
Abstract
Cardiac hypertrophy is a life-threatening disorder and is frequently observed in patients with chronic kidney disease (CKD). Much attention has been focused on the derangement in hormonal factors, including aldosterone and FGF23, as novel causes of cardiac hypertrophy in CKD. Plasma aldosterone concentrations are elevated as renal function declines. Although aldosterone antagonists are available for the treatment of hypertension with cardiac hypertrophy, concern remains regarding the possible occurrence of serious hyperkalemia. Alternatively, certain types of calcium channel blockers suppress aldosterone synthesis or exert blocking action for mineralocorticoid receptors and could halt the progression of cardiac dysfunction. Recently, FGF23 is shown to be elevated as CKD progresses and may be responsible for the development of cardiac hypertrophy and heart failure. Furthermore, FGF23 not only inhibits the renal expression of angiotensin converting enzyme 2 but also enhances renin gene transcription, both of which could accelerate renin-angiotensin-aldosterone system. Although the increase in serum phosphate concentrations is a pivotal stimulus for FGF23 production, recent studies suggest that reduced iron status and elevated aldosterone levels, frequently seen in patients with CKD or on dialysis, might also contribute to the elevation in serum FGF23 levels. Conversely, phosphate binders and appropriate iron status could reduce serum FGF23, potentially leading to the alleviation of cardiac hypertrophy and heart failure. In conclusion, novel therapeutic strategies associated with aldosterone and FGF23 may confer a benefit in the management of cardiac disorders in CKD.
Similar content being viewed by others


Background
Chronic kidney disease (CKD) is a life-threatening disorder and relentlessly progresses to end-stage kidney disease requiring renal replacement therapy. A growing body of evidence has been accumulated that CKD is closely associated with increased risk of cardiovascular events and death. Go et al. [1] demonstrated that the event rate inversely parallels the level of renal function. Likewise, cardiovascular events are associated with the reduction in glomerular filtration rate (GFR) among Japanese population [2, 3]. Cardiovascular disease observed in CKD includes a variety of disorders such as heart failure and cardiac hypertrophy. Since cardiac hypertrophy per se is more prevalent as renal function deteriorates and reflects an ominous outcome with increased mortality [4, 5], the alleviation of this disorder would offer improved survival to CKD patients.
Although hemodynamic derangement such as systemic hypertension and volume overload plays a major role in the development of cardiac hypertrophy, several lines of recent studies indicate that humoral factors also contribute to the development of cardiac hypertrophy. For example, angiotensin II is a well-known factor that exerts direct hypertrophic action on cardiomyocytes [6, 7]. Further evidence has accrued that aldosterone, a traditional hormone regulating serum electrolyte balance, not only induces renal glomerular hypertension [8] but also causes cardiac hypertrophy [9, 10] and heart failure [11]. Because such humoral factors are often elevated in CKD [12,13,14,15], the strategy to counter the action of these factors would improve various perturbed conditions in CKD and is currently proposed as a milestone treatment of CKD, particularly with the use of renin-angiotensin system (RAS) inhibitors [10, 11, 16, 17].
It is well recognized that CKD is accompanied by a variety of disturbance of the internal milieu, including electrolyte disorders. Despite impaired ability of renal excretory function, serum phosphate levels remain relatively unchanged until GFR falls below half of the normal level. Recent studies disclose that fibroblast growth factor 23 (FGF23) contributes importantly to the regulation of the serum phosphate concentration by inhibiting the phosphate reabsorption in the proximal tubule, which mitigates the tendency toward phosphate retention in CKD [18, 19]. Although this mechanism teleologically serves to act as an adaptive regulatory factor to maintain serum phosphate levels constant, further deterioration of CKD causes the elevation in serum phosphate concentrations, which hence would stimulate FGF23 production. Furthermore, of importance is the finding that elevated serum FGF23 concentrations are associated with the development of cardiac disorders [20,21,22,23]. Thus, apparently homeostatic mechanism for phosphate metabolism may act to aggravate cardiac disease, leading to cardiac hypertrophy and heart failure.
In this review, we survey the role of aldosterone in the development of cardiac hypertrophy in CKD and evaluate the therapeutic strategy for aldosterone blockade. Furthermore, recent findings regarding the effect of FGF23 on cardiac hypertrophy as well as the modulatory factors or therapeutic tools affecting FGF23 production are also discussed.
Aldosterone in CKD
Canonical concept of aldosterone is typically referred to as the hormone regulating electrolyte metabolism in the epithelial cell, i.e., sodium and potassium balance in renal tubular cells, intestinal epithelial cells, and sweat glands. Among these, renal distal and collecting tubules are the major target sites of action of aldosterone, where aldosterone facilitates sodium reabsorption through epithelial sodium channels (ENaC) and concomitantly potassium excretion mainly through ROMK channels [24]. Furthermore, aldosterone upregulates Na/K/ATPase that extrudes sodium from the cell to interstitial spaces as well as augments the uptake of potassium into the cell [25]. The overall action of aldosterone comprises volume expansion, hypertension, and the decrease in serum potassium levels, as are evident in primary aldosteronism.
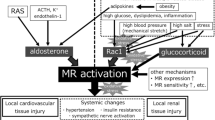
Plasma aldosterone levels are regulated by a couple of factors (Fig. 1). Angiotensin II and ACTH represent major regulatory hormones stimulating aldosterone synthesis. Furthermore, high serum potassium levels induce aldosterone release from adrenal cortical gland. In addition, previous studies demonstrated that plasma aldosterone concentrations were elevated in CKD [15, 26,27,28,29,30] and were correlated inversely with renal function [15] (Fig. 1). Although high potassium obviously constitutes a stimulus for aldosterone release in CKD, reduced aldosterone excretion is reported as a possible cause of elevated plasma aldosterone concentrations [31]. Alternatively, Wesson and Simoni [27] showed that acid retention during kidney failure induced aldosterone production and elevated plasma aldosterone concentrations. Furthermore, Hosoya et al. [15] have recently demonstrated that the expression of the aldosterone-producing enzyme CYP11B2 in the adipose tissue of 5/6-nephrectomized rats is upregulated, leading to increased tissue aldosterone content. Finally, aldosterone breakthrough phenomenon (i.e., an elevation in plasma aldosterone levels 3–6 months after the administration of RAS inhibitors) could be a possible mechanism for aldosterone dysregulation [32, 33]. Taken together, these mechanisms may act in concert to enhance aldosterone activity in CKD.

Tissue and plasma aldosterone levels in CKD. Besides multiple factors stimulating aldosterone synthesis and release, novel mechanisms contribute to the elevated aldosterone levels in CKD. Impaired renal function is associated with reduced aldosterone excretion [31] and acid retention, the latter causing enhanced aldosterone production [27, 28]. Furthermore, the expression of CYP11B2, an aldosterone-producing enzyme, in the adipose tissue of 5/6-nephrectomized rats is upregulated, resulting in increased tissue aldosterone content [15]. Finally, the continued use of ACE inhibitors or ARB may cause paradoxical increases in plasma aldosterone concentrations through aldosterone breakthrough phenomenon [32, 33]. These mechanisms would act in concert to elevate plasma aldosterone concentrations as renal function declines. The schematic bar graphs and line graphs depicted therein are constructed de novo from Hosoya et al. [15]
The kidney plays an important role in potassium homeostasis. In subjects with intact kidney function, approximately 90% of potassium is excreted from the kidney where aldosterone contributes importantly to potassium excretion, and only 10% of potassium is secreted from the intestine. In patients with end-stage kidney disease, including dialysis patients, however, potassium secretion from the large intestine is considerably increased [29, 34, 35], in which KCa1.1 (BK) channels are largely involved [36]. Whereas aldosterone is shown to enhance the colonic BK channel activity [37, 38] and contributes at least partly to the increased colonic potassium excretion [39], there are also reported several studies showing a modest role of aldosterone in colonic potassium secretion in both predialysis [29, 40] and dialysis CKD patients [35]. It requires further discussion whether aldosterone blockade causes perilous levels of hyperkalemia in dialysis patients with end-stage kidney disease (see below).
