
Abstract
Background
Trichoderma reesei represents a model system for investigation of plant cell wall degradation and its connection to light response. The cyclic adenosine monophosphate pathway (cAMP pathway) plays an important role in both physiological outputs, being crucial for regulation of photoreceptor function as well as for cellulase regulation on different carbon sources. Phosphorylation of photoreceptors and of the carbon catabolite repressor CRE1 was shown in ascomycetes, indicating a relevance of protein kinase A in regulation of the target genes of these transcription factors as well as an impact on regulation of induction specific genes. Moreover, the cAMP pathway impacts growth and development.
Results
Here, we investigated gene regulation by the catalytic subunit of protein kinase A (PKAc1) upon growth on cellulose. We found distinct gene sets for regulation upon growth in light and darkness with an overlap of only 13 genes. PKAc1 regulates metabolic genes as well as transport and defense functions. The overlap of gene regulation by PKAc1 with the genes representing the cAMP dependent regulatory output of the photoreceptor ENV1 indicates an involvement of PKA in this pathway, which counteracts its effects by contrasting regulation. Moreover, we found considerable overlap with the gene sets regulated under cellulase inducing conditions and by the carbon catabolite repressor CRE1. Our analysis also showed that PKAc1 regulates the genes of the SOR cluster associated with the biosynthesis of sorbicillinoids. The homologue of gin4, encoding a CAMK type kinase, which is regulated by PKAc1, CRE1 and YPR2 showed a moderate impact on trichodimerol production. We isolated trichodimerol as representative sorbicillin compound and established a method for its quantification in large sample sets using high performance thin layer chromatography (HPTLC), which can be broadly applied for secondary metabolite screening of mutants or different growth conditions. Due to the high expression levels of the SOR cluster under conditions of sexual development we crosschecked the relevance of PKAc1 under these conditions. We could show that PKAc1 impacts biosynthesis of trichodimerol in axenic growth and upon mating.
Conclusions
We conclude that PKAc1 is involved in light dependent regulation of plant cell wall degradation, including carbon catabolite repression as well as secondary metabolism and development in T. reesei.
Similar content being viewed by others

Background
Trichoderma reesei is one of the most prolific enzyme producers in industry worldwide and plays a crucial role in expression of plant cell wall degrading enzymes for second generation biofuel production [1, 2]. Therefore, its enzyme system is studied in detail as are the complex regulation systems supporting high efficiency production of homologous and heterologous proteins [3]. Regulation of plant cell wall degrading enzymes is induced in response to carbon sources such as cellulose, lactose and sophorose, the natural inducer, and genes encoding the major enzymes for this purpose are coregulated [3,4,5,6]. Numerous transcription factors are involved in adjustment of transcript levels of plant cell wall degrading enzymes to environmental conditions, which can act positively or negatively [7]. In T. reesei the most important transcription factors are XYR1 [8] and ACE3 [9], which are essential for cellulase gene expression, ACE1 [10], which negatively affects cellulases and CRE1 [11], the carbon catabolite repressor, which represses cellulase formation in the presence of easily metabolizable carbon sources. Besides regulation at the transcriptional level, recent analyses revealed that cellulase expression is also regulated at a posttranscriptional level in dependence of nutrient sensing via the heterotrimeric G-protein pathway [12].
In addition to the carbon source, also other environmental cues influence enzyme expression [13]. Especially light was shown to considerably impact cellulase gene expression in T. reesei and the gene set specific for inducing conditions is different in light and darkness [12, 14]. Light response in T. reesei is predominantly mediated by the photoreceptor complex consisting of BLR1 and BLR2 (blue light regulator 1 and 2) as well as by the photoreceptor ENV1 [15]. While BLR1 and BLR2 are GATA-type transcription factors, ENV1 is assumed to act on this complex by protein–protein interaction adjusting its function. All three components impact cellulase gene expression as well as transcript levels of numerous CAZyme encoding genes in T. reesei [16, 17]. The functions of ENV1 in T. reesei further extend to sexual and asexual development, growth and stress response [14].
Cyclic AMP (cAMP) is a central second messenger in the cellular function of all organisms and highly conserved across the kingdoms of life. The cAMP pathway crucially regulates morphology and nutrient sensing in fungi [18, 19].
The genome of T. reesei harbors an adenylate cyclase gene, an adenylate cyclase associated gene, two genes encoding catalytic subunits of protein kinase A (pkac1 and pkac2), one regulatory subunit encoding gene and two phosphodiesterase genes [2]. Protein kinase A is the main target of cAMP signaling. PKAc1 was found to be the major catalytic subunit of protein kinase A in N. crassa [20] and plays an important role in circadian rhythmicity as well as stabilization of the photoreceptor complex WCC and the protein FREQUENCY (FRQ1) [21,22,23]. For Aspergillus nidulans a function of PkaA was found in biomass formation in complete media and on cellulose, endocellulase activity, expression of xylanases and cellulases [24]. Additionally, CreA is differentially phosphorylated depending on the presence of PkaA, resulting in altered characteristics for import in the nucleus [25]. A positive role of the cAMP pathway as well as the heterotrimeric G-protein pathway on cellulase gene expression and secondary metabolism was further shown in Chaetomium globosum [26, 27]. Besides functions in development, a role of the cAMP pathway in secondary metabolism was shown for Aspergillus fumigatus, Pestalotiopsis microspora and Fusarium graminearum [28,29,30].
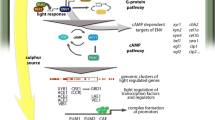
In T. reesei a broad physiological impact of the cAMP and the machinery adjusting its abundance is known [31,32,33,34]. The diverse functions of this pathway as shown in T. reesei and other fungi (Fig. 1) make protein kinase A an attractive target for investigation. The positive effect of cAMP on formation of endoglucanases in T. reesei has been described early in research on cellulase regulation [35] and confirmed with investigation of adenylate cyclase and protein kinase A [36]. Thereby, the cAMP level is modulated in response to the carbon source and is higher in the presence of the natural cellulase inducing compound sophorose than it is in the presence of cellulose, lactose, cellobiose or glucose, which corresponds to highest levels of cellulase gene expression on this carbon source [37]. The heterotrimeric G-protein pathway is one of the most important nutrient sensing pathways in fungi. In agreement with a carbon source dependent adjustment of cAMP levels, the G-protein alpha subunits GNA1 and GNA3 both influence intracellular cAMP content of T. reesei upon growth on cellulose [38, 39]. Interestingly, the impact of GNA1 and GNA3 as well as of pkac1 and acy1 is different in light and darkness under inducing conditions [36, 38, 39].

