
Abstract
Background
Von Hippel–Lindau (VHL) syndrome is a dominantly inherited multisystem cancer syndrome caused by a heterozygous mutation in the VHL tumor suppressor gene. Previous studies suggested that similar populations of Caucasian and Japanese patients have similar genotype or phenotype characteristics. In this comprehensive study of East Asian patients, we investigated the genetic and clinical characteristics of patients with VHL syndrome.
Methods
To create a registry of clinical characteristics and mutations reported in East Asian patients with VHL syndrome, we conducted a comprehensive review of English language and non-English language articles identified through a literature search. Publications in Japanese or Chinese language were read by native speakers of the language, who then performed the data extraction.
Results
Of 237 East Asian patients with VHL syndrome, 154 unique kindreds were identified for analysis. Analyzed by kindred, missense mutations were the most common (40.9%, 63/154), followed by large/complete deletions (32.5%, 50/154) and nonsense mutations (11.7%, 18/154). Compared with a previously reported study of both East Asian and non-East Asian patients, we found several key differences. First, missense and frameshift mutations in the VHL gene occurred less commonly in our population of East Asian patients (40.9% vs. 52.0%; P = 0.012 and 8.4% vs. 13.0%; P < 0.001, respectively). Second, large/complete deletions were more common in our population of East Asian patients (32.5% vs. 10.5%; P < 0.001). Third, phenotypically, we observed that, in our population of East Asian patients with VHL syndrome, the incidence of retinal capillary hemangioblastoma was lower, whereas the incidence of renal cell carcinoma was higher.
Conclusions
Evidence suggests that the genotypic and phenotypic characteristics of East Asian patients with VHL syndrome differ from other populations. This should be considered when making screening recommendations for VHL syndrome in Asia.
Similar content being viewed by others

Background
Von Hippel–Lindau (VHL) syndrome is an inherited multisystem cancer syndrome with an estimated incidence of 1 in 36,000 live births [1]. Those affected are predisposed to a spectrum of benign and malignant tumors of the central nervous system, retina, kidney, adrenal gland, pancreas, reproductive adnexal organ, and inner ear [2, 3].
Von Hippel–Lindau syndrome is transmitted in an autosomal dominant fashion and has a penetrance reported to exceed 90% by 60 years of age [4]. It is caused by germline mutations in the VHL tumor suppressor gene, located on the short arm of chromosome 3. The gene consists of 3 exons and encodes 2 protein products: a full-length 30-kDa form (p30) composed of 213 amino acids and a 19-kDa form (p19). The VHL protein (pVHL) is the substrate-binding subunit of an E3 ubiquitin ligase complex and has an alpha domain and a beta domain [5]. Through its beta domain, pVHL binds directly to hypoxia-inducible factor (HIF) [6] and targets the HIF alpha subunit for oxygen-dependent proteolysis [7].
Interfamilial differences in pheochromocytoma (PCC) susceptibility have been recognized, and a clinical classification of VHL syndrome was created based on the risk of developing PCC: VHL patients with type 1 disease are at low risk, whereas those with type 2 disease are at high risk [8, 9]. The cancer phenotypes of type 2 disease have been further subdivided based on the likelihood of developing renal cell carcinoma (RCC): type 2A and type 2B have a low and high likelihood, respectively; patients with type 2C disease develop PCCs only, without developing RCCs or hemangioblastomas [2]. Over 900 mutations of the VHL gene have been described [10]. The mutations accounting for type 1 disease include missense mutations (43%), frameshift mutations (17%), nonsense mutations (13%), partial/complete deletions (10%), splice (9%), and in-frame deletion/insertions (8%) [11]. Most mutations associated with type 2 disease are missense mutations [9, 11].
In the present study, we consolidated published data on genetic and clinical information about VHL syndrome in Asia to further understand the spectrum of VHL syndrome, focusing on differences between East Asian and Western patient populations.
Patients and methods
Local patient selection
Patients were identified through the clinical cancer genetics service in the National Cancer Centre Singapore. Before informed consent was provided to obtain blood samples for genetic testing, patients were provided genetic counseling by specialized clinical cancer geneticists. Patients and their families with confirmed VHL mutations were selected for the study and registered in a database. Institutional review board approval was obtained for this study.