Non-epithelial action of aldosterone
In addition to conventional action in epithelial cells, aldosterone is demonstrated to exert non-epithelial action in various organs. In the study evaluating the role of aldosterone in patients with heart failure, treatment with a mineralocorticoid receptor blocker (spironolactone) on top of conventional therapies (i.e., ACE inhibitors plus diuretics) conferred a profound benefit in terms of survival rate [11]. Since this clinical trial (i.e., RALES), a novel idea has emerged that aldosterone constitutes a critical determinant of cardiac function in patients with heart failure. Similarly, Zannad et al. [41] showed that a more selective aldosterone blocker, eplerenone, reduced the risk of death and hospitalization among patients with systolic heart failure and mild symptoms. It has also been demonstrated that aldosterone causes detrimental effects on vascular tension (enhanced vascular tone) [42] and glucose metabolism (insulin resistance) [15], both of which could influence mortality and morbidity.
Besides the critical role of aldosterone in survival rates in heart failure [11, 41], accumulating evidence indicates that aldosterone is responsible for the development of cardiac hypertrophy and impaired cardiac function. In primary aldosteronism in which overproduction of aldosterone causes hypertension, left ventricular (LV) hypertrophy can be induced independently of systemic hypertension [43, 44]. Furthermore, in patients with CKD, plasma aldosterone concentrations are inversely associated with GFR [15, 30] and parallel LV mass index [30]. Alternatively, aldosterone blockade with spironolactone elicited a regression of LV hypertrophy despite no changes in systemic blood pressure in diabetic patients with microalbuminuria [10] (Table 1). Furthermore, spironolactone treatment significantly ameliorated LV mass index and/or cardiac function not only in patients with early stage CKD [45, 46] but also in patients with advanced CKD on hemodialysis [47,48,49] or peritoneal dialysis [50,51,52]. More importantly, Matsumoto et al. [53] elegantly demonstrated that 3-year treatment with spironolactone reduced the death rate from cardiovascular or cerebrovascular events in patients on hemodialysis in Dialysis Outcomes Heart Failure Aldactone Study (DOHAS). In concert, these observations suggest that aldosterone is responsible substantially for the development of cardiac hypertrophy and remodeling in humans.
Although aldosterone has a great impact on cardiovascular organs, multiple signal transduction systems appear to be involved in the non-epithelial action of aldosterone. Several lines of experimental studies show that aldosterone promotes the signal transduction of ERK pathways [54, 55] and generates reactive oxygen species [56] and inflammation [57], all of which cause cardiac hypertrophy and remodeling. Very recently, it has been demonstrated that aldosterone upregulates TNF receptor-associated factor 3 interacting protein 2 (TRAF3IP2), which serves as an upstream regulator of multiple signaling components, including I kappa B kinase, JNK and c-Jun, and then stimulates the production of IL-18, IL-6, and CTGF [58, 59]. These results hence indicate a pivotal role of TRAF3IP2 and the multiple subordinate signal transduction pathways described above in mediating the aldosterone-induced adverse cardiac effects. Of interest, the mineralocorticoid receptor pathway is also stimulated by Rac1 activation through salt loading [60] and obesity [61] as a ligand-independent modulator without alterations in systemic aldosterone status. Furthermore, the Rac1-mediated activation of the mineralocorticoid receptor in the myocardium is responsible for the development of heart failure [62]. It follows therefore that mineralocorticoid receptor activation, whether a ligand-dependent or not, stimulates the downstream signaling pathways associated with growth and inflammation and induces cardiac hypertrophy and impaired contractility.
Aldosterone blockade in CKD
As indicated above, aldosterone blockade reduces cardiac hypertrophy and improves cardiac function in patients on hemodialysis [47,48,49] and peritoneal dialysis [50,51,52] (Table 1). Furthermore, DOHAS trial clearly demonstrates that aldosterone blockade by spironolactone prevents cardiovascular events and ameliorates the survival rate in hemodialysis patients [53]. Caveat is in order, however, because of the potential risk for hyperkalemia when the blockers are given to CKD patients. Thus, potassium secretion from the intestine is increased in patients with advanced CKD or on dialysis therapy [29, 34, 35], and mineralocorticoid receptor blockers are reported to suppress this mechanism [38]. Indeed, several studies have reported that the administration of spironolactone is associated with increased incidence of hyperkalemia in patients on maintenance hemodialysis therapy [63,64,65] although pronounced hyperkalemia (serum potassium ≥ 6 mEq/L) does not occur commonly [66]. Notably, there are also reported a substantial number of studies showing that spironolactone does not cause significantly higher levels of serum potassium in patients on hemodialysis [47,48,49] or peritoneal dialysis therapy [50, 51, 67, 68], when compared with placebo (Table 1). Taken together, the use of aldosterone blockers is teleologically reasonable in terms of cardiovascular protection, although the adverse effect of these blockers might hamper the wide-spread use of this type of agents in patients with CKD.
Alternatively, there exist several types of antihypertensive agents that act to inhibit aldosterone synthesis and/or release (Table 2) [69]. It is now well established that certain types of calcium channel blockers, including efonidipine [70], benidipine [71], azelnidipine [72], and cilnidipine [73], inhibit aldosterone synthesis in adrenocortical cells. Furthermore, clinical studies show that these agents decrease serum aldosterone levels in hypertensive patients [74,75,76,77,78,79]. Of interest, these calcium channel blockers are endowed with the ability to inhibit not only L-type but also T-type (efonidipine, benidipine, azelnidipine) or N-type calcium channels (benidipine, cilnidipine), whereas conventional calcium channel blockers such as nifedipine and amlodipine exert inhibitory action solely on L-type calcium channels [80,81,82]. Additionally, several calcium channel blockers, including nifedipine and benidipine, are shown to compete with aldosterone for mineralocorticoid receptor binding and block aldosterone activity [83, 84] (Table 2). Thus, divergent inhibitory action on calcium channel subtypes (i.e., L-, T-, and N-type calcium channels) and distinct antagonistic action on mineralocorticoid receptors inherent in certain calcium channel blockers could provide additive cardiovascular benefits in CKD patients. Indeed, in our preliminary study, we found that T-type calcium channel blockers (efonidipine, benidipine, azelnidipine) and nifedipine reduced LV mass index more markedly than other calcium channel blockers in hemodialysis patients (unpublished observation). This presumption, however, requires further investigations showing that these calcium channel blockers could contribute to the prevention of cardiac hypertrophy.
FGF23 and phosphate in CKD
FGF23 is identified as a glycoprotein hormone that has been discovered as a member of the FGF family [85]. The subsequent investigations have unveiled an important role of FGF23 in the homeostatic mechanism of serum phosphate levels [18, 19]. The conventional hypothesis, i.e., “trade-off theory” [86, 87], where secondary hyperparathyroidism is assumed to play a central role in phosphate metabolism in CKD, therefore, has been updated by the introduction of FGF23 to the concept of the phosphate metabolism in CKD.
