity influence the gut microbial community composition of the Australian sea lion (Neophoca cinerea). Appl Environ Microbiol. 2016;82:3440–9.
Nelson TM, Rogers TL, Carlini AR, Brown MV. Diet and phylogeny shape the gut microbiota of Antarctic seals: a comparison of wild and captive animals. Environ Microbiol. 2013;15:1132–45.
Ahasan MS, Waltzek TB, Huerlimann R, Ariel E. Comparative analysis of gut bacterial communities of green turtles (Chelonia mydas) pre-hospitalization and post-rehabilitation by high-throughput sequencing of bacterial 16S rRNA gene. Microbiol Res. 2018;207:91–9.
Pflughoeft KJ, Versalovic J. Human microbiome in health and disease. Annu Rev Pathol. 2012;7:99–122.
Ren T, Grieneisen LE, Alberts SC, Archie EA, Wu M. Development, diet and dynamism: longitudinal and cross-sectional predictors of gut microbial communities in wild baboons. Environ Microbiol. 2016;18:1312–25.
Wehrle BA. Intergenerational lizard lounges do not explain variation in the gut microbiomes of green iguanasMaster of Science in Biology: California State University; 2013.
Colston TJ. The reptile gut microbiome: its role in host evolution and community assembly: PhD. University of Mississippi; 2017.
Ghanbari M, Kneifel W, Domig KJ. A new view of the fish gut microbiome: advances from next-generation sequencing. Aquaculture. 2015;448:464–75.
Bowen BW, Karl SA. Population genetics and phylogeography of sea turtles. Mol Ecol. 2007;16:4886–907.
Bolten AB. Variation in sea turtle life history patterns: neritic vs. oceanic developmental stages. In: Lutz P, Musick J, Wyneken J, editors. The biology of sea turtles. Boca Raton: CRC Press; 2003. p. 243–58.
Amorocho DF, Reina RD. Feeding ecology of the East Pacific green sea turtle, Chelonia mydas agassizii at Gorgona National Park in Colombia. Endanger Species Res. 2007;3:43–51.
Limpus CJ. In: Fien L, editor. A biological review of Australian marine turtle species. 5. Flatback turtle, Natator depressus (Garman). Queensland: Queensland Environmental Protection Agency; 2008. p. 1–53.
Muegge BD, Kuczynski J, Knights D, Clemente JC, Gonzalez A, Fontana L, et al. Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science. 2011;332:970–4.
Raulo A. Social behaviour and gut microbiota in wild red-bellied lemurs (Eulemur rubriventer) - in search of the role of immunity in the evolution of socialityMaster of Science: University of Helsinki; 2015.
Fogel AT. The gut microbiome of wild lemurs: a comparison of sympatric Lemur catta and Propithecus verreauxi. Folia Primatol (Basel). 2015;86:85–95.
Dill-McFarland KA, Weimer PJ, Pauli JN, Peery MZ, Suen G. Diet specialization selects for an unusual and simplified gut microbiota in two- and three-toed sloths. Environ Microbiol. 2016;18:1391–402.
Pope PB, Mackenzie AK, Gregor I, Smith W, Sundset MA, McHardy AC, et al. Metagenomics of the Svalbard reindeer rumen microbiome reveals abundance of polysaccharide utilization loci. PLoS One. 2012;7:e38571.
Ishaq SL, Wright A-DG. Insight into the bacterial gut microbiome of the north American moose (Alces alces). BMC Microbiol. 2012;12:212.
Miyake S, Ngugi DK, Stingl U. Diet strongly influences the gut microbiota of surgeonfishes. Mol Ecol. 2015;24:656–72.
Sullam KE, Essinger SD, Lozupone CA, O'Connor MP, Rosen GL, Knight R, et al. Environmental and ecological factors that shape the gut bacterial communities of fish: a meta-analysis. Mol Ecol. 2012;21:3363–78.
Nielsen S, Wilkes Walburn J, Verges A, Thomas T, Egan S. Microbiome patterns across the gastrointestinal tract of the rabbitfish Siganus fuscescens. PeerJ. 2017;5:e3317.
Givens CE, Ransom B, Bano N, Hollibaugh JT. Comparison of the gut microbiomes of 12 bony fish and 3 shark species. Mar Ecol Prog Ser. 2015;518:209–23.
Davis JP, Youssef NH, Elshahed MS. Assessment of the diversity, abundance, and ecological distribution of members of candidate division SR1 reveals a high level of phylogenetic diversity but limited morphotypic diversity. Appl Environ Microbiol. 2009;75:4139–48.