Literature search strategy
On June 15, 2012, we performed a systematic search on PubMed using the following search strategy: “(von Hippel Lindau Disease OR Familial Cerebello-Retinal Angiomatosis OR Angiomatoses, Familial Cerebello-Retinal OR Angiomatosis, Familial Cerebello-Retinal OR Cerebello-Retinal Angiomatoses, Familial OR Cerebello-Retinal Angiomatosis, Familial OR Familial Cerebello Retinal Angiomatosis OR Familial Cerebello-Retinal Angiomatoses OR von Hippel–Lindau Syndrome OR Syndrome, von Hippel–Lindau OR von Hippel Lindau Syndrome OR Lindau Disease OR Lindau’s Disease OR Lindau’s Diseases OR Lindaus Disease OR Cerebelloretinal Angiomatosis, Familial OR Angiomatoses, Familial Cerebelloretinal OR Angiomatosis, Familial Cerebelloretinal OR Cerebelloretinal Angiomatoses, Familial OR Familial Cerebelloretinal Angiomatoses OR Familial Cerebelloretinal Angiomatosis OR Hippel–Lindau Disease OR Hippel Lindau Disease) AND (China OR Hong Kong OR Chinese OR Japan OR Japanese OR Korea OR Korean OR Taiwan OR Republic of China OR Taiwanese).”
All available articles were reviewed and included for analysis if both genotype and phenotype information were reported. Articles in the Chinese and Japanese languages were reviewed by native speakers. All data were input into a standardized template. We extracted the following data: (i) genetic information—sequence variation at the DNA and protein levels [12], and type of variation (e.g., missense); (ii) clinical information—diagnosis of central nervous system hemangioblastoma, retinal capillary hemangioblastoma (RCH), PCC, pancreatic cyst or tumor, renal cyst or tumor, endolymphatic sac tumor, or papillary cystadenoma of the epididymis or broad ligament; (iii) patient demographics—age at first diagnosis, sex, nationality, race, and familial or sporadic inheritance; and (iv) article publication data—journal, year of publication, and authors.
The open source program Mutalyzer was used to check the protein consequences of mutations [13]. To avoid duplication of data, if the same sequence variation was reported at the DNA level, we compared patient nationality, sex, age of first diagnosis, and disease manifestations to determine the likelihood of duplicate data. The entire dataset is included in the Additional file 1: Table S1.
Statistical analysis
The data from all VHL families were analyzed (Additional file 1: Table S1). SPSS 16.0 software (SPSS Inc., Chicago, IL, USA) was used for statistical analysis. Clinical characteristics were compared with the Chi square test, and P values less than 0.05 were considered statistically significant.
Results
Literature review
The described search strategy of VHL syndrome in an East Asian population identified 347 articles. Of the total of 237 East Asian VHL patients, 154 kindreds were identified for analysis based on available genotype and phenotype information.
Local patient selection
Ten affected individuals from 7 families in Singapore were identified.
Analysis of mutations
In our study of East Asian patients, we calculated the frequency of each mutation and then compared this with the dataset reported by Nordstrom-O’Brien et al. [11], which included both Asian and non-Asian patients. Analyzed by similarity, the most common mutation was missense (63/154, 40.9%), followed by large/complete deletions (50/154, 32.5%) and nonsense mutation (18/154, 11.7%). Frameshift, splice, and in-frame deletions/insertions occurred in 8.4% (13 of 154), 4.5% (7 of 154), and 1.3% (2 of 154) of patients, respectively (Table 1).
In our study, missense mutations in the VHL gene were the most common; this was also the most common class of mutation reported by Nordstrom-O’Brien et al. [11]. Missense mutations were more common in the series reported by Nordstrom-O’Brien et al. [11], which comprised both East Asian and non-East Asian patients, than in our cohort (52.0% vs. 40.9%, P = 0.012). Frameshift mutations were more common in the series reported by Nordstrom-O’Brien et al. [11], as compared with our series of East Asian patients (13.0% vs. 8.4%, P < 0.001). In contrast, large/complete deletions were more common in our series (32.5% vs. 10.5%, P < 0.001).