Serum FGF23 levels have been shown to rise at early stages of CKD. Several studies demonstrate that serum FGF23 is elevated even prior to the stage when serum parathyroid hormone rises [19, 88]. Although the precise cellular mechanisms for the release and synthesis of FGF23 remain fully undetermined, the elevation in serum phosphate and parathyroid hormone constitute determinants that trigger the release of FGF23 from osteocytes and osteoblasts (Fig. 2). Because FGF23 is a potent phosphaturic hormone that inhibits phosphate reabsorption through Na/P cotransporter 2a/c in the proximal tubule [18, 86, 89], FGF23 would serve to mitigate hyperphosphatemia entailing impaired renal function. FGF23 also suppresses vitamin D activity by inhibiting renal 1α-hydroxylase (the enzyme that converts 25-hydroxyvitamin D3 to its active form) and stimulating 24-hydroxylase (the enzyme degrading to inactive form) [90], leading to the decrease in phosphate and calcium absorption from the intestine. Although FGF23 can inhibit parathyroid hormone production [88, 91], the effects of suppressed vitamin D activity along with decreased Ca levels would govern the serum parathyroid hormone level more robustly in CKD, which results in elevated parathyroid hormone levels characteristics of the hormonal profiles seen in CKD patients [92, 93].

Schematic diagram illustrating the possible relationship of the hormonal factors associated with cardiac disorders in CKD. In CKD, both aldosterone and FGF23 are elevated, which directly and/or indirectly causes cardiac hypertrophy and heart failure. There is reported substantial interaction between aldosterone and FGF23, directly from aldosterone to FGF23 and indirectly from FGF23 to aldosterone through circulating volume status. Additionally, elevated FGF23 inhibits vitamin D activity and angiotensin converting enzyme 2 (ACE2) expression, both of which could result in augmentation in renin-angiotensin-aldosterone system. Aldosterone blockade by mineralocorticoid receptor antagonists and T-/N-type Ca channel blockers could mitigate cardiac hypertrophy. Phosphate binders not containing Ca reduce FGF23 and could potentially ameliorate cardiac hypertrophy, while those containing iron exert FGF23-lowering action not only through reducing serum phosphate but also through iron supplementation. NCC, sodium chloride cotransporter; ACE2, angiotensin converting enzyme 2; RAS, renin-angiotensin system; PTH, parathyroid hormone. The numbers in brackets denote the references cited in the text
FGF23 and sodium in CKD
In addition to the phosphaturic action in renal proximal tubules, FGF23 is found to exert sodium retaining action in distal tubular segments. Thus, Andrukhova et al. [94] have recently demonstrated that FGF23 upregulates the sodium chloride cotransporter (NCC) in distal tubules, which conceivably results in systemic volume expansion and hypertension (Fig. 2). This finding encompasses an important issue because FGF23 could cause the suppression of RAS due to systemic volume expansion and subsequently decrease plasma aldosterone levels [95]. In contrast, a positive correlation between FGF23 and aldosterone concentrations is also reported in patients with CKD and heart failure [96]. In this regard, CKD is demonstrated to be associated with decreased renal expression of Klotho [97, 98]. Since the action of FGF23 on NCC requires the integrity of the FGF receptor/Klotho complex [94], the ability of FGF23 to promote sodium retention and the subsequent development of hypertension may depend on intact FGF receptor/Klotho complex activity. Indeed, the observation that plasma aldosterone levels are elevated in advanced CKD [12,13,14,15, 26, 27, 29] suggests the diminished ability of elevated FGF23 to induce volume expansion, possibly due to reduced Klotho expression in the kidney.
FGF23 and RAS/aldosterone
Recent investigations reveal a cross talk between FGF23 and RAS/aldosterone [99] (Fig. 2). Dai et al. [100] demonstrated that FGF23 suppressed the renal expression of angiotensin converting enzyme 2 (ACE2), the enzyme mediating the conversion mainly from angiotensin II to angiotensin-(1-7), in FGF23-transgenic mice. The suppression of ACE2 activity results in the decrease in vasodilatory angiotensin-(1-7) and the increase in angiotensin II [101, 102]. It is inferred therefore that in CKD, where plasma FGF23 is elevated, altered balance between angiotensin II and angiotensin-(1-7) might play a part in hypertension and volume retention. Furthermore, accumulating evidence indicates that vitamin D inhibits RAS by downregulating renin gene transcription [99, 103, 104]. Because FGF23 suppresses vitamin D activity, elevated plasma FGF23 would augment RAS and possibly aldosterone as well and may play a role in the development of hypertension in CKD.
Finally, the interaction between FGF23 and aldosterone merits comment. Imazu et al. [96] showed that serum FGF23 levels correlated with plasma aldosterone concentrations in patients with CKD and heart failure. An in vitro study also demonstrates that FGF23 transcription is upregulated by aldosterone and is inhibited by an aldosterone receptor blocker (eplerenone) in cultured osteoblasts [105]. Although clinical evidence endorsing a causative role of aldosterone in FGF23 production remains insufficient, these findings are consistent with the notion that aldosterone contributes to the enhanced FGF23 production in CKD (Fig. 2).
FGF23 and cardiac hypertrophy
Cardiac hypertrophy is a critical complication that is frequently observed in CKD [106, 107]. Cardiac hypertrophy develops beginning at early stages of CKD and is quite common in patients on dialysis therapy. In addition to the traditional determinants causing cardiac hypertrophy, including hypertension, renin-angiotensin system [6, 7], and chronic anemia [108], aldosterone is also established as a crucial factor for cardiac hypertrophy [9, 10, 15, 30, 43]. Furthermore, parathyroid hormone is suggested as a cause of cardiac hypertrophy in dialysis patients [109, 110], although contradictory results are also reported [111, 112]. Of interest, mineralocorticoid receptor blockade reduces serum parathyroid hormone levels in normal subjects as well as in patients with CKD and heart failure, suggesting that aldosterone stimulates parathyroid hormone production [113, 114]. Alternatively, parathyroid hormone enhances the aldosterone secretion from adrenal cortex [115, 116]. Clinical implications of these interactions in the development of cardiac hypertrophy, however, remain undetermined [117].
More recently, much attention has been focused on the role of FGF23 since this substance not only participates in the phosphate homeostasis but also induces cardiac hypertrophy (Table 3) [21, 22, 118,119,120,121,122]. Thus, Gutierrez et al. [21] discovered that there existed a close relationship between serum FGF23 levels and LV mass index in patients with CKD. Faul et al. [22] also demonstrated that LV mass index was increased as serum FGF23 levels were elevated. This relationship was also observed in patients on maintenance hemodialysis. Finally, intravenous injection of FGF23 caused cardiac hypertrophy in mice, and the administration of an FGF receptor antagonist (PD173074) prevented the development of the CKD (i.e., 5/6 nephrectomy)-induced cardiac hypertrophy. Of importance, elevated serum FGF23 levels are causally linked to reduced ejection fraction [22]. Collectively, these observations provide conclusive evidence for the role of FGF23 in the development of cardiac disorders in CKD.
Although a growing body of evidence has been accumulated regarding the role of FGF23 in cardiac hypertrophy in CKD, the mechanism responsible for the cardiac disorder remains undetermined fully. In experimental models of mice, Faul et al. [22] demonstrated that the FGF23-induced cardiac hypertrophy was abrogated by a phospholipase Cγ inhibitor (U73122) and a calcineurin inhibitor (cyclosporine A), but not by a MAP kinase inhibitor (PD98059), a PI3 kinase inhibitor (wartmannin), or an Akt inhibitor (A6730). Furthermore, pan FGF receptor blockade by PD173074 reduced LV mass and the cardiac expression of genes associated with LV hypertrophy [123], and the receptor involved was identified as FGF receptor 4 [124]. These findings lend support to the premise that FGF23-induced cardiac hypertrophy is mediated by the FGF receptor 4/PLCγ/calcineurin pathway.