Ghotra SK. Novel bacterial lineages in the uncultured candidate division SR1Masters of Science: San José State University; 2014.
Solden L, Lloyd K, Wrighton K. The bright side of microbial dark matter: lessons learned from the uncultivated majority. Curr Opin Microbiol. 2016;31:217–26.
Hug LA, Baker BJ, Anantharaman K, Brown CT, Probst AJ, Castelle CJ, et al. A new view of the tree of life. Nat Microbiol. 2016;1:16048.
Campbell JH, O'Donoghue P, Campbell AG, Schwientek P, Sczyrba A, Woyke T, et al. UGA is an additional glycine codon in uncultured SR1 bacteria from the human microbiota. PNAS. 2013;110:5540–5.
Takeshita T, Suzuki N, Nakano Y, Yasui M, Yoneda M, Shimazaki Y, et al. Discrimination of the oral microbiota associated with high hydrogen sulfide and methyl mercaptan production. Sci Rep. 2012;2:215.
Griffen AL, Beall CJ, Campbell JH, Firestone ND, Kumar PS, Yang ZK, et al. Distinct and complex bacterial profiles in human periodontitis and health revealed by 16S pyrosequencing. ISME J. 2012;6:1176–85.
Moeller AH, Li Y, Mpoudi Ngole E, Ahuka-Mundeke S, Lonsdorf EV, Pusey AE, et al. Rapid changes in the gut microbiome during human evolution. PNAS. 2014;111:16431–5.
Davenport ER, Sanders JG, Song SJ, Amato KR, Clark AG, Knight R. The human microbiome in evolution. BMC Biol. 2017;15:127.
Schlebusch CM, Malmström H, Günther T, Sjödin P, Coutinho A, Edlund H, et al. Southern African ancient genomes estimate modern human divergence to 350,000 to 260,000 years ago. Science. 2017;358:652–5.
Jessop TS, Hamann M, Limpus CJ. Body condition and physiological changes in male green turtles during breeding. Mar Ecol Prog Ser. 2004;276:281–8.
Kohl KD, Amaya J, Passement CA, Dearing MD, McCue MD. Unique and shared responses of the gut microbiota to prolonged fasting: a comparative study across five classes of vertebrate hosts. FEMS Microbiol Ecol. 2014;90:883–94.
Eichmiller JJ, Hamilton MJ, Staley C, Sadowsky MJ, Sorensen PW. Environment shapes the fecal microbiome of invasive carp species. Microbiome. 2016;4:44.
Gulino LM, Ouwerkerk D, Kang AY, Maguire AJ, Kienzle M, Klieve AV. Shedding light on the microbial community of the macropod foregut using 454-amplicon pyrosequencing. PLoS One. 2013;8:e61463.
Rafferty A, Evans R, Scheelings T, Reina R. Limited oxygen availability in utero may constrain the evolution of live birth in reptiles. Am Nat. 2013;181:245–53.
Fadrosh DW, Ma B, Gajer P, Sengamalay N, Ott S, Brotman RM, et al. An improved dual-indexing approach for multiplexed 16S rRNA gene sequencing on the Illumina MiSeq platform. Microbiome. 2014;2:6.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–6.
Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–1.
Ashelford KE, Chuzhanova NA, Fry JC, Jones AJ, Weightman AJ. At least 1 in 20 16 S rRNA sequence records currently held in public repositories is estimated to contain substantial anomalies. Appl Environ Microbiol. 2005;71:7724–36.
DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol. 2006;72:5069–72.
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–402.
Zakrzewski M, Proietti C, Ellis JJ, Hasan S, Brion M-J, Berger B, et al. Calypso: a user-friendly web-server for mining and visualizing microbiome-environment interactions. Bioinformatics. 2017;33:782–3.
McMurdie PJ, Holmes S. Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One. 2013;8:e61217.
Letunic I, Bork P. Interactive tree of life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics. 2007;23:127–8.
Revell LJ. Phytools: an R package for phylogenetic comparative biology (and other things). Methods Ecol Evol. 2012;3:217–23.
Diniz-Filho JAF, Santos T, Rangel TF, Bini LM. A comparison of metrics for estimating phylogenetic signal under alternative evolutionary models. Genet Mol Biol. 2012;35:673–9.
Acknowledgements
We thank Christine Biesgen and the Care for Port Hedland Group, the Heron Island Research Station, the Mon Repos Turtle Conservation volunteers, Justin Perrault and the staff of the Loggerhead Marinelife Centre, Joanne King and the Parks and Wildlife Service of Western Australia, Kate Hadden and the Tiwi Marine Rangers, Edgar Sanchez, Martha Hernandez, Hugo Acosta and the staff of the Playa Rancho Nuevo Research Station.
