
Abstract
Background
To investigate the effect of long-term febrile convulsions on gene expression in mesial temporal lobe epilepsy with hippocampal sclerosis (MTLE-HS) and explore the molecular mechanism of MTLE-HS.
Methods
Microarray data of MTLE-HS were obtained from the Gene Expression Omnibus database. Differentially expressed genes (DEGs) between MTLE-HS with and without febrile seizure history were screened by the GEO2R software. Pathway enrichment and gene ontology of the DEGs were analyzed using the DAVID online database and FunRich software. Protein–protein interaction (PPI) networks among DEGs were constructed using the STRING database and analyzed by Cytoscape.
Results
A total of 515 DEGs were identified in MTLE-HS samples with a febrile seizure history compared to MTLE-HS samples without febrile seizure, including 25 down-regulated and 490 up-regulated genes. These DEGs were expressed mostly in plasma membrane and synaptic vesicles. The major molecular functions of those genes were voltage-gated ion channel activity, extracellular ligand-gated ion channel activity and calcium ion binding. The DEGs were mainly involved in biological pathways of cell communication signal transduction and transport. Five genes (SNAP25, SLC32A1, SYN1, GRIN1, and GRIA1) were significantly expressed in the MTLE-HS with prolonged febrile seizures.
Conclusion
The pathogenesis of MTLE-HS involves multiple genes, and prolonged febrile seizures could cause differential expression of genes. Thus, investigations of those genes may provide a new perspective into the mechanism of MTLE-HS.
Similar content being viewed by others
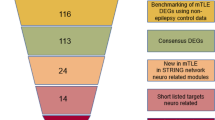


Background
Epilepsy is a disabling and frequent neurological disease that causes a significant burden on patients worldwide. Epilepsy is characterized by sudden attacks and recurrent seizures due to abnormal excessive neuronal discharges [1]. It can occur at any age and currently has affected about 70 million people worldwide [2]. Temporal lobe epilepsy is a focal type of epilepsy characterized by recurrent lesions in the temporal lobe, most commonly occurring in the medial lobe. It is related with a variety of mental phenomena, including hallucinations, cognitive impairment and emotional experience. Mesial temporal lobe epilepsy with hippocampal sclerosis (MTLE-HS) is one of the common epilepsy syndromes worldwide, which commonly arises from long-duration seizures, especially febrile status epilepticus, in the early life [3, 4]. Despite the great efforts to uncover the etiology, the underlying pathogenesis of MTLE-HS remains unclear. Current evidence suggests that MTLE-HS is closely related to febrile convulsion, with its pathogenesis involving epigenetic regulation and genetic susceptibility.
Gene chips are a valuable tool for analyzing expression of thousands of genes in an organism, screening gene targets for drugs, and predicting disease diagnosis. Accumulating slices of data have been generated by using gene chips and stored in public databases. In recent years, there has been an increased number of studies on epilepsy using bioinformatics approaches, which suggests that the integrated bioinformatics method is a powerful tool for mechanism-exploring studies. In this study, we set out to profile gene expression after febrile seizures in MTLE-HS based on microarray data from a public genome database using bioinformatics approaches.
Methods
Microarray data
Microarray data were obtained from the Gene Expression Omnibus (GEO) database, which is one of the largest and most comprehensive public genome databases containing array- and sequence-based data [5], by using keywords “temporal lobe epilepsy”, “febrile seizures”, and “febrile seizures”. Finally, the GSE28674 dataset (Agilent-014850 Whole Human Genome Microarray 4x44K G4112F version, Affymetrix GPL6480) was obtained and used in this study [6]. The GSE28674 dataset contained 6 samples of MTLE-HS with well-characterized antecedent prolonged febrile seizures, and 12 samples of no- MTLE-HS without previous febrile seizure.
DEG screening
GEO2R was used to screen differentially expressed genes (DEGs) between MTLE-HS with and without febrile seizure history using criteria of |log FC| (|log2Fold Change|) > 1 and adjusted P value < 0.05, where logFC = log (expr1)-log (expr2). The DEGs with logFC > 1 were considered to be up-regulated genes, and those with logFC<1 were considered with down-regulation.