Role of phosphate binders in FGF23 levels and iron metabolism
Great progress in the therapeutic modalities for phosphate binders offers more favorable management of serum phosphate levels in CKD. Following the established use of Ca carbonate, new (i.e., second generation) phosphate binders, including sevelamer, bixalomer, and lanthanum carbonate, have come into common use. Because of the physiological interaction between serum phosphate and FGF23, it is judiciously anticipated that phosphate binders should reduce serum FGF23 levels [125,126,127,128,129]. In this regard, Ca load is reported to be associated with an elevation in serum FGF23 levels [130, 131] (Fig. 2). Furthermore, a cross-sectional study shows that FGF23 levels correlate with serum Ca ion concentrations and Ca-phosphate product [132]. Thus, phosphate binders containing Ca (e.g., Ca carbonate) may have less ameliorating impact on serum FGF23 levels than those without Ca (e.g., sevelamer, lanthanum) in both pre-dialysis [133] and dialysis patients [125, 128, 129].
Although FGF23 constitutes an important determinant inducing cardiac hypertrophy, whether the improvement in serum FGF23 levels by phosphate binders exerts beneficial action on cardiac hypertrophy is a matter of controversy (Table 3). Thus, it has been reported that 40-week treatment with sevelamer decreases serum FGF23 levels but fails to reduce LV mass in patients with stage 3 CKD, though the inability to ameliorate LV mass might be attributable to the insufficient treatment period [134]. Furthermore, sevelamer and lanthanum are shown to reduce serum phosphate concentrations and ameliorate LV hypertrophy without changes in FGF23 levels in experimental animals [135, 136]. Of interest, a strong association between serum phosphate concentrations and LV mass is demonstrated in humans [137, 138] and animals [139], and hyperphosphatemia per se could be a potential risk factor causing cardiac hypertrophy [140, 141]. Accordingly, the ability of phosphate binders to alleviate cardiac hypertrophy may vary, depending on the responsiveness of serum FGF23 and/or phosphate to these drugs. Further investigations are required to clarify this important issue.
Recently, iron-containing phosphate binders, including ferric citrate hydrate and sucroferric oxyhydroxide, have been developed and are actually available in clinical practice [142, 143]. Because of the nature of iron loss during hemodialysis sessions, the use of this type of phosphate binders appears reasonable in such condition since iron is dissociated from the binders and then absorbed in part from the intestine. Of note, iron deficiency and the subsequent anemia are associated with the elevation in plasma FGF23 concentrations [95, 144,145,146,147] and the upregulation of bone FGF23 mRNA expression (Table 4) [147, 148]. An in vitro study also shows that osteocytes in the medium containing low iron produce greater FGF23 mRNA expression [149]. Conversely, oral [146, 150, 151], but not intravenous [152, 153], iron supplementation is associated with a decrease in serum FGF23 levels. Additionally, switching from sevelamer to ferric citrate hydrate has recently been shown to replenish iron status and reduce circulating FGF23 levels independently of serum phosphate concentrations in hemodialysis patients [154]. In concert, an appropriate level of iron status is required to suppress serum FGF23 levels and potentially to prevent the development of cardiac hypertrophy.
In this regard, hepcidin, a regulatory molecule that inhibits iron absorption in the duodenum and iron recruitment from the liver and the reticuloendothelial system, is elevated in CKD [155, 156]. Furthermore, serum hepcidin levels are tightly associated with serum phosphate concentrations [157], and the reduction in serum phosphate by lanthanum is causally correlated with the decrement in serum hepcidin levels [129]. These observations therefore suggest complex interaction between phosphate-FGF23 pathways and iron metabolism (Fig. 2).
Conclusions
Cardiac hypertrophy is a serious complication observed frequently in patients with CKD. Among multiple factors involved in cardiac disease, humoral factors, including aldosterone and FGF23, are gaining much attention as critical components responsible substantially for the development of cardiac hypertrophy and heart failure that lead to increased morbidity and mortality. Recent progresses in the therapeutic strategies using novel tools facilitate the management of CKD. Novel approaches from the standpoint of hormonal (aldosterone and FGF23) and mineral/electrolyte factors (phosphate) as well as iron status appear to be a promising strategy and could constitute a mainstay in the treatment of cardiovascular disorders in CKD.
Abbreviations
- ACE2:
-
Angiotensin converting enzyme 2
- ACTH:
-
Adrenocortical stimulating hormone
- CKD:
-
Chronic kidney disease
- FGF23:
-
Fibroblast growth factor 23
- GFR:
-
Glomerular filtration rate
- LV:
-
Left ventricle
- NCC:
-
Sodium chloride cotransporter
- RAS:
-
Renin-angiotensin system
- ROMK:
-
Renal outer medullary potassium channels
References
Go AS, Chertow GM, Fan D, et al. Cheonic kidney disease and the cardiovascular events, and hospitalization. N Engl J Med. 2004;351:1296–305.
Ninomiya T, Kiyohara Y, Tokuda Y, et al. Japan arteriosclerosis longitudinal Study Group. Impact of kidney disease and blood pressure on the development of cardiovascular disease: an overview from the Japanese Longitudinal Study. Circulation 2008; 118: 2694-2701.
Tanaka K, Watanabe T, Takeuchi A, et al. CKD-JAC Investigators. Cardiovascular events and death in Japanese patients with chronic kidney disease. Kidney Int 2017; 91:227-234.
Zoccali C, Benedetto FA, Mallamaci F, et al. Left ventricular mass monitoring in the follow-up of dialysis patients: prognostic value of left ventricular hypertrophy progression. Kidney Int. 2004;65:1492–8.
Dubin RF, Deo R, Bansal N, et al. Associations of conventional echocardiographic measures with incident heart failure and mortality: the chronic renal insufficiency cohort. Clin J Am Soc Nephrol. 2017;12:60–8.
Nishida M, Tanabe S, Maruyama Y, et al. Gα12/13- and reactive oxygen species-dependent activation of c-Jun NH2-terminal kinase and p38 MAPK by angiotensin receptor stimulation in rat neonatal cardiomyocytes. J Biol Chem. 20015;280:18434–41.
Yamazaki T, Komuro I, Shiojima I, et al. The renin-angiotensin system and cardiac hypertrophy. Heart. 1996;76(3 Suppl 3):33–5.
Arima S, Kohagura K, Xu HL, et al. Nongenomic vascular action of aldosterone in the glomerular microcirculation. J Am Soc Nephrol. 2003;14:2253–1163.
Brilla CG, Pick R, Tan LB, et al. Remodeling of the rat right and left ventricles in experimental hypertension. Circ Res. 1990;67:1355–64.
Sato A, Hayashi K, Naruse M, et al. Effectiveness of aldosterone blockade in patients with diabetic nephropathy. Hypertension. 2003;41:64–8.
Pitt B, Zannad F, Remme WJ, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. N Engl J Med. 1999;341:709–17.
Reubi FC, Weidmann P. Relationships between sodium clearance, plasma renin activity, plasma aldosterone, renal hemodynamics and blood pressure in essential hypertension. Clin Exp Hypertens. 1980;2:593–612.
Berle T, Katz FH, Henrich WL, et al. Role of aldosterone in the control of sodium excretion in patients with advanced chronic renal failure. Kidney Int. 1978;14:228–35.
Hene RJ, Boer P, Koomans HA, et al. Plasma aldosterone concentrations in chronic renal disease. Kidney Int. 1982;21:98–101.
Hosoya K, Minakuchi H, Wakino S, et al. Insulin resistance in chronic kidney disease is ameliorated by spironolactone in rats and humans. Kidney Int. 2015;87:749–60.