Model for the regulatory function of PKA in the cAMP pathway. Adenylate cyclase ACY impacts cAMP dependent function of PKA. The regulation of plant cell wall degrading enzymes by PKA is in parts mediated via phosphorylation of the carbon catabolite repressor CRE1 and the photoreceptor complex of BLR1 and BLR2. Function of the photoreceptor ENV1 on the BLR-complex overlaps with PKA regulation regarding enzyme production. The regulatory effect of ENV1 in part shows a cAMP level dependent output. PKA further positively impacts sexual development as well as production of secondary metabolites (for details see “Background”)
The light dependent functions of both the G-protein alpha subunits as well as of acy1 and pkac1 in T. reesei indicated a connection of the cAMP pathway to the light response pathway. Indeed, in a mutant lacking the photoreceptor ENV1, cAMP levels are strongly decreased. This phenotype is prevalent, even if GNA1 or GNA3 are constitutively activated in such a strain [40]. Additionally, genes regulated by ENV1 and ACY1, and hence likely reflecting the cAMP dependent output of the ENV1 regulon, show considerable overlap in light upon growth on cellulose [41]. Analysis of transcriptomes from different carbon sources suggests that this cAMP dependent regulatory output of ENV1 may be involved in substrate- or surface sensing [12]. The characteristic phenotype of strains lacking ENV1 (growth defect in light, altered sporulation [42, 43]) is reminiscent of both the perturbed growth of acy1 and pkac1 deletion strains and is hence likely to be cAMP related as well.
In this study we investigated the alterations in the transcriptome in dependence of the catalytic subunit 1 of protein kinase A (pkac1). Evaluation of the regulatory targets of PKAc1 showed functions in enzyme expression in light and darkness as well as modulation of secreted secondary metabolites upon growth on cellulose. Comparison of the PKAc1 regulome with those of known targets of PKA revealed considerable overlapping regulation, implicating the CAMK kinase GIN4 as potential target. Importantly, we also optimized a method for fungal secondary metabolite production by high performance thin layer chromatography (HPTLC) with a focus on large scale screening and metabolite quantification, using trichodimerol as representative compound.
Results
Transcriptome analysis of ∆pkac1
Previous studies showed that the cAMP pathway, including pkac1, is involved in regulation of cellulase gene expression in T. reesei upon growth on the cellulase inducing carbon source lactose [36]. Based on DNA–protein interaction studies and gene regulation analysis of the cellulase regulator xyr1, which is co-regulated with the cellobiohydrolase gene cbh1 under these conditions, it was concluded, that PKAc1 acts on a transcriptional regulator impacting XYR1, rather than on XYR1 itself [36].
We investigated the transcriptome of ∆pkac1 in comparison the the wildtype QM9414 upon growth on cellulose, which is more closely related to the natural substrate of T. reesei (Additional file 1). Due to the previously reported effect of light on the regulatory output of PKAc1 [36] and the functions of its homologues in N. crassa in regulation of the light response machinery as well as circadian rhythmicity [21, 22], we performed our experiments under controlled light conditions in constant light and constant darkness.
In order to validate the results of the transcriptome data we checked whether transcript levels of the wild-type correspond to known regulation patterns from previous studies [44]. The strongly light regulated genes env1 and hpp1 as well as TR_68924 and TR_123955 show the characteristic decrease in transcript levels in constant light compared to darkness. TR_123865 and TR_44278 show the characteristic increase in transcript abundance in light in the wild-type in our dataset. Hence we consider our transcriptome data valid and reliable.
PKAc1 impacts metabolism and transport functions
The regulatory targets of PKAc1 (indirect, as PKAc1 is not a transcription factor; Additional file 1) exert metabolic functions and comprise genes involved in signaling, transport and defense (Fig. 2a, b; Additional file 2). The genes regulated by PKAc1 in light are enriched in functions of transport (p-value 1.30E−04), particularly C-compound and carbohydrate transport (p-value 2.16E−05), nitrogen, sulphur and selenium metabolism (p-value 3.06E−4), C-compound and carbohydrate metabolism (p-value 9.7E−04) as well as in glycolysis and gluconeogenesis (p-value 2.68E−03). In darkness, the gene set regulated by PKAc is particularly enriched in glycogen catabolism (p-value 3.8E−04). Other metabolic functions as well as functions in energy supply are enriched to a lower extent (p-value < 5E−02).

Functional category analysis of genes regulated by PKAc1 in a light or b darkness upon growth on cellulose in liquid culture. Selected, particularly relevant categories are shown
Genes regulated by PKAc1 in darkness
Constant darkness represents a condition closely related to the conditions of industrial fermentation and is hence most interesting for studying T. reesei. We found 170 genes to be more than twofold regulated (p-value threshold 0.01) in constant darkness compared to wildtype (Additional file 1). Among the 128 genes downregulated in darkness, we found 6 CAZyme encoding genes including a predicted trehalase as well as eight transcription factor genes, of which TR_122523 was previously shown to positively influence plant cell wall degradation [9]. TR_123881 (SxlR), which is more than threefold downregulated in ∆pkac1, represses xylanase, but not cellulase activity [45].
Interestingly, under these conditions the recently described SOR cluster, which is responsible for biosynthesis of dihydrotrichotetronine on cellulose [46] and impacts sorbicilline formation upon growth on glucose [47], is downregulated as well. The consistently positive effect of PKAc1 on the polyketide synthases (pks) sor1 and sor2, the predicted flavoprotein monooxygenase gene sor5, the transporter sor4 as well as the transcription factor ypr2 (sor3) supports a regulation of this cluster via nutrient dependent phosphorylation. The second transcription factor gene in this cluster, ypr1, is not regulated by PKAc1 on cellulose, but positively regulated by the adenylate cyclase (ACY1) upon growth on cellulose in darkness [41]. For ypr2, the opposite is the case, it is not regulated by ACY1 [41]. Previously it was shown that the transcription factors YPR1 and YPR2 exert their functions in a different way on glucose and cellulose [46, 47]. Accordingly, carbon source dependent regulation of ypr1 and ypr2 is different between inducing and repressing carbon sources [12].
In darkness, only three genomic clusters of regulation were detected (Additional file 1), one of them being the SOR cluster on chromosome 5 of the recent genome sequence of QM6a [48].
The 43 genes upregulated in ∆pkac1 in darkness included the small, cellulose specific gene ooc1 [49], the polyketide synthase pks4g, which was shown to be responsible for the green spore coloration in T. reesei [50]. Additionally, transcript levels the gene represented by the model TR_64125, encoding a protein kinase were increased. Moreover, this gene set comprises the protein phosphatase gene pp1 (TR_120722), which is related to N. crassa histidine phosphatase pph-3 as well as two putative multicopper oxidases (TR_102820 and TR_124079) (Additional file 1). Of the two transcription factor genes upregulated in darkness in ∆pkac1, TR_112524 was screened for a function in plant cell wall degradation, which was however not detected [9].