Pathway enrichment and gene ontology (GO) analyses of DEGs
The Database for Annotation, Visualization and Integrated Discovery (DAVID) database (version 6.8), which provides systematic and comprehensive biological function annotation of genes (http://david.ncifcrf.gov) [7], and FunRich software were used to analyze the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway and GO enrichment of DEGs [8]. The KEGG database provides systematic functional information of genes. One of the characteristics of the KEGG database is to associate gene catalogs from genomes that have been completely sequenced with higher-level system functions at the cell, species, and ecosystem levels [9]. The GO database was used to analyze biological processes and annotation of genes [10]. In this study, the functional and biological implications of DEGs were analzyed with GO enrichment and KEGG pathway analysis using the DAVID online database and FunRich software. The cutoff criterion was set as P < 0.05.
Protein–protein interaction (PPI) network construction
The STRING database (version 11.0) was used to predict PPI networks of DEGs [11], and an interaction score > 0.4 was considered as statistically significant. The PPI networks were constructed by Cytoscape software [12] and the most significant module in the PPI network was identified using plug-in MCODE within Cytoscape [13] with criteria of MCODE score > 5, K-score = 2, Max depth = 100, degree cut-off = 2 and node score cut-off = 0.2. GO and KEGG pathway analyses of genes in the identified module were performed by using the DAVID online database.
Hub gene identification and analysis
Genes with the highest connectivity degree in the PPI network were identified as hub genes. The connectivity degree refers to the number of nodes connected to a specified node. The more nodes connected, the more likelihood the gene in a central position of the interaction network. Subsequently, DAVID online tool and Cytoscape were used to analyze the network and the biological process of hub genes.
Results
Identification of DEGs in MTLE-HS
Using criteria of |log FC| > 1 and adjusted P value < 0.05, 515 DEGs from the GSE28674 dataset were identified between samples of MTLE-HS with and without febrile seizure history, containing 25 down-regulated and 490 up-regulated genes in MTLE-HS with prolonged febrile seizure (Fig. 1, Additional file 1).

Differentially expressed genes (DEGs) in GSE28674
KEGG pathway and GO enrichment analyses of DEGs
GO cell component (CC) analysis revealed that the DEGs were mainly enriched in plasma membrane and synaptic vesicle and axon (Fig. 2a); GO molecular function (MF) analysis revealed that the DEGs were significantly enriched in voltage-gated ion channel activity, calcium ion binding and extracellular ligand-gated ion channel activity (Fig. 2b). GO biological process (BP) analysis revealed that the DEGs were mainly enriched in cell communication signal transduction and transport (Fig. 2c). The KEGG pathway enrichment analysis showed that the up-regulated DEGs were mainly enriched in Ras signaling pathway, retrograde endocannabinoid signaling, neuroactive ligand-receptor interaction, calcium signaling pathway and GABAergic synapse etc. (Table 1), while the downregulated DEGs were considered not statistically significant (P>0.05).

GO and KEGG pathway enrichment analysis of DEGs in MTLE-HS samples using FunRich and DAVID. a The cell component analysis of DEGs. b The molecular function of DEGs. c The biological process analysis of DEGs
PPI network construction and analysis
PPI data are often represented by a relationship graph where vertices represent proteins and the edges represent interactions between two proteins. The PPI network concerns interactions between proteins, which helps to discover core regulatory genes. Cytoscape was used to obtain the PPI network of DEGs (Fig. 3) and the most significant module (Fig. 4). The DAVID online tool was used to analyze the functions of the genes involved in this module. The results revealed that genes in the module were mainly enriched in synaptic vesicle cycle, nicotine addiction, amyotrophic lateral sclerosis and glutamatergic synapse (Table 2).

PPI network of DEGs

The most significant module identified by using the Cytoscape software
Hub gene selection and analysis
Five genes were selected as hub genes, including SNAP25, SLC32A1, SYN1, GRIN1 and GRIA1 (Fig. 5, Table 3). Heatmap showed that the hub genes were mainly expressed simultaneously in the fetal brain and adult frontal cortex (Fig. 6).