Sarafidis PA, Khosla N, Bakris GL. Antihypertensive therapy in the presence of proteinuria. Am J Kidney Dis. 2007;49:12–26.
Saruta T, Hayashi K, Ogihara T, et al. Effects of candesartan and amlodipine on cardiovascular events in hypertensive patients with chronic kidney disease: subanalysis of the CASE-J study. Hypertens Res. 2009;32:505–12.
Bowe AE, Finnegan R, Jan de Beur SM, et al. FGF-23 inhibits renal tubular phosphate transporter and is a PHEX substrate. Biochem Biophys Res Commun. 2001;284:977–81.
Evenepoel P, Meijers B, ViaeneL, et al. Fibroblast growth factor-23 in early chronic kidney disease: additional support in favor of a phosphate-centric paradigm for the pathogenesis of secondary hyperparathyroidism. Clin J Am Soc Nephrol. 2010;5:1268–76.
Holden RM, Beseau D, Booth SL, et al. FGF-23 is associated with cardiac troponin T and mortality in hemodialysis patients. Hemodial Int. 2012;16:53–8.
Gutierrez OM, Januzzi JL, Isakova T, et al. Fibroblast growth factor 23 and left ventricular hypertrophy in chronic kidney disease. Circulation. 2009;119:2545–52.
Faul C, Amaral AP, Oskouei B, et al. FGF-23 induces left ventricular hypertrophy. J Clin Invest. 2011;121:4393–408.
Hamano T, Sakaguchi Y, Fujii N, et al. Clinical features of CKD-MBD in Japan: cohort studies and registry. Clin Exp Nephrol. 2017;21(Suppl 1):9–20.
Wang WH, Giebisch G. Regulation of potassium (K) handling in the renal collecting duct. Pflugers Arch. 2009;458:157–68.
O’Neil RG. Aldosterone regulation of sodium and potassium transport in the cortical collecting duct. Semin Nephrol. 1990;10:365–74.
Gant CM, Laverman GD, Vogt L, et al. Renoprotective RAAS inhibition does not affect the association between worse renal function and higher plasma aldosterone levels. BMC Nephrol. 2017;18:370. https://doi.org/10.1186/s12882-017-0789-x.
Wesson DE, Simoni J. Acid retention during kidney failure induces endothelin and aldosterone production which lead to progressive GFR decline, a situation ameliorated by alkali diet. Kidney Int. 2010;78:1128–35.
Wesson DE, Simoni J, Broglio K, et al. Acid retention accompanies reduced GFR in humans and increases plasma levels of endothelin and aldosterone. Am J Phys. 2011;300:F830–7.
Martin RS, Panese S, Virginillo M, et al. Increased secretion of potassium in the rectum of humans with chronic renal failure. Am J Kidney Dis. 1986;8:105–10.
Mulè G, Nardi E, Guarino L, et al. Plasma aldosterone and its relationship with left ventricular mass in hypertensive patients with early-stage chronic kidney disease. Hypertens Res. 2015;38:276–83.
Williams GH, Bailey GL, Hamper GL, et al. Studies on the metabolism of aldosterone on chronic renal failure and anephric man. Kidney Int. 1973;4:280–8.
Otani H, Otsuka F, Inagaki K, et al. Aldosterone breakthrough caused by chronic blockade of angiotensin type 1 receptors in human adrenocortical cells: possible involvement of bone morphogenetic protein-6 actions. Endocrinology. 2008;149:2816–25.
Moranne O, Bakris G, Fafin C, et al. Determinants and changes associated with aldosterone breakthrough after angiotensin II receptor blockade in patients with type 2 diabetes with overt nephropathy. Clin J Am Soc Nephrol. 2013;8:1694–701.
Bastl C, Hayslett JP, Binder HJ. Increased large intestinal secretion of potassium in renal insufficiency. Kidney Int. 1977;12:9–16.
Sandle GI, Gaiger E, Tapster S, et al. Evidence for large intestinal control of potassium homeostasis in uraemic patients undergoing long-term dialysis. Clin Sci. 1987;73:247–52.
Mathialahan T, Maclennan KA, Sandle LN, et al. Enhanced large intestinal potassium permeability in end-stage renal disease. J Pathol. 2005;206:46–51.
Sorensen MV, Leipziger J. The essential role of luminal BK channels in distal colonic K+ secretion. J Med Investig. 2009;56(Suppl):301.
Sorensen MV, Matos JE, Sausbier M, et al. Aldosterone increases KCa1.1 (BK) channel-mediated colonic K+ secretion. J Physiol. 2008;586:4251–64.
Wilson DR, Ing TS, Metcafe-Gibson A, et al. The chemical composition of faeces in uraemia, as revealed by in vivo faecal dialysis. Clin Sci. 1968;35:197–209.
Sandle GI, Gaiger E, Tapster S, et al. Enhanced rectal potassium secretion in chronic renal insufficiency: evidence for large intestinal potassium adaptation in man. Clin Sci. 1986;71:393–401.
Zannad F, McMurray JJ, Krum H, et al. Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med. 2011;364:11–21.
Leopold JA, Dam A, Maron BA, et al. Aldosterone impairs vascular reactivity by decreasing glucose-6-phosphate dehydrogenase activity. Nat Med. 2007;13:189–97.
Ori Y, Chagnac A, Korzets A, et al. Regression of left ventricular hypertrophy in patients with primary aldosteronism/low-renin hypertension on low-dose spironolactone. Nephrol Dial Transplant. 2013;28:1787–93.
Catena C, Colussi G, Nait F, et al. Aldosterone and the heart: still an unresolved issue? Front Endocrinol. 2014;5:168. https://doi.org/10.3389/fendo.2014.00168.eCollection 2014.
Edwards NC, Steeds RP, Stewart PM, et al. Effect of spironolactone on left ventricular mass and aortic stiffness in early-stage chronic kidney disease: a randomized controlled trial. J Am Coll Cardiol. 2009;54:505–12.
Edwards NC, Ferro CJ, Kirkwood H, et al. Effect of spironolactone on left ventricular systolic and diastolic function in patients with early stage chronic kidney disease. Am J Cardiol. 2010;106:1505–11.
Feniman-De-Stefano GM, Zanati-Basan SG, De Stefano LM, et al. Spironolactone is secure and reduces left ventricular hypertrophy in hemodialysis patients. Ther Adv Cardiovasc Dis. 2015;9:158–67.
Taheri S, Mortazavi M, Shahidi S, et al. Spironolactone in chronic hemodialysis patients improves cardiac function. Saudi J Kidney Dis Transpl. 2009;20:392–7.
Lin C, Zhang Q, Zhang H, et al. Long-term effects of low-dose spironolactone on chronic dialysis patients: a randomized placebo-controlled study. J Clin Hypertens (Greenwich). 2016;18:121–8.
Ito Y, Mizuno M, Suzuki Y, et al. Long-term effects of spironolactone in peritoneal dialysis patients. J Am Soc Nephrol. 2014;25:1094–102.
Taheri S, Mortazavi M, Pourmoghadas A, et al. A prospective double-blind randomized placebo-controlled clinical trial to evaluate the safety and efficacy of spironolactone in patients with advanced congestive heart failure on continuous ambulatory peritoneal dialysis. Saudi J Kidney Dis Transpl. 2012;23:507–12.
Hausmann MJ, Liel-Cohen N. Aldactone therapy in a peritoneal dialysis patients with decreased left ventricular function. Nephrol Dial Tranplant. 2002;17:2035–6.
Matsumoto Y, Mori Y, Kageyama S, et al. Spironolactone reduces cardiovascular and cerebrovascular morbidity and mortality in hemodialysis patients. J Am Coll Cardiol. 2014;63:528–36.