Genes regulated by PKAc1 in light
Upon growth in light with cellulose as carbon source, 225 genes were regulated compared to wild-type, 126 showed decreased transcript levels and 99 increased transcript levels in ∆pkac1 (Additional file 1). Among the 126 genes downregulated in light, we found 9 CAZyme encoding genes including gph1 encoding a glycogen phosphorylase regulated by Hog1 under stress conditions in Candida albicans [51] and the light dependent induction of which is positively regulated by ENV1. Additionally, this gene set comprises the gene encoding the mannitol dehydrogenase lxr1 [52, 53] which resides in a gene cluster regulated by the photoreceptor ENV1 [17]. We also found downregulation of 5 genes associated to secondary metabolism, of them 4 cytochrome P450 encoding genes and 8 transcription factor genes of unknown function. The transcription factor encoding TR_54703 was tested for a positive function on plant cell wall degradation, but no alteration in enzyme expression was found [9].
Of the 6 transporter encoding genes regulated by PKAc1 in light, there is also ste6, encoding the putative pheromone transporter, which is downregulated in ∆pkac1. This is in agreement with the previously reported positive effect of the cAMP pathway on sexual development [36].
For 99 genes we detected upregulation in ∆pkac1 in light. This geneset comprised 6 CAZyme including cip2, cel3d and egl5/cel45a, 9 genes involved in sulphur metabolism, among them 5 taurine dioxygenases and 3 methionine permeases. Moreover, we found 2 transcription factor encoding genes including vib1 and TR_3449 as well as 3 transporters including MFS hexose transporter CLP1, which transports cellodextrins in N. crassa [54].
Interestingly, of the genes regulated by PKAc in light, cip2 and the glycoside hydrolase family 30 xylanase TR_69276, which is assumed to have both endoxylanase and endoglucanase functionality [55] encode proteins that are listed among the three most important limiting proteins for hydrolysis of pretreated corn stover (PCS), a typical substrate for industrial enzyme production [55].
Light independent targets of PKAc1
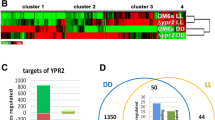
Of the 382 genes regulated by PKAc1 upon growth on cellulose, only 13 genes showed regulation in both light and darkness, suggesting a considerable light dependent relevance for PKAc1 (Fig. 3a). The gene encoding the regulatory subunit of protein kinase A, pkar1, the CAZyme encoding gene TR_120198, the catalase gene cat8 and the glycogen phosphorylase gene gph1 are downregulated by PKAc1 in light and darkness. Of the two transcription factor genes regulated light-independently by PKAc1 (TR_105520 and TR_122523), TR_122523 influences cellulase regulation in T. reesei [9]. Contrasting regulation in light and darkness was found for TR_81122 (upregulated in light and downregulated in darkness) and TR_109378 is several fold upregulated in light and darkness in ∆pkac1.

Genes regulated by PKAc1 and their correlation with patterns in other strains. a Overlap between genes regulated by PKAc1 in light (LL) and darkness (DD) is shown. b Overlap between induction specific genes [12] and genes regulated by PKAc1 is shown. Regulation in darkness (DD) is shown in blue, regulation in light is shown in yellow. c Overlap between genes regulated by PKAc1 and CRE1 [46] in light. d Overlap between genes regulated by PKAc1 and CRE1 [46] in darkness. e Overlapping regulation between PKAc1 and BLR1 [17] in light. f Overlapping regulation between the gene set reflecting the cAMP dependent output of the photoreceptor ENV1 [41] and regulation by PKAc1 in light. Genes consistently upregulated in the compared gene sets a, c–f are shown in green, those downregulated are shown in red. If a contrasting regulation was observed, the situation in ∆pkac1 is shown in the figure (third column) with upregulation in yellow and downregulation in blue
Induction specific genes regulated by PKAc1
Protein kinase A impacts diverse important physiological pathways in fungi (Fig. 1), often by phosphorylation of important transcription factors including photoreceptors [21] and the carbon catabolite repressor CRE1 [25]. Due to the relevance of PKAc1 in cellulase regulation [24, 36], a more general impact on induction specific genes is also expected. Therefore, we evaluated available transcriptomes from growth of T. reesei on cellulose in light and darkness for overlapping or contrasting regulation indicating functions in the same pathway.
Induction of plant cell wall degrading enzymes and more specifically of cellulases happens in response to distinct carbon sources, mainly representing plant cell wall components [56]. We were therefore interested if the core set of induction specific genes (regulation on the inducing carbon sources cellulose, lactose and sophorose versus glucose and glycerol, in the presence of which no cellulases are induced; [12]) in light and darkness is subject to regulation by PKAc1.
Of the 170 PKAc targets (indirect) in darkness, 48 overlapped with genes regulated in an induction specific manner [12], most of which were regulated in the opposite direction in ∆pkac1 (positive vs. negative regulation; contrasting) (Additional file 3) (Fig. 3b). These overlapping genes were enriched in the function of catalase reaction (p-value 8.45e−04), but comprised also genes associated with stress response, metabolism including C-compound and carbohydrate metabolism and transport functions.
Among the 225 genes regulated by PKAc1 in light, 91 genes overlapped with the previously reported genes specifically regulated under inducing conditions [12] (Fig. 2c). As seen in darkness, in many cases the regulation in ∆pkac1 was contrasting that under inducing conditions (Additional file 3). The function of catalase reaction was also enriched among the genes regulated by PKAc1 in light (p-value 3.12E−03) as were those involved in secondary metabolism (p-value 9.16E−03). Additionally, 37 genes associated with metabolism, particularly with C-compound and carbohydrate metabolism, amino acid metabolism, detoxification and transport functions were detected in this gene set.
The overlap of induction specific genes and those regulated by PKAc1 is limited to only six genes between light and darkness. Consequently, regulation of induction specific genes by PKAc is light dependent with distinct target gene sets. The contrasting regulation both in light and in darkness suggests that particularly induction specific up-regulation under the respective conditions is dependent on the function of PKAc1.
Overlapping targets with CRE1
The carbon catabolite repressor CRE1 shows the potential for cAMP dependent phosphorylation in its protein sequence. Moreover, an influence on phosphorylation of CRE1 was recently shown for PKA in A. nidulans [25]. Therefore we screened the respective dataset for gene regulation by CRE1 in constant light and constant darkness upon growth on cellulose [46] for overlapping regulation with PKAc1 (Additional file 4).