Hub genes in the most significant module

Heatmap of hub genes constructed using the FunRich software
Discussion
Febrile convulsion is one of the most common forms of morbidity in childhood, with an incidence rate of 2–5% in children worldwide, but it rarely occurs before 6 months or after 3 years of age. The febrile convulsions generally have better prognosis, but it can be transformed into epilepsy [14]. Early febrile convulsion may be related to the occurrence of later epilepsy and HS. Brain damage caused by early febrile convulsions may become the basis of late epilepsy and lead to the formation of HS [15]. Clinical long-term febrile seizure and the febrile status epilepticus often result in acute hippocampal damage and develop into hippocampal atrophy [16]. MTLE-HS is closely related to febrile seizures. The hippocampus may have abnormal formation and ultrastructure after a prolonged febrile seizure. However, whether the HS is a result or a cause of febrile seizures is still under debate [17]. HS caused by febrile convulsions is an important cause of drug-refractory temporal lobe epilepsy, posing a great economic and psychological burden on patients. However, the pathogenesis of HS caused by febrile convulsions may be that in the developing stage of brain, the myelin sheath is not completely generated, and the excitatory/inhibitory neurotransmitters have not reached a balance. Febrile convulsions in infants and young children can lead to neuronal loss and gliosis in the temporal lobe and hippocampus, a condition named as Ammon’s sclerosis. Autopsy results showed that the incidence of HS in patients without epilepsy was 9–10%, and the incidence of epilepsy was up to 30% [15].
The basic histopathological features of MTLE-HS found in surgically-removed specimens and autopsies of patients with intractable epilepsy include the following aspects: (1) loss of neurons; (2) adaptive changes in structure, such as germination, nerve formation and particle cell dispersion; (3) glial hyperplasia and glial cell function changes; (4) loss of blood-brain barrier integrity; and (5) neuroinflammation [18]. These characteristics have also been confirmed in the TLE animal model [19]. As an acquired epilepsy, the pathogenesis of MTLE-HS is very complex. The imbalance between excitation and inhibition of neurons leads to seizures, which is mainly related to the changes of ion channel, neurotransmitters and glial cells. Ion channel is the basis for the regulation of excitability in vivo, and the polymorphisms of genes encoding ion channels can affect the function of ion channels, thus increasing the genetic susceptibility to epilepsy. At present, it is believed that idiopathic epilepsy is an ion channel disease. Current studies have focused on the following aspects: (1) The hypothesis of neural network reconstruction: prolonged or repeated seizures lead to neuronal degeneration, glial proliferation, interneurons increasing, mossy fibers sprouting, increased synapses or false synaptic formation, thus leading to the remodeling of the hippocampal neural network structure, resulting in increased spontaneous electrical activity; (2) Altered ion channels, which could result in increased neuronal excitability; and (3) genetic factors. Among the above hypotheses, the neural network reconstruction has attracted increasing attention [20]. It is thought that the neural network remodeling can be divided into two aspects, the morphological change, such as neuron degeneration, glial proliferation and moss fiber budding; and the functional change, such as changes in the molecular level of functional proteins in hippocampal cells, which may affect the spontaneous discharge of neural networks after remodeling [21].
In recent years, a large number of new mutations of genes and population susceptibility genes have been identified. It is generally accepted that the temporal lobe epilepsy is influenced by both genetic and environmental factors. The genes related to epilepsy mainly include: (1) Neuronal ion channel-like genes KCNQ2, KCNQ3, KCNT1, KCNA1, SCN1A, CACNA1A and SCN2A [22]; (2) God-grade metatransposome-like genes GABRA1, GABRG2, CHRNA4 and CHRNB2 [23, 24]; (3) Energy metabolism genes mt-tRNA Lys and mt CSTB [25]; and (4) Other genes like LGI1 [26]. MTLE-HS is closely correlated to febrile seizures. The hippocampus may develop abnormalities in formation and ultrastructure after a prolonged febrile seizure. However, it remains unclear if HS is the result or a cause of febrile seizures. Therefore, it is important to identify potential biomarkers for diagnosis and determine whether febrile seizures efficiently cause HS. Microarray technology allows us to explore the MTLE-HS genetic alterations, and identify new biomarkers of disorders.