Fiebeler A, Haller H. Participation of the mineralocorticoid receptor in cardiac and vascular remodeling. Nephron Physiol. 2003;94:47–50.
Okoshi MP, Yan X, Okoshi K, et al. Aldosterone directly stimulates cardiac hypertrophy. J Card Fail. 2004;10:511–8.
Park YM, Park MY, Suh YL, et al. NAD(P)H oxidase inhibitor prevents blood pressure elevation and cardiovascular hypertrophy in aldosterone-infused rats. Biochem Biol Res Commun. 2004;313:812–7.
Brown NJ. Contribution of aldosterone to cardiovascular and renal inflammation and fibrosis. Nat Rev Nephrol. 2013;9:459–69.
Somanna NK, Yariswamy M, Garagliano JM, et al. Aldosterone-induced cardiomyocyte growth, and fibroblast migration and proliferation are mediated by TRAF2IP2. Cell Signal. 2015;27:1928–38. https://doi.org/10.1016/j.cellsig.2015.07.001.
Sakamuri SS, Valente AJ, Siddesha JM, et al. TRAF3IP2 mediates aldosterone/salt-induced cardiac hypertrophy and fibrosis. Mol Cell Endocrinol. 2016;429:84–92.
Shibata S, Nagase M, Yoshida S, et al. Modification of mineralocorticoid receptor function by Rac1 GTPase: implication in proteinuric kidney function. Nat Med. 2008;14:1370–6.
Nagase M, Fujita T. Mineralocorticoid receptor activation in obesity hypertension. Hypertens Res. 2009;32:649–57.
Ayuzawa N, Nagase M, Ueda K, et al. Rac1-mediated activation of mineralocorticoid receptor in pressure overload-induced cardiac injury. Hypertension. 2016;67:99–106.
Papadimitriou M, Vyzantiadis A, Milionis A, et al. The effect of spironolactone in hypertensive patients on regular haemodialysis and after renal allotransplantation. Life Support Syst. 1983;1:197–205.
Flevari P, Kalogeropoulou S, Drakou A, et al. Spironolactone improves endothelial and cardiac autonomic function in non heart failure hemodialysis patients. J Hypertens. 2013;31:1239–44.
Walsh M, Manns B, Garg AX, et al. The safety of eplerenone in hemodialysis patients: a noninferiority randomized controlled trial. Clin J Am Soc Nephrol. 2015;10:1602–8.
Tawada M, Suzuki Y, Sakata F, et al. Mineralocorticoid receptor antagonists in dialysis patients. Ren Replace Ther. 2016;2:64. https://doi.org/10.1186/s41100-016-0077-4, 2016.
Yelken B, Gorgulu N, Gursu M, et al. Effects of spironolactone on residual renal function and peritoneal function in peritoneal dialysis patients. Adv Perit Dial. 2014;30:5–10.
Vazquez-Rangel A, Soto V, Escalona M, et al. Spironolactone to prevent peritoneal fibrosis in peritoneal dialysis patients: a randomized controlled trial. Am J Kidney Dis. 2014;63:1072–4.
Homma K, Hayashi K, Yamaguchi S, et al. Renal microcirculation and calcium channel subtypes. Cur Hypertens Rev. 2013;9:182–6.
Imagawa K, Okayama S, Takaoka M, et al. Inhibitory effect of efonidipine on aldosterone synthesis and secretion in human adrenocarinoma (H295R) cells. J Cardiovasc Pharmacol. 2006;47:133–8.
Akizuki O, Inayoshi A, Kitayama T, et al. Blockade of T-type voltage-dependent Ca2+ channels by benidipine, a dihydropyridine calcium channel blocker, inhibits aldosterone production in human adrenocortical cell line NCl-H295R. Eur J Pharmacol. 2008;584:424–34.
Isaka T, Ikeda K, Takada Y, et al. Azelnidipine inhibits aldosterone synthesis and secretion in human adrenocortical cell line NCl-H295R. Eur J Pharmacol. 2009;605:49–52.
Aritomi S, Wagatsuma H, Numata T, et al. Expression of N-type calcium channels in human adrenocortical cells and their contribution to corticosteroid synthesis. Hypertens Res. 2011;34:193–201.
Tsutamoto T, Tanaka T, Nishiyama K, et al. Long-term effect of efonidipine therapy on plasma aldosterone and left ventricular mass index in patients with essential hypertension. Hypertens Res. 2009;32:670–4.
Nakano N, Ishimitsu T, Takahashi T, et al. Effects of efonidipine, an L- and T-type calcium channel blocker, on the renin-angiotensin-aldosterone system in chronic hemodialysis patients. Int Heart J. 2010;51:188–92.
Abe M, Okada K, Maruyama N, et al. Benidipine reduces albuminuria and plasma aldosterone in mild-to-moderate stage chronic kidney disease with albuminuria. Hypertens Res. 2011;34:268–73.
Nakamura T, SugayaT KY, et al. Azelnidipine reduces urinary protein excretion and urinary liver-type fatty acid binding protein in patients with hypertensive chronic kidney disease. Am J Med Sci. 2007;333:321–6.
Konoshita T, Makino Y, Kimura T, et al. A new-generation N/L-type calcium channel blocker leads to less activation of the renin-angiotensin system compared with conventional L type calcium channel blocker. J Hypertens. 2010;28:2156–60.
Abe M, Maruyama N, Suzuki H, et al. L/N-type calcium channel blocker cilnidipine reduces plasma aldosterone, albuminuria, and urinary liver-type fatty acid binding protein in patients with chronic kidney disease. Heart Vessel. 2012;28:480–9.
Furukawa T, Nukada T, Namiki Y, et al. Five different profiles of dihydrppyridines in blocking T-type Ca(2+) channel subtypes (Ca(v)3.1(alfa(1G)), Ca(v)3.2(alfa(1H)), and Ca(v)3.3(alfa(1I))) expressed in Xenopus oocytes. Eur J Pharmacol. 2009;613:100–7.
Hayashi K, Wakino S, Sugano N, et al. Ca2+ channel subtypes and pharmacology in the kidney. Circ Res. 2007;100:342–53.
Hayashi K, Ozawa Y, Saruta T, Epstein M. Renal hemodynamic effects of calcium antagonists. In: Epstein M, editor. Calcium Antagonists in Clinical Medicine. 3rd ed. Philadelphia: Hanley & Belfus; 2002. p. 559–78.
Dietz JD, Du S, Bolten CW, et al. A number of marketed dihydropyridine calcium channel blockers have mineralocorticoid receptor antagonist activity. Hypertension. 2008;51:742–8.
Kosaka H, Hirayama K, Yoda N, et al. The L-, N-, and T-type triple calcium channel blocker benidipine acts as an antagonist of mineralocorticoid receptor, a member of nuclear receptor family. Eur J Pharmacol. 2010;635:49–55.
Shimada T, Mizutani S, Muto T, et al. Cloning and characterization of FGF 23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci U S A. 2001;98:6500–5.
Felsenfeld AJ, Levine BS, Rodriguez M. Pathophysiology of calcium, phosphorus, and magnesium dysregulation in chronic kidney disease. Semin Dial. 2015;28:564–77.
Bricker NS. On the pathogenesis of the uremic state. An exposition of the “trade-off hypothesis”. N Engl J Med. 1972;286:1093–9.
Isakova T, Wahl P, Vargas GS, et al. Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int. 2011;79:1370–8.
Tomoe Y, Segawa H, Shiozawa K, et al. Phosphaturic action of fibroblast growth factor 23 in Npt2 null mice. Am J Physiol. 2010;298:F1341-50.