Of the 225 genes regulated by PKAc in light, 99 were also regulated by CRE1 in light (Fig. 3c). Among these 99 genes, 46 were downregulated in ∆pkac1 and in ∆cre1 in light. Fifty-one genes were upregulated in ∆pkac1 and in ∆cre1 in light. They comprise cip1 and cel3d, several putative permeases and genes involved in sulphur metabolism. However, two genes were upregulated in ∆pkac1 and downregulated in ∆cre1 in light.
In darkness, 48 genes were downregulated in ∆pkac1 and ∆cre1 (Fig. 3d), among them four CAZyme encoding genes, the catalase gene cat2 and the superoxidedismutase gene sod1. Twenty-one genes were upregulated in ∆pkac1 and ∆cre1, which comprise ooc1 and pks4 as well as the putative multicopperoxidase gene TR_124079. Only two genes showed contrasting regulation including the regulator of G-protein signaling (RGS) gene rgs2 (TR_72259). These overlapping regulation patterns (Fig. 3c, d) support the hypothesis of a positive effect of phosphorylation by protein kinase A in the function of CRE1.
Overlapping targets with photoreceptors
In N. crassa, a function for protein kinase A (PKA) in phosphorylation dependent regulation of the photoreceptor complex (white collar complex; WCC) activity was shown and PKA was found to serve as a priming kinase for the casein kinase dependent phosphorylation of the WCC components [57]. The T. reesei homologues of the WC photoreceptor complex, BLR1 and BLR2 [16], comprise putative cAMP dependent phosphorylation sites (data not shown). Hence, deletion of pkac1 may in part cause similar gene regulation as lack of functionality or deletion of blr1 or blr2.
Comparison of gene regulation by light in ∆pkac1 with that in ∆blr1 and ∆blr2 [17] showed 98 of the 225 genes regulated by PKAc1 in light to be regulated by BLR1 in light as well (Fig. 3e) (Additional file 4). They include 15 CAZyme genes, 10 genes involved in sulfur metabolism, 10 transcription factor encoding genes including vib1, which was recently shown to regulate cellulase gene expression [58] and 9 genes encoding transporters.
Most of the genes overlapping between BLR1 targets and PKAc1 targets in light were consistently regulated in both mutants, but in some cases also contrasting regulation was observed (Fig. 3e). Of the 76 genes down regulated in ∆blr1, 22 were up-regulated in ∆pkac1. Interestingly, these genes comprise four CAZyme encoding genes including cip2, cel3d and egl5 as well as the hexose transporter gene clp1, which is located next to cel3d in the genome.
Among the 22 genes up regulated in ∆blr1, 7 were down-regulated in ∆pkac1. They include three transcription factors (TR_120975, TR_122523 and TR_105220). TR_122523 was previously shown to positively influence cellulase gene expression and to be co-expressed with many hemicellulase genes [9].
Many of the genes with regulation in ∆pkac1 and ∆blr1 in light are also regulated in ∆blr2 (Additional file 4). Ninety of the 225 genes were regulated by both. Again 39 of the genes down regulated in ∆blr2 showed contrasting regulation in ∆pkac1, with 7 genes involved in sulphur metabolism. Up-regulated genes in ∆blr2, which are down-regulated in ∆pkac1 comprise the same transcription factor genes showing contrasting regulation with ∆blr1. Protein kinase A was shown to inhibit the activity of the photoreceptor complex by acting as a priming kinase in N. crassa [21]. Hence deletion of protein kinase A would lead to increased photoreceptor activity. Our findings show a complex picture in this respect. Positive regulation of transcription factors by the photoreceptor complex BLR1–BLR2 (up-regulation in the mutants) occurs in the wildtype. Under these conditions, inhibition of the photoreceptor complex (to some extent) by PKA is operative. Deletion of pkac1 should alleviate the negative effect on BLR1 and BLR2 activity and the regulation by the photoreceptor complex should remain or even increase. This is the case for two transcription factor genes (TR_71823 and TR_105980). However, we also saw the opposite effect for three transcription factors (TR_120975, TR_105220 and TR_122523), potentially reflecting an indirect effect of the presence of pkac1 on a downstream component of the regulatory cascade of BLR1 and BLR2.
Functional category analysis of genes with altered transcript levels in ∆pkac1, ∆blr1 and ∆blr2 in light, indicating regulation by an effect of PKAc1 on activity of the photoreceptor complex showed enrichment (p-value threshold < 5e−02) in nitrogen, sulfur and selenium metabolism (p-value 8.48e−3), C-compound and carbohydrate transport (p-value 8.19e−04) and detoxification involving cytochrome P450 (p-value 3.0e−03) (Additional file 2).
The cAMP dependent output of ENV1
Deletion of env1 causes strongly decreased cAMP levels and a severe growth defect in light [40, 43]. Comparison of the transcriptional targets of ENV1 with those of the adenylate cyclase ACY1, revealed an overlap of 31 genes up-regulated in light and 114 genes down regulated in light, which represent the cAMP dependent regulatory targets of the photoreceptor ENV1. No genes with contrasting regulation in the two mutants were found [41]. In part these genes also correlate with genes implicated in surface sensing in T. reesei [12].
Of the respective gene set down regulated in light in ∆env1 and ∆acy1, 29 of 114 genes were also regulated by PKAc1, but surprisingly all of them were upregulated in ∆pkac1 (Fig. 3f) (Additional file 4). They include the CAZyme encoding genes cip1 and egl5, four genes involved in sulphur metabolism as well as the transcription factor vib1. Also the 8 genes which overlap with the 31 genes up-regulated in ∆acy1 and ∆env1 in light, show contrasting regulation in ∆pkac1. We conclude that part of the cAMP dependent regulatory output of ENV1 in light is mediated by PKAc1, including the important transcription factor gene vib1. The overlapping regulation also with the photoreceptors BLR1 and BLR2 emphasizes the light dependent function of VIB1 in light dependent substrate sensing and cellulase regulation, which remains to be studied in detail.
GIN4 moderately influences trichodimerol levels
Due to the positive influence of PKAc1 on SOR cluster genes, we checked regulated genes overlapping with the CRE1 regulon to select a potential regulator of sorbicillin production. TR_64125, encoding a homologue of the CAMK kinase GIN4 is up-regulated in ∆pkac1 (2.8fold) and in ∆cre1 (20-fold) in darkness. Additionally, transcript abundance of this gene is more than twofold significantly decreased in a strain lacking the SOR-cluster transcription factor YPR2 under the same conditions [59].