In this study, the microarray dataset GSE28674 was analyzed to identify DEGs between samples of MTLE-HS with and without febrile seizure history. Totally 515 DEGs were obtained, consisting of 25 down-regulated and 490 up-regulated genes. The up-regulated genes were mainly enriched in Ras signaling pathway, GABAergic synapse, retrograde endocannabinoid signaling, neuroactive ligand-receptor interaction and calcium signaling pathway, while the down-regulated DEGs were considered not statistically significant (P>0.05). Previous studies have reported that the Ras proteins act as molecular switches for signaling pathways and play a critical role in regulating differentiation and growth, cell proliferation, survival, or cytoskeletal dynamism. Activated Ras regulates a variety of cellular functions through phosphatidylinositol 3-kinase (PI3K), Raf and Ral guanine nucleotide-dissociation stimulator [27]. A previous study has demonstrated that the reduced expression of Ras-GRF1 may be involved in the pathogenesis of temporal lobe epilepsy [28]. GABA as the most abundant inhibitory neurotransmitter in the central nervous system of mammalian, can bind to three major classes of receptors: GABAA, GABAB, and GABAC receptors upon release. Fast GABA responses are mediated by chloride channel opening through GABAA and GABAC receptors, while slower GABA responses are mediated by G-protein activation and its effect on the second messenger systems through GABAB receptors [29, 30]. Several studies have demonstrated that the GABAergic synapses are closely related to various types of epilepsy, such as febrile seizures, idiopathic generalized epilepsies and early infantile epileptic encephalopathy [31, 32]. In addition, recent studies have further confirmed that the retrograde endocannabinoid signaling and calcium signaling play a major role in epileptogenesis [33, 34]. Therefore, we hypothesized that the above signaling pathways mediate the relationships between febrile convulsion and HS.
Comprehensive analyses of gene expression profiles allows us to identify potential molecular biomarkers. In this study, we selected 5 DEGs as hub genes, including SNAP25, SLC32A1, SYN1, GRIN1 and GRIA1, and they may play an important role in the process of HS caused by febrile convulsions.
SNAP25 is a 206-amino-acid protein, and its gene is located on chromosome 20. The SNAP25 protein is located at the presynaptic end of neurons, contributing to SNARE complex assembly that plays a crucial role in calcium-dependent exocytosis of synaptic vesicles, ensuring the effective release of neurotransmitters and the propagation of action potentials [35]. SNAP25 was first considered as a neuron-specific gene specifically expressed in the hippocampus of mice, and plays a crucial role in the functions of specific neuronal systems. SNAP25 is associated with proteins involved in vesicle docking and membrane fusion, regulates protein recovery on plasma membrane through its interactions with CENPF, and modulates the gating characteristics of the delayed rectifying voltage-dependent potassium channel KCNB1 in pancreatic β cells [36, 37]. The expression of SNAP25 is also correlated with presynaptic congenital myasthenic syndromes and congenital myasthenic syndrome-18 [38, 39]. Several studies have shown that SNAP25 also participates in epilepsy, and decreased SNAP25 expression will increase the susceptibility to epilepsy [40]. It has been reported that SNAP25 expression is significantly upregulated in infantile seizures [41]. In addition, a decrease in phosphorylation of SNAP-25 and dysregulated palmitoylation of SNAP-25 have been observed in rat hippocampus during seizures [42]. The GO annotations of the SNAP25 gene included SNAP receptor activity and calcium-dependent protein binding, and the related pathways to this gene are neurotransmitter release cycle and innate immune system.