Schiavi SC, Kumar R. The phosphatonin pathway: new insights in phosphate homeostasis. Kidney Int. 2004;65:1–14.
Ben-Dov IZ, Galitzer H, Lavi-Moshayoff V, et al. The parathyroid is a target organ for FGF23 in rats. J Clin Invest. 2007;117:4003–8.
Kovesdy CP, Quarles LD. FGF23 from bench to bedside. Am J Phys. 2016;310:F1168–74.
Ritter CS, Slatopolsky E. Phosphate toxicity in CHD: the killer among UA. Clin J Am Soc Nephrolo. 2016;11:1088–100.
Andrukhova O, Slavic S, Smorodchenko A, et al. FGF23 regulates renal sodium handling and blood pressure. EMBO Mol Med. 2014;6:744–59.
Tsai MH, Leu JG, Fang YW, et al. High fibroblast growth factor 23 levels associated with low hemoglobin levels in patients with chronic kidney disease stages 3 and 4. Medicine. 2016;95:e3049. https://doi.org/10.1097/MD.0000000000003049.
Imazu M, Takahama H, Asanuma H, et al. Pathophysiological impact of serum fibroblast growth factor 23 in patients with non-ischemic cardiac disease and early chronic kidney disease. Am J Phys. 2014;307:H1504–11.
Hu MC, Kuro-o M, Moe OW. Klotho and chronic kidney disease. Contrib Nephrol. 2013;180:47–63.
de Seigneux S, Courbebaisse M, Rutkowski JM, Wilhelm-Bals A, et al. Proteinuria increases plasma phosphate by altering its tubular handling. J Am Soc Nephrol. 2015;26:1608–18.
De Borst MH, Vervloet MG, ter Wee PM, et al. Cross talk between the renin-angiotensin-aldosterone system and vitamin D-FGF23-klotho in chronic kidney disease. J Am Soc Nephrol. 2011;22:1603–9.
Dai B, David V, Martin A, et al. A comparative transcriptome analysis identifying FGF23 regulated genes in the kidney of a mouse CKD model. PLoS One. 2012;7:e44161. https://doi.org/10.1371/journal.pone.0044161.
Santos RA, Rerreira AJ. Angiotensin-(1-7) and the renin-angiotensin system. Curr Opin Nephrol Hypertens. 2007;16:122–8.
Varaqic J, Ahmad S, Nagata S, Ferrario CM. ACE2: angiotensin II/angiotensin-(1-7) balance in cardiac and renal injury. Curr Hypertens Rep. 2014;16:420. https://doi.org/10.1007/s11906-014-0420-5.
Li YC, Kong J, Wei M, et al. 1,25-Dihydroxyvitamin D(3) is a negative endocrine regulator of the renin-angiotensin system. J Clin Invest. 2002;110:229–38.
Freundlich M, Quiroz Y, Zhang Z, et al. Suppression of renin-angiotensin gene expression in the kidney by paricalcitol. Kidney Int. 2008 Dec;74(11):1394–402. https://doi.org/10.1038/ki.2008.408.
Zhang B, Umbach AT, Chen H, et al. Up-regulation of FGF23 release by aldosterone. Biochem Biophy Res Commun. 2016;470:384–90.
Di Lullo L, Gorini A, Russo D, et al. Left ventricular hypertrophy in chronic kidney disease patients: from pathophysiology to treatment. Cardiorenal Med. 2015;5:254–66.
Nardi E, Palermo A, Mule G, et al. Left ventricular hypertrophy and geometry in hypertensive patients with chronic kidney disease. J Hypertens. 2009;27:633–41.
Hayashi T, Joki N, Tanaka Y, et al. Anaemia and early phase cardiovascular events on haemodialysis. Nephrology (Carlton). 2015;20(Suppl 4):1–6.
Randon RB, Rohde LE, Comerlato L, et al. The role of secondary hyperparathyroidism in left ventricular hypertrophy of patients under chronic hemodialysis. Braz J Med & Biol Res. 2005;38:1409–16.
Strozecki P, Adamowicz A, Nartowicz E, et al. Parathormone, calcium, phosphorus and left ventricular structure and function in normotensive hemodialysis patients. Ren Fail. 2001;23:115–26.
London GM, De Vernejoul MC, Fabiani F, et al. Secondary hyperparathyroidism and cardiac hypertrophy in hemodialysis patients. Kidney Int. 1997;32:900–7.
Wang AY, Fang F, Chan J, et al. Effect of paricalcitol on left ventricular mass and function in CKD-the OPERA trial. J Am Soc Nephrol. 2014;25:175–86.
Hassan M, Qureshi W, Sroujieh LS, et al. Interplay of parathyroid hormone and aldosterone antagonist in prevention of heart failure hospitalizations in chronic kidney disease. J Renin-Angiotensin-Aldosterone Syst. 2014;15:278–85.
Brown JM, Williams JS, Luther JM, et al. Human interventions to characterize novel relationships between the renin-angiotensin-aldosterone system and parathyroid hormone. Hypertension. 2014;63:273–80.
Olgaard K, Lewin E, Bro S, et al. Enhancement of the stimulatory effect of calcium on aldosterone secretion by parathyroid hormone. Miner Electrolyte Metab. 1994;20:309–14.
Mazzocchi G, Aragona F, Malendowicz LK, et al. PTH and PTH-related peptide enhance steroid secretion from human adrenocortical cells. Am J Physiol Endocrinol Metab. 2001;280:E209–13.
Tomaschitz A, Ritz E, Pieske B, et al. Aldosterone and parathyroid hormone interactions as mediators of metabolic and cardiovascular disease. Metabolism Clin & Exp. 2014;63:20–31.
Hsu HJ, Wu MS. Fibroblast growth factor 23: a possible cause of left ventricular hypertrophy in hemodialysis patients. Am J Med Sci. 2009;337:116–22.
Seifert ME, de Las FL, Ginsberg C, et al. Left ventricular mass progression despite stable blood pressure and kidney function in stage 3 chronic kidney disease. Am J Nephrol. 2014;39:392–9.
Sarmento-Dias M, Santos-Araújo C, Poínhos R, Oliveira B, et al. Fibroblast growth factor 23 is associated with left ventricular hypertrophy, not with uremic vasculopathy in peritoneal dialysis patients. Clin Nephrol. 2016;85:135–41.
Jovanovich A, Ix JH, Gottdiener J, et al. Fibroblast growth factor 23, left ventricular mass, and left ventricular hypertrophy in community-dwelling older adults. Atherosclerosis. 2013;231:114–9.
Tanaka S, Fujita S, Kizawa S, et al. Association between FGF23, α-Klotho, and cardiac abnormalities among patients with various chronic kidney disease stages. PLoS One. 2016;11(7):e0156860. https://doi.org/10.1371/journal.pone.0156860. eCollection 2016
Di Marco GS, Reuter S, Kentrup D, et al. Treatment of established left ventricular hypertrophy with fibroblast growth factor receptor blockade in an animal model of CKD. Nephrol Dial Transplant. 2014;29:2028–35.
Grabner A, Amaral AP, Schramm K, et al. Activation of cardiac fibroblast growth factor receptor 4 causes left ventricular hypertrophy. Cell Metab. 2015;22:1020–32.
Koiwa F, Kazawa JJ, Tokumoto A, et al. Sevelamer hydrochloride and calcium bicarbonate reduce serum fibroblast growth factor 23 levels in dialysis patients. Ther Apher Dial. 2005;9:336–9.
Gonzalez-Parra E, Gonzalez-Casaus ML, Galan A, et al. Lanthanum carbonate reduces FGF23 in chronic kidney disease stage 3 patients. Nephrol Dial Transplant. 2011;26:2567–71.