Analysis of a deletion strain of T. reesei GIN4 for production of trichodimerol representing the compounds associated with the SOR cluster showed only a moderate alteration of trichodimerol levels (Fig. 4a). Lack of A. nidulans Gin4 in the genome causes earlier sexual development [60]. Hence we analyzed mating behaviour of ∆gin4, which revealed no significant difference to the wildtype in timing for fruiting body formation or morphology (Fig. 4b).

Influence of gin4 on trichodimerol production and fruiting body development. a Trichodimerol production in cellulose liquid culture in total darkness of ∆gin4 relative to wildtype (QM6a∆mus53) (p-value = 0.075). b Sexual development of ∆gin4 and QM6a with FF1 after 7 and 14 days
Establishing a method for large scale screening of fungal secondary metabolite patterns
We found that PKAc1 is involved in regulation of the SOR cluster at the transcript level. Previously, genes of this cluster were shown to be among the most strongly transcribed genes under conditions of sexual development, considerably exceeding the transcript levels on cellulose [61]. Therefore, we aimed to cross-check the impact of PKAc1 on production of sorbicillin compounds under mating conditions. Deletion of pkac1 causes delayed fruiting body formation in T. reesei [36] and N. crassa [62], indicating a positive effect of the cAMP pathway on sexual development, although it is not essential for mating [36].
First, we aimed to isolate a representative sorbicillin compound as a reference in our analyses. Then we optimized secondary metabolite extraction and the previously tested method of high performance thin layer chromatography (HPTLC) to establish a method for large scale screening of samples from diverse fungal strains and environmental conditions.
HPTLC allows for analysis of patterns of secreted metabolites of multiple samples (tens to even hundreds within reasonable time), which can be directly visually compared without employing complex and costly metabolomics tools like mass spectrometry. Reliable identification and quantification of individual compounds based on authentic standards is possible [63, 64]. The provided workflow also allows extraction of liquid samples from culture filtrates—ideally related to the produced biomass. Consequently, we consider HPTLC as the optimal system for screening of mutant libraries and the evaluation of diverse conditions for selection of samples for in depth analysis by mass spectrometry.
PKAc1 impacts trichodimerol production
Crossing plates containing the wild-type strains FF1 and FF2 [65], both sexually fertile, were extracted with a focus on potential sorbicillinoids. After chromatographic purification, NMR analysis confirmed the identity of the sorbicillin derivative trichodimerol, production of which is associated with the SOR cluster [46]. Trichodimerol was produced under conditions of sexual development and therefore reflects chemical communication events between the two (potential) mating partners. Hence, trichodimerol can be used as a reference in high performance thin layer chromatography (HPTLC) analyses for secondary metabolite production during sexual development.
To investigate secondary metabolite production upon sexual development we grew ∆pkac1 alone and in the presence of a mating partner. Additionally, we tested the response of a wild-type strain to the encounter of ∆pkac1. Wildtype combinations were used as controls (Fig. 5a–f). This analysis showed that already in the absence of a mating partner, secondary metabolite production of ∆pkac1 was decreased compared to wildtype (Fig. 5a–c, arrows) including an impact on potential sorbicillin derivatives. For the dimeric sorbicillin derivative trichodimerol (Fig. 5g) as representative of sorbinillinoids (Fig. 5a–f, triangles) a decrease by 32.4% (± 6.7%, p-value 0.012) was detected (Fig. 5h). The reaction of both wildtype and ∆pkac1 to the presence of CBS999.97 MAT1-1 as mating partner was only subtle (Fig. 5a–c, arrows). In contrast, the reaction of the fully fertile strain CBS999.97 MAT1-2 to the parental strain of ∆pkac1, QM9414 was very clear compared to axenic growth of this strain (Fig. 5d–f, arrows). Also potential sorbicillin compounds were concerned (Fig. 5f) and trichodimerol showed a positive trend. Deletion of pkac1 almost abolished this response and resulted in a lower abundance of trichodimerol compared to axenic growth by 30.1% (± 9.3%, p-value 0.028) (Fig. 5i). Consequently, the association of the SOR cluster and its products with chemical communication during sexual development is supported as well as a role of PKAc1 in this process.

Detection and quantification of trichodimerol. a–f High performance thin layer chromatography (HPTLC) analysis of ∆pkac1. Triangles show trichodimerol. Arrows highlight major differences between samples. Secondary metabolite patterns of ∆pkac1 and wildtype QM9414 under asexual and crossing conditions (a–c) and reaction of CBS1-1 after 14 days (d–f) on 2% MEX at 22 °C, LD. Visualization: a, d fluorescence at 366 nm, b, e visible light, c, f visible light with low saturation for better illustration. Analyses were done in three biological replicates with tree pooled plates per replicate. Replicates for HPTLC analysis were consistent and are provided in Additional file 5. g Trichodimerol. HR ESI–MS m/z 497.2164 [M+H]+ (calcd for C28H33O8, 497.2175), m/z 519.1994 [M+Na]+ (calcd for C28H32O8Na, 519.1995); 1H NMR (600 MHz, CD3OD): δH = 7.29 (1H, dd, J = 14.9 Hz, J = 10.9 Hz, H-9), 6.39 (1H, dd, J = 15.0 Hz, J = 10.9 Hz, H-10), 6.35 (1H, d, J = 14.9 Hz, H-8), 6.24 (1H, dq, J = 15.0 Hz, J = 7.0 Hz, H-11), 3.11 (1H, s, H-1), 1.92 (3H, d, J = 7.0 Hz, H-12), 1.40 (3H, s, H-14), 1.38 (3H, s, H-13); 13C NMR (150 MHz, CD3OD): δC = 201.3 (s, C-5), 175.8 (s, C-7), 144.1 (d, C-9), 140.8 (d, C-11), 132.7 (d, C-10), 120.2 (d, C-8), 105.7 (s, C-3), 104.6 (s, C-6), 80.3 (s, C-2), 60.9 (s, C-4), 58.6 (d, C-1), 21.7 (q, C-13), 19.8 (q, C-14), 18.7 (q, C-12). Numbering of protons and carbons is shown. All data in agreement with those reported earlier for this compound [79]. h Quantification of trichodimerol in axenic growth in the parental strain QM9414 (Q) and in ∆pkac1 (P) compared to interaction with the fully fertile strain CBS999.97 MAT1-1 (C) under conditions favoring sexual development (corresponds to HPTLC data on panel D). i Quantification of trichodimerol in axenic growth in CBS999.97 MAT1-1 compared to interaction with QM9414 and ∆pkac1 under conditions favoring sexual development (corresponds to HPTLC data on panel c). Error bars reflect standard deviations, *p-value < 0.05 and **p-value < 0.01
Discussion
The cAMP pathway represents one of the most important signaling checkpoints in living organisms, with protein kinase A being a central component of the signal transmission machinery. Due to its function in adjustment of the levels of the secondary messenger cAMP, this pathway was postulated to be a coincidence detector [66] or a means to integrate signals from different sources. The components of this pathway as well as cAMP itself were previously shown to impact cellulase regulation [67] in a concentration dependent manner [35]. Additionally, considerable light dependent effects in correlation with the cAMP pathway were observed including altered membrane potential, intracellular levels of ATP and cAMP and an increase of oxygen consumption [68]. cAMP dependent protein kinase A is an important output pathway of altered cAMP levels and represents one of the factors mediating the physiological effects of changing cAMP concentrations in the cell.