SLC32A1, also known as vesicular inhibitory amino acid transporter or vesicular GABA transporter, is the only member of the SLC32 family and belongs to the eukaryote-specific superfamily of H+ coupled amino acid transporters, which also includes mammalian SLC36 and SLC38 transport protein. SLC32A1 is expressed in synaptic vesicles of GABAergic and glycinergic neurons, and in some endocrine cells, which can exchange protons with GABA or glycine. Although having a similar function in the loading of vesicular neurotransmitters, SLC32A1 is not associated with the vesicular glutamate transporter (VGLUT, SLC17) or the vesicular monoamine transporter/vesicular acetylcholine transporter (VMAT/VACHT, SLC18) [43]. SLC32A1 is a complete membrane protein involved in synaptic vesicle uptake of GABA and glycine [44]. Studies have shown that GABA is closely related to febrile seizures, and mutated GABA receptors have the characteristics of temperature-dependent transport, suggesting that patients with mutated GABA genes are more prone to FS. When high-frequency stimulation of the perforant path was performed on tissue samples from MTLE patients with HS, the inhibitory effect of GABA generated by dentate cells was weaker than that of patients without HS. It has been reported that the decrease of GABAergic current in dentate granule cells of the HS patients is due to the decreased GABA reuptake [45]. In the study of hippocampal specimens from temporal lobe epilepsy patients, in the presence of GABAA receptors blockade, a lower dose of dicholine is required to induce discharges in tissues with HS, reflecting the hyperexcitability in the moss fiber region [46]. Moss fiber sprouting is a common manifestation of brain development, but sprouting is also found in adult tissues with seizures. This plasticity may mainly manifest as a repair response to the loss of hippocampal neurons, but may eventually promote the occurrence of epilepsy. Moss fiber sprouting is considered as a key factor in repeated episodes of MTLE-HS. Under normal circumstances, less than 1% of mossy fibers have axon branches that enter the molecular layer, but in HS, mossy fiber side branches widely project into the molecular layer of the dentate gyrus, and make excitatory synaptic contacts with the apical dendrites and granular cell spines, essentially creating a local short circuit that promotes neuronal synchronization. This process is thought to be caused by hippocampal epilepsy and neuron loss [47].
Synapsin I (SYN1) is a neuronal phosphoprotein and is associated with the membranes of small synaptic vesicles. It is also a marker of synapse development, with its increased expression reflecting increases of synapses, synaptic connections and synaptic transmission [48]. Synapsins may play important roles in synaptogenesis, neuronal development, and synaptic neurotransmission and plasticity, and has been shown to be correlated with epilepsy [49]. Studies have demonstrated that SYN1 plays an important role in neurotransmitter release, axon formation and synapse formation in epileptic mouse model [50]. SYN1 also regulates the connection of synaptic vesicles to the cytoskeleton [51] and is involved in the development of neurons and the formation of synaptic contacts between neurons [52].
The human GRIN1 gene is located at chromosome 9q34.3 and contains 22 exons with a total length of about 31 kb. GRIN1 protein regulates the formation of synapses at early development, the maintenance of synapses plasticity, and the number of neurons and their connections. GRIN1 is a glutamate ionotropic receptor. GRIA1 is glutamate ionotropic receptor as well, which is associated with depression and cerebral cortical dysplasia. GRIA1 is related to transport to the Golgi and subsequent modification and vesicle-mediated transport [53]. Abnormalities of the glutamate system can affect neuronal plasticity and cause neurotoxicity [54]. It has been reported that GRIN1 is highly correlated with infantile spasms [55]. Seizure-caused damage and loss of neurons are mainly attributed to the excitotoxicity, transmission of glutamatergic neurotransmitters, and excessive Na+ and Ca2+, which lead to increased osmotic pressure and intracellular generation of free radicals, eventually leading to necrosis.
From the above analysis, it is reasonable to assume that the five hub genes have pivotal functions in febrile convulsion and HS. However, as the results of this study are based on bioinformatics, further studies exploring the functions of these genes are required.
Conclusion
In this study, we identified 515 DEGs in samples of MTLE-HS with versus without febrile seizure history. These DEGs had molecular functions in voltage-gated ion channel activity, extracellular ligand-gated ion channel activity and calcium ion binding, and they were involved in pathways of cell communication signal transduction and transport. We also identified five hub genes (SNAP25, SLC32A1, SYN1, GRIN1, and GRIA1) that were significantly expressed in the MTLE-HS with prolonged febrile seizures. The bioinformatics analysis in this study may help us to identify potential biomarkers and explore the potential mechanisms of MTLE-HS with prolonged febrile seizures.
Availability of data and materials
The datasets used in this study are available.
Abbreviations
- DAVID:
-
Database for Annotation, Visualization and Integrated Discovery
- DEGs:
-
Differentially expressed genes
- GABA:
-
Gamma aminobutyric acid
- GEO:
-
Gene Expression Omnibus
- GO:
-
Gene ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- MTLE-HS:
-
Mesial temporal lobe epilepsy with hippocampal sclerosis
- PPI:
-
Protein-protein interaction network
- PI3K:
-
Phosphatidylinositol 3-kinase
- SYN1:
-
Synapsin I
References
Hedrich UBS, Koch H, Becker A, Lerche H. Epileptogenesis and consequences for treatment. Nervenarzt. 2019;90(8):773–80.