Shigematsu T, Negi S, for the COLC Research Group. Combined therapy with lanthanum carbonate and calcium carbonate for hyperphosphatemia decreases serum FGF-23 independently of calcium and PTH (COLC Study). Nephrol Dial Transpant. 2012;27:1050–4.
Lin HH, Liou HH, Wu MS, et al. Long-term sevelamer treatment lowers serum fibroblast growth factor 23 accompanied with increasing serum Klotho levels in chronic haemodialysis patients. Nephrology (Carlton). 2014;19:672–8.
Chang YM, Tsai SC, Shiao CC, et al. Effects of lanthanum carbonate and calcium carbonate on fibroblast growth factor 23 and hepcidin levels in chronic hemodialysis patients. Clin Exp Nephrol. 2016; https://doi.org/10.1007/s10157-016-1362-9.
Rodriguez-Ortiz ME, Lopez I, Munoz-Castaneda JR, et al. Calcium deficiency reduces circulating levels of FGF23. J Am Soc Nephrol. 2012;23:1190–7.
David V, Dai B, Martin A, et al. Calcium regulates FGF-23 expression in bone. Endocrinology. 2013;154:4469–82.
Yasin A, Liu D, Chau L, et al. Fibroblast growth factor-23 and calcium phosphate product in young chronic kidney disease patients: a cross-sectional study. BMC Nephrol. 2013;14:39. https://doi.org/10.1186/1471-2369-14-39.
Siriano S, Ojeda R, Rodriguez M, et al. The effect of phosphate binders, calcium and lanthanum carbonate on FGF23 levels in chronic kidney disease patients. Clin Nephrol. 2013;80:17–22.
Chue CD, Townend JN, Moody WE, et al. Cardiovascular effects of sevelamer in stage 3 CKD. J Am Soc Nephrol. 2013;24:842–52.
Maizel J, Six I, Dupont S, et al. Effects of sevelamer treatment on cardiovascular abnormalities in mice with chronic renal failure. Kidney Int. 2013;84:491–500.
Yamazaki-Nakazawa A, Mizobuchi M, Ogata H, et al. Correction of hyperphosphatemia suppresses cardiac remodeling in uremic rats. Clin Exp Nephrol. 2014;18:56–64.
Chue CD, Edwards NC, Moody WE, et al. Serum phosphate is associated with left ventricular mass in patients with chronic kidney disease: a cardiac magnetic resonance study. Heart. 2012;98:219–24.
Dhingra R, Gona P, Benjamin EJ, et al. Relations of serum phosphorus levels to echocardiographic left ventricular mass and incidence of heart failure in the community. Eur Heart Fail. 2010;12:812–8.
Nakamura H, Tokumoto M, Mizobuchi M, et al. Novel markers of left ventricular hypertrophy in uremia. Am J Nephrol. 2010;31:292–302.
Achinger SG, Ayus JC. Left ventricular hypertrophy: is hyperphosphatemia among dialysis patients a risk factor? J Am Soc Nephrol. 2006;17(12 Suppl 3):S255–61.
Liu YL, Huang CC, Chang CC, et al. Hyperphosphate-induced myocardial hypertrophy through the GATA-4/NFAT-3 signaling pathway is attenuated by ERK inhibitor treatment. Cardiorenal Med. 2015;5:79–88. https://doi.org/10.1159/000371454.
Yagil Y, Fadem SZ, Kant KS, et al. Managing hyperphosphatemia in patients with chronic kidney disease on dialysis with ferric citrate: latest evidence and clinical usefulness. Ther Adv Chronic Dis. 2015;6:252–63.
Floege J, Covic AC, Ketteler M, et al. Long-term effects of the iron-based phosphate binder, sucrferric oxyhydroxide, in dialysis patients. Nephrol Dial Transplant. 2015;30:1037–46.
Bozentowicz-Wikarek M, Kocelak P, Owczarek A, et al. Plasma fibroblast growth factor 23 concentration and iron status. Does the relationship exist in the early elderly population? Clin Biochem. 2015;48:431–6.
Lewerin C, Ljunggren O, Nilsson-Ehle H, et al. Low serum iron is associated with high serum intact FGF23 in elderly men. The Swedish MrOS study. Bone. 2017;98:1–8.
Braithwaite V, Prentice AM, Doherty C, et al. FGF23 is correlated with iron status but not with inflammation and decreases after iron supplementation: a supplementation study. Int J Pediatr Endocrinol. 2012;2012(1):27. https://doi.org/10.1186/1687-9856-2012-27.
David V, Martin A, Isakova T, et al. Inflammation and functional iron deficiency regulate fibroblast growth factor 23 production. Kidney Int. 2016;89:135–46.
Hanudel MR, Chua K, Rappaport M, et al. Effects of dietary iron intake and chronic kidney disease on fibroblast growth factor 23 metabolism in wild-type and hepcidin knockout mice. Am J Phys. 2016;311:F1369–77.
Farrow EG, Yu X, Summers LJ, et al. Iron deficiency drives an autosomal dominant hypophosphatemic ricket (ADHR) phenotype in fibroblast growth factor-23 knock-in mice. Proc Natl Acad Sci U S A. 2011;108:E1146–55.
Deger SM, Erten Y, Pasaoglu OT, et al. The effects of iron on FGF23-mediated Ca-P metabolism in CKD patients. Clin Exp Nephrol. 2013;17:416–23. https://doi.org/10.1007/s10157-012-0725-0.
Yamashita K, Mizuiri S, Nishizawa Y, et al. Oral iron supplementation with sodium ferrous citrate reduces the serum intact and c-terminal FGF23 levels of maintenance hemodialysis patients. Nephrology (Carlton). 2016. https://doi.org/10.1111/nep.12909.
Takeda Y, Komaba H, Goto S, et al. Effect of intravenous saccharated ferric oxide on serum FGF23 and mineral metabolism in hemodialysis patients. Am J Nephrol. 2011;33:421–6. https://doi.org/10.1159/000327019. Epub 2011 Apr 19.
Gravesen E, Hofman-Bang J, Mace ML, et al. High dose intravenous iron, mineral homeostasis and intact FGF23 in normal and uremic rats. BMC Nephrol. 2013;27(14):281. https://doi.org/10.1186/1471-2369-14-281.
Iguchi A, Kazama JJ, Yamamoto S, et al. Administration of ferric citrate hydrate decreases circulating FGF23 levels independently of serum phosphate levels in hemodialysis patients with iron deficiency. Nephron. 2015;131:161–6.
Babitt JL, Lin HY. Mechanisms of anemia in CKD. J Am Soc Nephrol. 2012;23:1631–4.
Tsuchiya K, Nitta K. Hepcidin is a potential regulator of iron status in chronic kidney disease. Ther Apher Dial. 2013;17:1–8. https://doi.org/10.1111/1744-9987.12001.
Carvalho C, Isakova T, Collerone G, et al. Hepcidin and disordered mineral metabolism in chronic kidney disease. Clin Nephrol. 2011;76:90–8.
Acknowledgements
Not applicable
Funding
There is no funding to be disclosed.
Availability of data and materials
Not applicable
Author information
Authors and Affiliations
Contributions
KH and TS designed and wrote the manuscript. YS and SI collected the literature and discussed the contents of the manuscript with TS and KH. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Hayashi, K., Suzuki, T., Sakamaki, Y. et al. Cardiac hypertrophy in chronic kidney disease—role of Aldosterone and FGF23. Ren Replace Ther 4, 10 (2018). https://doi.org/10.1186/s41100-018-0152-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41100-018-0152-0