We analyzed the distribution of exon deletions (Table 2) and compared this with the data collected by McNeill et al. [14] via the VHL Registry at Birmingham Women’s Hospital and the Department of Medical and Molecular Genetics, University of Birmingham, United Kingdom. No difference was found in the distribution of exon deletions. The most common mutation was a large deletion of exons 1, 2, and 3. Of those patients who had deletion of a single exon (22 of 50 similar mutations), exon 3 was the most commonly involved exon. No significant difference was observed in the distribution of exon deletions or in the risk of developing central nervous system hemangioblastoma, RCC, or RCH, irrespective of whether there was complete deletion of the VHL gene or incomplete deletion of the VHL gene (Table 3).
The most common single amino acid mutation was c.499 C>T (p.Arg167Trp), which was present in 4.5% (7 of 154) of patients (Fig. 1).

Frequency of point mutations in the von Hippel–Lindau (VHL) gene in an East Asian cohort
We found that 89.5% (17 of 19) of East Asian patients who developed PCC had an underlying missense mutation. The most frequently mutated residues in patients with PCC were Arg167 (36.8%, 7 of 19) and Arg161 (15.8%, 3 of 19),
Phenotype
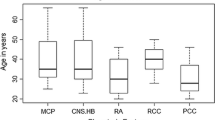
In our study of East Asian patients, we observed that the incidence of RCH was 27.9% (43 of 154). RCH was present in 38.1% (24 of 63) East Asian VHL kindreds with missense mutations, 2.5% (3 of 24) with complete exon deletions, and 19.2% (5 of 26) with exon deletions.
The overall frequency of RCC in this cohort was 57.8% (89 of 154). The frequency of RCC was 55.6% (10 of 18) in the families with nonsense mutations compared with 63.5% (40 of 63) in those with missense mutations (P = 0.541). A smaller proportion of families with frameshift mutations—46.2% (6 of 13 families)—developed RCC.
Discussion
This study consolidated all published reports about VHL syndrome in East Asian patients for whom both genetic and clinical information were available, with which we could better understand the spectrum of VHL syndrome.
PCCs associated with VHL syndrome
We found that 89.5% (17 of 19) of East Asian kindreds who developed PCC had an underlying missense mutation. This is consistent with the results in earlier studies that identified a missense mutation in the VHL gene in 83.5% to 96% of VHL patients who developed PCC [9, 11, 15]. The remaining 10.5% of patients in our study who developed PCC had a frameshift mutation. Again, these results are similar to those reported by Nordstrom-O’Brien et al. [11], who found that 7% of VHL type 2 families had frameshift mutations.
The most frequently mutated residues in this East Asian cohort were Arg167 (36.8%) and Arg161 (15.8%), which are both surface missense mutations. The Arg167 residue has been reported by Stebbins et al. [5] to have a structural role in stabilizing the H1 helix and the α-β domain interface. Forman et al. [16] used bioinformatics tools to determine that mutations at residues 161, 164, and 166 are at conserved sites with elongin C interactions and thus interfere with the elongin C interface. In our cohort, 3 of 4 matched patients who had the mutation Arg161Gln developed PCC at ages 10, 18, and 33 years. None of the 4 matched patients with a truncating mutation at Arg161 had PCC, which is consistent with our findings that most patients who develop PCC carry a missense mutation.
In our series, 12.3% of patients had type 2 VHL syndrome. In 1995, the Clinical Research Group for VHL in Japan reviewed data from Japan and reported that 9% of the evaluated patients had type 2 VHL syndrome [9]. A more recent nationwide epidemiologic survey of VHL syndrome in Japan reported a higher incidence of 15.1% for PCC [17]. Although improved screening may account for increased detection of PCC, another possibility for the increased incidence is age-dependent penetrance. The mean age at diagnosis of the first manifestation of VHL syndrome is between 24.7 and 26.3 years [4, 18], but almost complete penetrance is observed only by age 60 [1]. Thus, in a given population, some of the clinical manifestations that would develop over a lifetime may not have manifested yet.
RCHs associated with VHL syndrome
The overall frequency of RCH in this East Asian cohort was 27.9% (43 of 154). In the literature, there is much variability in the reported frequency of RCH. Ong et al. [18] and Maher et al. [4] found the frequency of RCH to be 73% and 59%, respectively (Table 4). In a large cohort enrolled between 1988 and 2005 at the National Cancer Institute (National Institutes of Health, Bethesda, Maryland, USA), 890 patients with clinically definite VHL syndrome were screened, among them 335 (37.6%) were found to have ocular involvement with RCH [19].