Beghi E, Giussani G. Aging and the epidemiology of epilepsy. Neuroepidemiology. 2018;51(3–4):216–23.
Patterson KP, Baram TZ, Shinnar S. Origins of temporal lobe epilepsy: febrile seizures and febrile status epilepticus. Neurotherapeutics. 2014;11(2):242–50.
Baulac M. MTLE with HS in adult as a syndrome. Rev Neurol (Paris). 2015;171(3):259–66.
Edgar R, Domrachev M, Lash AE. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30(1):207–10.
Bando SY, Silva FN, Costa Lda F, Silva AV, Pimentel-Silva LR, Castro LH, et al. Complex network analysis of CA3 transcriptome reveals pathogenic and compensatory pathways in refractory temporal lobe epilepsy. PLoS One. 2013;8(11):e79913.
Huang DW, Sherman BT, Tan Q, Collins JR, Alvord WG, Roayaei J, et al. The DAVID gene functional classification tool: a novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 2007;8(9):R183.
Pathan M, Keerthikumar S, Chisanga D, Alessandro R, Ang CS, Askenase P, et al. A novel community driven software for functional enrichment analysis of extracellular vesicles data. J Extracell Vesicles. 2017;6(1):1321455.
Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. The gene ontology consortium. Nat Genet. 2000;25(1):25–9.
Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47(D1):D607–D13.
Smoot ME, Ono K, Ruscheinski J, Wang PL, Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011;27(3):431–2.
Bandettini WP, Kellman P, Mancini C, Booker OJ, Vasu S, Leung SW, et al. MultiContrast delayed enhancement (MCODE) improves detection of subendocardial myocardial infarction by late gadolinium enhancement cardiovascular magnetic resonance: a clinical validation study. J Cardiovasc Magn Reson. 2012;14:83.
Darkins A, Polkey CE. The relationship of transient hemiparesis following febrile convulsions in infancy to subsequent temporal lobectomy for intractable seizures. J Neurol Neurosurg Psychiatry. 1985;48(6):551–5.
Harvey AS, Grattan-Smith JD, Desmond PM, Chow CW, Berkovic SF. Febrile seizures and hippocampal sclerosis: frequent and related findings in intractable temporal lobe epilepsy of childhood. Pediatr Neurol. 1995;12(3):201–6.
VanLandingham KE, Heinz ER, Cavazos JE, Lewis DV. Magnetic resonance imaging evidence of hippocampal injury after prolonged focal febrile convulsions. Ann Neurol. 1998;43(4):413–26.
Heuser K, Cvancarova M, Gjerstad L, Tauboll E. Is temporal lobe epilepsy with childhood febrile seizures a distinctive entity? A comparative study. Seizure. 2011;20(2):163–6.
Maroso M, Balosso S, Ravizza T, Liu J, Aronica E, Iyer AM, et al. Toll-like receptor 4 and high-mobility group box-1 are involved in ictogenesis and can be targeted to reduce seizures. Nat Med. 2010;16(4):413–9.
Marchi N, Lerner-Natoli M. Cerebrovascular remodeling and epilepsy. Neuroscientist. 2013;19(3):304–12.
Thom M. Review: HS in epilepsy: a neuropathology review. Neuropathol Appl Neurobiol. 2014;40(5):520–43.
Taylor PN, Han CE, Schoene-Bake JC, Weber B, Kaiser M. Structural connectivity changes in temporal lobe epilepsy: spatial features contribute more than topological measures. Neuroimage Clin. 2015;8:322–8.
Lauxmann S, Boutry-Kryza N, Rivier C, Mueller S, Hedrich UB, Maljevic S, et al. An SCN2A mutation in a family with infantile seizures from Madagascar reveals an increased subthreshold Na(+) current. Epilepsia. 2013;54(9):e117–21.