Funding
This research was funded by the Holsworth Wildlife Endowment as well as an Australian Postgraduate Award.
Author information
Authors and Affiliations
Contributions
TFS was responsible for experimental design, collecting samples and analysing and interpreting data. MK and RDR were responsible for experimental design, and interpreting data. RM and TTHV were responsible for bioinformatics as well as data interpretation All authors were major contributors in writing the manuscript and have approved the final document.
Corresponding author
Ethics declarations
Ethics approval
This study was approved by the Biological Sciences Animal Ethics Committee of Monash University (approval 14,694). Green sea turtles and loggerhead turtles were sampled under the permit WITK17730216 from the Queensland Department of Environment and Heritage Protection, Australia. Hawksbill turtles and flatback turtles were sampled under the permit 01–000121-1 from the Department of Parks and Wildlife Western Australia. Olive ridley turtles were sampled under the permit 60,298 from the Parks and Wildlife Commission Northern Territory, Australia. Leatherback turtles were sampled under the permit MTP-18-205A from the Florida Fish and Wildlife Conservation Commission, USA and samples were imported into Australia under the CITES permit PWS2018-AU-000738 and quarantine permit 0003027044. Kemp’s ridley turtles were sampled under the permit 00663/19 from the Secretaría de Medio Ambiente y Recursos Naturales, Mexico, exported under the CITES permit MX100214, and imported into Australia under the CITES permit PWS2019-AU-000981 and quarantine permit 0003027044.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1.
Sequencing results for all sea turtle species in this investigation
Additional file 2: Table S2.
Pairwise Wilcoxon rank sum test comparisons between species for Observed OTUs. Numbers represent corrected (Holm) p-values for multiple comparisons. Significant values are indicated by bold text. Table S3. Pairwise Wilcoxon rank sum test comparisons between species for Chao1. Numbers represent corrected (Holm) p-values for multiple comparisons. Significant values are indicated by bold text. Table S4. Pairwise Wilcoxon rank sum test comparisons between species for Shannon index. Numbers represent corrected (Holm) p-values for multiple comparisons. No significance was observed between Shannon index for any inter-species comparisons
Additional file 3: Table S5.
Pairwise Adonis comparisons of beta diversity for all combinations of microbiota composition in sea turtles. Both the p, and adjusted (Holm) p-values for multiple comparisons are reported. All pairwise comparisons were significantly different. For microbial composition of species and how they relate to other species refer to Fig. 3
Additional file 4: Table S6
. Moran’s I and Abouheif’s Cmean calculations for alpha diversity and their correlation to sea turtle phylogeny. The observed value of I (Obs), is the expected value under the null hypothesis of no correlation. Positive values indicate that the data is spatially clustered in some way. Other values represented in this table include the standard-deviation of the observed I (Std.Obs), and the alternative hypothesis (alter) which has been set to “greater” meaning that the p-value is estimated as a number of random values equal to, or greater than the observed, + 1. No significance was detected for any diversity measure.
Additional file 5: Fig. S1
Graphical representation of Moran’s I calculations for alpha diversity. Bars represent phylogenetic tree branches, the diamond represents the observed value, and the y-scale is frequency.
Additional file 6: Fig. S2
Graphical representation of Abouheif’s Cmean calculations for alpha diversity. Bars represent phylogenetic tree branches, the diamond represents the observed value, and the y-scale is frequency.
Additional file 7: Table S7.
Moran’s I and Abouheif’s Cmean calculations for bacterial phyla and their correlation to sea turtle phylogeny. Significant values are indicated by bold text.
Additional file 8: Fig. S3
Graphical representation of Moran’s I calculations for microbial composition. Bars represent phylogenetic tree branches, the diamond represents the observed value, and the y-scale is frequency.
Additional file 9: Fig. S4
Graphical representation of Abouheif’s Cmean calculations for microbial composition. Bars represent phylogenetic tree branches, the diamond represents the observed value, and the y-scale is frequency.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
{kind=link}
Cite this article
Scheelings, T.F., Moore, R.J., Van, T.T.H. et al. Microbial symbiosis and coevolution of an entire clade of ancient vertebrates: the gut microbiota of sea turtles and its relationship to their phylogenetic history. anim microbiome 2, 17 (2020). https://doi.org/10.1186/s42523-020-00034-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s42523-020-00034-8