We found that 38.1% (24 of 63) of East Asian VHL kindreds with missense mutations developed RCH, which is consistent with the findings of Mettu et al. [20], who found that 38.6% of 412 VHL patients with missense mutations had ocular VHL syndrome. In contrast, the prevalence of RCHs was lower in patients with exon deletions in the present study. RCHs were present in 12.5% (3 of 24) with complete exon deletions and in 19.2% (5 of 26) with exon deletions. This is similar to earlier findings that the prevalence of RCHs was lower in patients with complete deletions [21]. Interestingly, we found that RCHs were identified less frequently in East Asians; this may be because exon deletions are more common in East Asian patients with VHL syndrome. However, ascertainment bias in this series cannot be excluded since a comprehensive standard evaluation of affected individuals was not possible.
RCCs associated with VHL syndrome
The overall frequency of RCC in this cohort was 57.8% (89 of 154), which was higher than that reported by Ong et al. [18] and Maher et al. [4] (35% and 28%, respectively). Previous studies suggested that truncating and frameshift mutations confer a higher risk of developing RCC compared with missense mutations (75% vs. 57%, P = 0.04) [22]. In this study, no significant difference was found: 55.6% (10 of 18 families) with nonsense mutations developed RCC compared with 63.5% (40 of 63 families) with missense mutations (P = 0.541). A smaller proportion of families with frameshift mutations—46.2% (6 of 13 families)—developed RCC. In this series, nearly two-thirds of patients with nonsense or missense mutations were subsequently diagnosed with RCC. For such patients, screening for RCC needs to be prioritized.
Strengths and limitations
To minimize publication-related bias, articles in non-English language journals identified through the literature review were reviewed by native Chinese or Japanese speakers.
However, this study still had several limitations. An asymptomatic person can be identified as late as 67 years old, and only after careful screening [4]. As such, there may be under-estimation of the number of affected individuals, as some clinical manifestations may have been missed if they were not screened. Another limitation was that East Asian VHL patients who did not undergo genetic testing and lacked an identified genetic mutation were excluded from the analysis. Furthermore, duplicate data could not be completely excluded since patient identifiers were removed in reports, although we did our best to match identities across publications. The other constraint to accurately determining genotype-phenotype association was that the clinical workup of patients was not always disclosed; thus, when a person was not reported to have a particular manifestation, we could not determine whether this is because the investigations returned negative or the patient was asymptomatic and not investigated. We acknowledge that restricting our study selection to the PubMed database is a possible limitation; however, we do not expect bias in our conclusions, given the breadth of PubMed and that we included non-English language articles in our analysis.
Conclusions
Evidence suggests that the genotypic and phenotypic characteristics of East Asian patients with VHL syndrome differ from other populations. This should be considered when making screening recommendations for VHL syndrome in Asia.
Patients with VHL syndrome are predisposed to a spectrum of benign and malignant tumors. As such, affected people should be thoroughly evaluated at regular intervals. However, since time and fiscal constraints remain very real considerations, physician’s familiarity with the genotypic and phenotypic profile of patients with VHL syndrome would enable judicious prioritization of screening.
References
Maher ER, Iselius L, Yates JR, Littler M, Benjamin C, Harris R, et al. Von Hippel-Lindau disease: a genetic study. J Med Genet. 1991;28(7):443–7.
Lonser RR, Glenn GM, Walther M, Chew EY, Libutti SK, Linehan WM, et al. von Hippel-Lindau disease. Lancet. 2003;361(9374):2059–67.
Chan CC, Collins AB, Chew EY. Molecular pathology of eyes with von Hippel-Lindau (VHL) disease: a review. Retina. 2007;27(1):1–7.
Maher ER, Yates JR, Harries R, Benjamin C, Harris R, Moore AT, et al. Clinical features and natural history of von Hippel-Lindau disease. Q J Med. 1990;77(283):1151–63.
Stebbins CE, Kaelin WG Jr, Pavletich NP. Structure of the VHL-ElonginC-ElonginB complex: implications for VHL tumor suppressor function. Science. 1999;284(5413):455–61.