Rozycka A, Dorszewska J, Steinborn B, Lianeri M, Winczewska-Wiktor A, Sniezawska A, et al. Association study of the 2-bp deletion polymorphism in exon 6 of the CHRFAM7A gene with idiopathic generalized epilepsy. DNA Cell Biol. 2013;32(11):640–7.
Arlier Z, Bayri Y, Kolb LE, Erturk O, Ozturk AK, Bayrakli F, et al. Four novel SCN1A mutations in Turkish patients with severe myoclonic epilepsy of infancy (SMEI). J Child Neurol. 2010;25(10):1265–8.
Hypponen J, Aikia M, Joensuu T, Julkunen P, Danner N, Koskenkorva P, et al. Refining the phenotype of Unverricht-Lundborg disease (EPM1): a population-wide Finnish study. Neurology. 2015;84(15):1529–36.
Michelucci R, Pulitano P, Di Bonaventura C, Binelli S, Luisi C, Pasini E, et al. The clinical phenotype of autosomal dominant lateral temporal lobe epilepsy related to reelin mutations. Epilepsy Behavior. 2017;68:103–7.
Skaper SD. The neurotrophin family of neurotrophic factors: an overview. Methods Mol Biol. 2012;846:1–12.
Zhu Q, Wang L, Xiao Z, Xiao F, Luo J, Zhang X, et al. Decreased expression of Ras-GRF1 in the brain tissue of the intractable epilepsy patients and experimental rats. Brain Res. 2013;1493:99–109.
Chalifoux JR, Carter AG. GABAB receptor modulation of synaptic function. Curr Opin Neurobiol. 2011;21(2):339–44.
Luscher B, Fuchs T, Kilpatrick CL. GABAA receptor trafficking-mediated plasticity of inhibitory synapses. Neuron. 2011;70(3):385–409.
Padgett CL, Slesinger PA. GABAB receptor coupling to G-proteins and ion channels. Adv Pharmacol. 2010;58:123–47.
Dejanovic B, Lal D, Catarino CB, Arjune S, Belaidi AA, Trucks H, et al. Exonic microdeletions of the gephyrin gene impair GABAergic synaptic inhibition in patients with idiopathic generalized epilepsy. Neurobiol Dis. 2014;67:88–96.
Meng F, You Y, Liu Z, Liu J, Ding H, Xu R. Neuronal calcium signaling pathways are associated with the development of epilepsy. Mol Med Rep. 2015;11(1):196–202.
von Ruden EL, Jafari M, Bogdanovic RM, Wotjak CT, Potschka H. Analysis in conditional cannabinoid 1 receptor-knockout mice reveals neuronal subpopulation-specific effects on epileptogenesis in the kindling paradigm. Neurobiol Dis. 2015;73:334–47.
Chang JY, Stamer WD, Bertrand J, Read AT, Marando CM, Ethier CR, et al. Role of nitric oxide in murine conventional outflow physiology. Am J Physiol Cell Physiol. 2015;309(4):C205–14.
Zhao N, Hashida H, Takahashi N, Sakaki Y. Cloning and sequence analysis of the human SNAP25 cDNA. Gene. 1994;145(2):313–4.
Karmakar S, Sharma LG, Roy A, Patel A, Pandey LM. Neuronal SNARE complex: a protein folding system with intricate protein-protein interactions, and its common neuropathological hallmark, SNAP25. Neurochem Int. 2019;122:196–207.
Shen XM, Selcen D, Brengman J, Engel AG. Mutant SNAP25B causes myasthenia, cortical hyperexcitability, ataxia, and intellectual disability. Neurology. 2014;83(24):2247–55.
Engel AG. Congenital Myasthenic syndromes in 2018. Curr Neurol Neurosci Rep. 2018;18(8):46.
Corradini I, Donzelli A, Antonucci F, Welzl H, Loos M, Martucci R, et al. Epileptiform activity and cognitive deficits in SNAP-25(+/−) mice are normalized by antiepileptic drugs. Cereb Cortex. 2014;24(2):364–76.
Wang J, Wang J, Zhang Y, Yang G, Shang AJ, Zou LP. Proteomic analysis on infantile spasm and prenatal stress. Epilepsy Res. 2014;108(7):1174–83.