Besides an influence on regulation of several CAZyme encoding genes, our data showed that PKAc1 also impacts glycogen metabolism. Interestingly, several genes involved in metabolism of energy reserves like glycogen or trehalose are downregulated in ∆pkac1, for example the glycoside hydrolase family 31 encoding TR_82235 and the putative trehalase TR_123456. The glycosylphosphorylase gene gph1, (glycosyltransferase family 35; TR_120198), is down regulated as well in light and darkness in ∆pkac1. This gene is located between the G-protein alpha subunit gene gna3 and the MAPkinase gene tmk3 [38] and is regulated by light and ENV1 in T. reesei [69]. Moreover, growth on glycogen as carbon source is decreased in a mutant lacking ENV1, but not in wildtype in light [69]. The regulatory output of ENV1 is in part mediated by adjustment of cAMP levels [40, 41], particularly in light. Also gene regulation in ∆pkac1 overlaps with the gene set representing the cAMP dependent output of ENV1. Intriguingly, all these genes show contrasting regulation in ∆pkac1 compared to ∆env1 and ∆acy1 (Fig. 2g). Consequently, the cAMP dependent output of ENV1 involves the function of PKAc1 as well. Moreover, the previously reported involvement of the cAMP pathway in energy supply of the cell and adjustment of reserve carbohydrates is in agreement with the function of PKAc1, which positively influences genes involved in degradation of glycogen and trehalose.
Besides the connection of PKAc1 to the photoreceptor ENV1, also the considerable overlap of gene regulation by PKAc1 with the components of the photoreceptor complex BLRC, BLR1 and BLR2, strengthens the importance of PKA in dealing with altered light conditions in T. reesei.
One of the most interesting genes in this PKAc1 mediated, cAMP dependent output of ENV1 is the regulator gene vib1. In N. crassa, VIB1 links glucose signaling to carbon catabolite expression and is required for cellulase gene expression, due to its impact on the transcription factor CLR2 [70]. VIB1 impacts expression of genes involved in metabolism and energy and may hence mediate the effects we saw for PKAc1. Accordingly, VIB1 is a key regulator of cellulase gene expression in T. reesei [58]. However, T. reesei CLR2 does not have a striking function in cellulase regulation in T. reesei [9], indicating rewiring of the pathway.
Secondary metabolism in T. reesei is known to be modulated in light upon growth on cellulose [46]. We isolated trichodimerol as a representative of sorbicillins, which are enhanced under conditions of sexual development [61] and demonstrate its regulation by PKAc1. Together with the refined HPTLC method for quantification, we thereby provide a means to screen large numbers of samples for modifications in trichodimerol production, which can also serve as a representative for metabolites associated with the SOR cluster including sorbicillins in general. This method is valuable as a prescreening for in depth analysis of selected samples by mass spectrometry. In addition, the presented method is applicable for extraction of secondary metabolites from supernatants of liquid cultures in relation to the biomass produced by the organism.
The genes associated with secondary metabolism regulated by PKAc1 comprise the particularly interesting secondary metabolite cluster responsible for biosynthesis of sorbicillinoids (SOR cluster). Ypr2, encoding one of the transcription factors regulating the SOR cluster, is regulated by light [59], by PKAc1 in darkness and by BLR1 and BLR2 in light. Previously we found a positive feedback cycle for regulation of the SOR cluster in darkness, which may involve the function of YPR2 [46]. Since the genes of the SOR cluster are considerably upregulated under mating conditions [61] and the production of sorbicillins including trichodimerol and dihydrotrichotetronine on cellulose is low, we chose to evaluate the role of PKAc1 in secondary metabolism in combination with sexual development. Therefore we isolated trichodimerol as a representative for the SOR cluster.
Besides a decrease in trichodimerol production and biosynthesis of putative sorbicillinoids in general we also found that in ∆pkac1 communication with a putative mating partner on the plate was altered. Interestingly, the response to a wildtype strain, that was elicited in the fully fertile CBS999.97 strain in a confrontation assay, was not observed with ∆pkac1. In addition to a certain defect in sexual development in ∆pkac1 as shown previously [36], this signaling alteration represents a further function of PKAc1 in development.
Materials and methods
Strains and culture conditions
QM9414 (ATCC26921) and ∆pkac1 [36] were used for transcriptome analysis. FF1 and FF2 [65] were used for analysis of development and CBS999.97 MAT1-1 [71] was used to study secondary metabolite production and chemical communication. For transcriptome analysis, strains were grown in 200 ml Mandels Andreotti minimal medium [72] with 1% (w/v) cellulose (Alfa Aesar, Karlsruhe, Germany) as carbon source in constant light (1800 lx) or constant darkness at 28 °C on a rotary shaker (200 rpm) for 72 h. Dark grown cultures were harvested under red safety light (darkroom light, Philips PF712E) to prevent any influence of a light pulse on transcript levels.
For HPTLC analysis, strains were cultivated on 2% malt extract agar medium at 22 °C, LD (12 h light, 12 h darkness) at 1700 lx until harvest after 14 days. Petri dishes were inoculated near the margin with agar slices of 0.5 × 4 cm from fully grown cultures to guarantee an even confrontation line.
Transcriptome analysis
For transcriptome analysis, custom arrays were used with the gene expression service as provided by Roche-NimbleGen (Madison, USA). Therefore, total RNA was isolated and quality controlled as described previously [40, 44]. Data of two biological replicates are deposited at NCBI Gene Expression Omnibus (GEO accession number GSE131419). Bioinformatic analysis was done using the PARTEK Genomics Suite 6.6 (St. Louis, USA) applying a threshold of twofold regulation and a p-value of 0.01 (false discovery rate (FDR) corrected; ANOVA statistics). Functional category analysis was done using the FUNGIFUN2 online tool [73]. Genomic clustering was evaluated using the open source software REEF [74] and adjusted manually.