Ohh M, Park CW, Ivan M, Hoffman MA, Kim TY, Huang LE, et al. Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Nat Cell Biol. 2000;2(7):423–7.
Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399(6733):271–5.
Leung SK, Ohh M. Playing tag with HIF: the VHL story. J Biomed Biotechnol. 2002;2(3):131–5.
Chen F, Kishida T, Yao M, Hustad T, Glavac D, Dean M, et al. Germline mutations in the von Hippel-Lindau disease tumor suppressor gene: correlations with phenotype. Hum Mutat. 1995;5(1):66–75.
Gergics P, Patocs A, Toth M, Igaz P, Szucs N, Liko I, et al. Germline VHL gene mutations in Hungarian families with von Hippel-Lindau disease and patients with apparently sporadic unilateral pheochromocytomas. Eur J Endocrinol. 2009;161(3):495–502.
Nordstrom-O’Brien M, van der Luijt RB, van Rooijen E, van den Ouweland AM, Majoor-Krakauer DF, Lolkema MP, et al. Genetic analysis of von Hippel-Lindau disease. Hum Mutat. 2010;31(5):521–37.
den Dunnen JT, Antonarakis SE. Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum Mutat. 2000;15(1):7–12.
Wildeman M, van Ophuizen E, den Dunnen JT, Taschner PE. Improving sequence variant descriptions in mutation databases and literature using the Mutalyzer sequence variation nomenclature checker. Hum Mutat. 2008;29(1):6–13.
McNeill A, Rattenberry E, Barber R, Killick P, MacDonald F, Maher ER. Genotype-phenotype correlations in VHL exon deletions. Am J Med Genet A. 2009;149A(10):2147–51.
Zbar B, Kishida T, Chen F, Schmidt L, Maher ER, Richards FM, et al. Germline mutations in the von Hippel-Lindau disease (VHL) gene in families from North America, Europe, and Japan. Hum Mutat. 1996;8(4):348–57.
Forman JR, Worth CL, Bickerton GR, Eisen TG, Blundell TL. Structural bioinformatics mutation analysis reveals genotype-phenotype correlations in von Hippel-Lindau disease and suggests molecular mechanisms of tumorigenesis. Proteins. 2009;77(1):84–96.
Shuin T, Yao M, Shinohara N, Yamasaki I, Tamura K. Clinical status of Von Hippel-Lindau disease associated pheochromocytoma in Japan: a national epidemiologic survey. Nihon Hinyokika Gakkai Zasshi. 2012;103(3):557–61 (in Japanese).
Ong KR, Woodward ER, Killick P, Lim C, Macdonald F, Maher ER. Genotype-phenotype correlations in von Hippel-Lindau disease. Hum Mutat. 2007;28(2):143–9.
Wong WT, Agron E, Coleman HR, Tran T, Reed GF, Csaky K, et al. Clinical characterization of retinal capillary hemangioblastomas in a large population of patients with von Hippel-Lindau disease. Ophthalmology. 2008;115(1):181–8.
Mettu P, Agron E, Samtani S, Chew EY, Wong WT. Genotype-phenotype correlation in ocular von Hippel-Lindau (VHL) disease: the effect of missense mutation position on ocular VHL phenotype. Invest Ophthalmol Vis Sci. 2010;51(9):4464–70.
Wong WT, Agron E, Coleman HR, Reed GF, Csaky K, Peterson J, et al. Genotype-phenotype correlation in von Hippel-Lindau disease with retinal angiomatosis. Arch Ophthalmol. 2007;125(2):239–45.
Gallou C, Chauveau D, Richard S, Joly D, Giraud S, Olschwang S, et al. Genotype-phenotype correlation in von Hippel-Lindau families with renal lesions. Hum Mutat. 2004;24(3):215–24.
Authors’ contributions
MW, YHC, and HB collected the data from articles in the English, Chinese, and Japanese languages, respectively. JN, TT, and MHT contributed patient data. MHT, MW, and HLT designed the study, analyzed the data, and wrote the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Wong, M., Chu, YH., Tan, H.L. et al. Clinical and molecular characteristics of East Asian patients with von Hippel–Lindau syndrome. Chin J Cancer 35, 79 (2016). https://doi.org/10.1186/s40880-016-0141-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40880-016-0141-z