Kataoka M, Kuwahara R, Matsuo R, Sekiguchi M, Inokuchi K, Takahashi M. Development- and activity-dependent regulation of SNAP-25 phosphorylation in rat brain. Neurosci Lett. 2006;407(3):258–62.
Gasnier B. The SLC32 transporter, a key protein for the synaptic release of inhibitory amino acids. Pflugers Arch. 2004;447(5):756–9.
Jellali A, Stussi-Garaud C, Gasnier B, Rendon A, Sahel JA, Dreyfus H, et al. Cellular localization of the vesicular inhibitory amino acid transporter in the mouse and human retina. J Comp Neurol. 2002;449(1):76–87.
Williamson A, Patrylo PR, Spencer DD. Decrease in inhibition in dentate granule cells from patients with medial temporal lobe epilepsy. Ann Neurol. 1999;45(1):92–9.
Franck JE, Pokorny J, Kunkel DD, Schwartzkroin PA. Physiologic and morphologic characteristics of granule cell circuitry in human epileptic hippocampus. Epilepsia. 1995;36(6):543–58.
Buckmaster PS. Does mossy fiber sprouting give rise to the epileptic state?[J]. Adv Exp Med Biol. 2014;813:161–8.
Hedegaard C, Kjaer-Sorensen K, Madsen LB, Henriksen C, Momeni J, Bendixen C, et al. Porcine synapsin 1: SYN1 gene analysis and functional characterization of the promoter. FEBS Open Bio. 2013;3:411–20.
Fassio A, Patry L, Congia S, Onofri F, Piton A, Gauthier J, et al. SYN1 loss-of-function mutations in autism and partial epilepsy cause impaired synaptic function. Hum Mol Genet. 2011;20(12):2297–307.
Li L, Chin LS, Shupliakov O, Brodin L, Sihra TS, Hvalby O, et al. Impairment of synaptic vesicle clustering and of synaptic transmission, and increased seizure propensity, in synapsin I-deficient mice. Proc Natl Acad Sci U S A. 1995;92(20):9235–9.
Schiebler W, Jahn R, Doucet JP, Rothlein J, Greengard P. Characterization of synapsin I binding to small synaptic vesicles. J Biol Chem. 1986;261(18):8383–90.
Valtorta F, Iezzi N, Benfenati F, Lu B, Poo MM, Greengard P. Accelerated structural maturation induced by synapsin I at developing neuromuscular synapses of Xenopus laevis. Eur J Neurosci. 1995;7(2):261–70.
Barkus C, Sanderson DJ, Rawlins JN, Walton ME, Harrison PJ, Bannerman DM. What causes aberrant salience in schizophrenia? A role for impaired short-term habituation and the GRIA1 (GluA1) AMPA receptor subunit. Mol Psychiatry. 2014;19(10):1060–70.
Platzer K, Lemke JR. GRIN1-related neurodevelopmental disorder. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2020. 2019.
Paciorkowski AR, Thio LL, Dobyns WB. Genetic and biologic classification of infantile spasms. Pediatr Neurol. 2011;45(6):355–67.
Acknowledgements
Not applicable.
Funding
The study was supported by the Sanming Project of Medicine in Shenzhen (No.SZSM201911003)National Natural Science Foundation of China (No. 81571266, 81771405).
Author information
Authors and Affiliations
Contributions
Liemin Zhou conceived the idea to this paper; Yinchao Li collected and analyzed the data, and drafted the paper. Chengzhe Wang, Peiling Wang and Xi Li participated in the information registration and performed the statistical analysis. The authors read and approved the final manuscript.
Authors’ information
Liemin Zhou, Chief physician, Doctoral tutor, Director of Epilepsy Center of the 7th Affiliated Hospital, Sun Yat-sen Universit.
Yinchao Li, Masters of the 7th Affiliated Hospital, Sun Yat-sen University.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
We consent the publication in Acta Epileptologica.
Competing interests
We do not have any competing interests.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, Y., Wang, C., Wang, P. et al. Effects of febrile seizures in mesial temporal lobe epilepsy with hippocampal sclerosis on gene expression using bioinformatical analysis. Acta Epileptologica 2, 20 (2020). https://doi.org/10.1186/s42494-020-00027-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s42494-020-00027-9