Construction and analysis of ∆gin4
Deletion of gin4 (TR_64125) was performed as described previously [75] using yeast mediated recombination for vector construction and protoplast transformation for homologous integration into QM6a∆mus53 [76] and the hygromycin phosphotransferase cassette as selection marker. Successful deletion of gin4 was confirmed by PCR with primers binding within the deleted region. DNA integrity appropriate for PCR amplification of the samples representing gin4 was ensured using standard primers amplifying the tef1 gene. Sexual development was investigated under standard conditions [71, 77]. ∆gin4 was grown on minimal medium with cellulose as carbon source as described above in constant darkness. Trichodimerol production in culture filtrates was analyzed by mass spectrometry using internal standards as described previously [46, 78].
High-performance thin-layer chromatographic (HPTLC) analysis and sample preparation
Analysis was done according to Bazafkan et al. [65] with modifications. The workflow for preparation of analytical samples from fungal cultures grown on agar medium is shown in Fig. 6. All samples were measured in three biological replicates from three pooled plates each. Agar slices of 2 cm2 (0.5 × 4 cm) were collected near the confrontation zone in crossings and from the corresponding area in asexual cultures. Metabolites were extracted from collected agar slices in 15 ml centrifugation tubes by addition of 5 ml 50% acetone in water and supersonication for 15 min. Thereafter 2 ml chloroform (CHCl3) were added, the tube shaken by hand and centrifuged at 1.000g for 1 min for phase separation. Organic phase was collected in glass vials for evaporation. CHCl3 extraction was repeated two times. Dry extracts were re-collected in 140 µl CHCl3 and 5 µl applied to HPTLC analysis. Samples were spotted onto a normal phase silica gel plate (HPTLC silica gel 60 F254s, glass, 200 × 100 mm, Merck, Darmstadt, Germany, #1.1.5696.0001) with an automatic TLC sampler (ATS 4, CAMAG, Muttenz, Switzerland) with 4.5 mm band length and 5.5 mm track distance for 30 samples per plate. As mobile phase a mixture of water extracted CHCl3 and 1 mM trifluoroacetic acid in methanol 7:1 (v/v) was used. Developing was done in an automated developing chamber (ADC2, CAMAG, Muttenz, Switzerland) at a relative humidity of 11% and a migration distance of 70 mm. Metabolite patterns were analyzed at 254 nm, 366 nm and visible light with a TLC visualizer (CAMAG, Muttenz, Switzerland) before and after derivatization with p-anisaldehyde:sulfuric acid reagent. Scanning densitometry was done with a TLC scanner (Scanner 3, CAMAG, Muttenz, Switzerland) at various wavelengths (254, 290, 345, 366, 420, 470 and 520 nm) before derivatization. Results were recorded and evaluated using the software visionCATS 2.0 (CAMAG, Muttenz, Switzerland).

Workflow of sample preparation for high performance thin layer chromatography (HPTLC) analysis from fungal cultures grown on agar medium
Detection and quantification of trichodimerol using HPTLC
Trichodimerol was identified in secondary metabolite samples by comparing the Rf-value and UV/vis spectra (200–800 nm; Scanner 3, CAMAG, Muttenz, Switzerland) to those of the isolated reference compound. Trichodimerol was quantified relative to wildtype. A linear calibration based on peak height at 420 nm was obtained from combined wildtype replicates and two dilutions (1:3; n = 3). Significance was evaluated by t-test in RStudio (compare_means, ggpubr).
Isolation of trichodimerol
Well grown FF1 and FF2 plates containing 3% (w/v) malt extract agar were extracted with an excess amount of a mixture of chloroform and acetone (1:1) and sonication for 15 min. The solvent mixture was filtered and further purified by two times liquid–liquid extraction with water. The solvents of the organic phase were evaporated and the crude extract stored at − 20 °C until processing.
Size-exclusion column chromatography of 126 mg crude extract over Sephadex LH-20® [GE Healthcare; 750 mm column length, 12 mm diameter, coupled with C-640 UV detector (Büchi)] eluted isocratically with methanol yielded 16 mg of impure trichodimerol. Final purification of this fraction was accomplished by preparative thin layer chromatography (TLC) using silica gel 60 glass plates (PLC Silica gel 60 F254, 0.5 mm thickness; Merck, Darmstadt, Germany) developed in dichloromethane/methanol (98:2). This step afforded 7.2 mg trichodimerol.
NMR spectroscopy
For NMR spectroscopic measurements trichodimerol was dissolved in CD3OD (~ 3.0 mg in 0.7 mL) and transferred into 5 mm high precision NMR sample tubes. All spectra were measured on a Bruker DRX-600 at 600.25 MHz (1H) or 150.94 MHz (13C) and performed using the Topspin 3.5 software (Bruker, Rheinstetten, Germany). Measurement temperature was 298 K ± 0.05 K. 1D spectra were recorded by acquisition of 32 k data points and after zero filling to 64 k data points and Fourier transformation spectra were performed with a range of 7200 Hz (1H) and 32,000 Hz (13C), respectively. To determine the 2D COSY, TOCSY, NOESY, HMQC, and HMBC spectra 128 experiments with 2048 data points each were recorded, zero filled and Fourier transformed to 2D spectra with a range of 6000 Hz (1H) and 24,000 Hz (HSQC) or 32,000 Hz (HMBC) (13C), respectively. Residual CD2HOD was used as internal standard for 1H NMR measurements (δH 3.34) and CD3OD for 13C NMR measurements (δC 49.0).
Mass spectrometry
Mass spectra were measured on a high resolution time-of-flight (hr-TOF) mass spectrometer (maXis, Bruker Daltonics) by direct infusion electrospray ionization (ESI) in positive ionization mode (mass accuracy ± 5 ppm). TOF MS measurements have been performed within the selected mass range of m/z 100–2500. ESI was made by capillary voltage of 4 kV to maintain a (capillary) current between 30 and 50 nA. Nitrogen temperature was maintained at 180 °C using a flow rate of 4.0 l min−1 and the N2 nebulizer gas pressure at 0.3 bar.
Availability of data and materials
All data generated during this study are included in this published article and its additional files. GenBank Accession numbers for datasets analyzed for this study are given in Methods and described in the respective cited articles.
References
Martinez D, Berka RM, Henrissat B, Saloheimo M, Arvas M, Baker SE, et al. Genome sequencing and analysis of the biomass-degrading fungus Trichoderma reesei (syn. Hypocrea jecorina). Nat Biotechnol. 2008;26(5):553–60.
Schmoll M, Dattenböck C, Carreras-Villasenor N, Mendoza-Mendoza A, Tisch D, Aleman MI, et al. The genomes of three uneven siblings: footprints of the lifestyles of three Trichoderma species. Microbiol Mol Biol Rev. 2016;80(1):205–327.
Bischof RH, Ramoni J, Seiboth B. Cellulases and beyond: the first 70 years of the enzyme producer Trichoderma reesei. Microb Cell Fact. 2016;15(1):106.
Bazafkan H, Tisch D, Schmoll M. Regulation of glycoside hydrolase expression in Trichoderma. In: Gupta VK, Schmoll M, Herrera-Estrella A, Upadhyay RS, Druzhinina I, Tuohy MG, editors. Biotechnology and biology of Trichoderma. Oxford: Elsevier; 2014. p. 291–307.
Foreman PK, Brown D, Dankmeyer L, Dean R, Diener S, Dunn-Coleman NS, et al. Transcriptional regulation of biomass-degrading enzymes in the filamentous fungus Trichoderma reesei. J Biol Chem. 2003;278(34):31988–97.
Margolles-Clark E, Ilmén M, Penttilä M. Expression patterns of ten hemicellulase genes of the filamentous fungus Trichoderma reesei on various carbon sources. J Biotechnol. 1997;57:167–79.
Benocci T, Aguilar-Pontes MV, Zhou M, Seiboth B, de Vries RP. Regulators of plant biomass degradation in ascomycetous fungi. Biotechnol Biofuels. 2017;10:152.
Stricker AR, Grosstessner-Hain K, Wurleitner E, Mach RL. Xyr1 (Xylanase Regulator 1) regulates both the hydrolytic enzyme system and D-xylose metabolism in Hypocrea jecorina. Eukaryot Cell. 2006;5(12):2128–37.
Häkkinen M, Valkonen MJ, Westerholm-Parvinen A, Aro N, Arvas M, Vitikainen M, et al. Screening of candidate regulators for cellulase and hemicellulase production in Trichoderma reesei and identification of a factor essential for cellulase production. Biotechnol Biofuels. 2014;7(1):14.
Saloheimo A, Aro N, Ilmen M, Penttila M. Isolation of the ace1 gene encoding a Cys(2)-His(2) transcription factor involved in regulation of activity of the cellulase promoter cbh1 of Trichoderma reesei. J Biol Chem. 2000;275(8):5817–25.
Brown NA, Ries LNA, Goldman GH. How nutritional status signalling coordinates metabolism and lignocellulolytic enzyme secretion. Fungal Genet Biol. 2014;72:48–63.
Stappler E, Dattenböck C, Tisch D, Schmoll M. Analysis of light- and carbon-specific transcriptomes implicates a class of G-protein-coupled receptors in cellulose sensing. mSphere. 2017;2(3):e00089-17.
Stappler E, Rodriguez-Iglesias A, Bazafkan H, Li G, Schmoll M. Relevance of signal transduction pathways for efficient gene expression in fungi. In: Schmoll M, Dattenböck C, editors. Gene expression systems in fungi: Advancements and applications. Cham: Springer International Publishing; 2016. p. 309–34.
Schmoll M. Regulation of plant cell wall degradation by light in Trichoderma. Fungal Biol Biotechnol. 2018;5:10.
Schmoll M, Esquivel-Naranjo EU, Herrera-Estrella A. Trichoderma in the light of day—physiology and development. Fungal Genet Biol. 2010;47(11):909–16.
Castellanos F, Schmoll M, Martinez P, Tisch D, Kubicek CP, Herrera-Estrella A, et al. Crucial factors of the light perception machinery and their impact on growth and cellulase gene transcription in Trichoderma reesei. Fungal Genet Biol. 2010;47(5):468–76.
Tisch D, Schmoll M. Targets of light signalling in Trichoderma reesei. BMC Genomics. 2013;14(1):657.
D’Souza CA, Heitman J. Conserved cAMP signaling cascades regulate fungal development and virulence. FEMS Microbiol Rev. 2001;25(3):349–64.
Turra D, Segorbe D, Di Pietro A. Protein kinases in plant-pathogenic fungi: conserved regulators of infection. Annu Rev Phytopathol. 2014;52:267–88.
Banno S, Ochiai N, Noguchi R, Kimura M, Yamaguchi I, Kanzaki S, et al. A catalytic subunit of cyclic AMP-dependent protein kinase, PKAC-1, regulates asexual differentiation in Neurospora crassa. Genes Genet Syst. 2005;80(1):25–34.
Huang G, Chen S, Li S, Cha J, Long C, Li L, et al. Protein kinase A and casein kinases mediate sequential phosphorylation events in the circadian negative feedback loop. Genes Dev. 2007;21(24):3283–95.
Liu X, Li H, Liu Q, Niu Y, Hu Q, Deng H, et al. Role for protein kinase A in the Neurospora circadian clock by regulating White Collar-independent frequency transcription through phosphorylation of RCM-1. Mol Cell Biol. 2015;35(12):2088–102.
Mehra A, Shi M, Baker CL, Colot HV, Loros JJ, Dunlap JC. A role for casein kinase 2 in the mechanism underlying circadian temperature compensation. Cell. 2009;137(4):749–60.
Brown N, de Gouvea P, Krohn N, Savoldi M, Goldman G. Functional characterisation of the non-essential protein kinases and phosphatases regulating Aspergillus nidulans hydrolytic enzyme production. Biotechnol Biofuels. 2013;6(1):91.
Ribeiro LFC, Chelius C, Boppidi KR, Naik NS, Hossain S, Ramsey JJJ, et al. Comprehensive analysis of Aspergillus nidulans PKA phosphorylome identifies a novel mode of CreA regulation. MBio. 2019;10(2):e02825-18.
Hu Y, Hao X, Chen L, Akhberdi O, Yu X, Liu Y, et al. Galpha-cAMP/PKA pathway positively regulates pigmentation, chaetoglobosin A biosynthesis and sexual development in Chaetomium globosum. PLoS ONE. 2018;13(4):e0195553.
Hu Y, Liu Y, Hao X, Wang D, Akhberdi O, Xiang B, et al. Regulation of the Galpha-cAMP/PKA signaling pathway in cellulose utilization of Chaetomium globosum. Microb Cell Fact. 2018;17(1):160.
Grosse C, Heinekamp T, Kniemeyer O, Gehrke A, Brakhage AA. Protein kinase A regulates growth, sporulation, and pigment formation in Aspergillus fumigatus. Appl Environ Microbiol. 2008;74(15):4923–33.
Park AR, Fu M, Shin JY, Son H, Lee YW. The Protein Kinase A pathway regulates zearalenone production by modulating alternative ZEB2 transcription. J Microbiol Biotechnol. 2016;26(5):967–74.
Yu X, Liu H, Niu X, Akhberdi O, Wei D, Wang D, et al. The Galpha1-cAMP signaling pathway controls conidiation, development and secondary metabolism in the taxol-producing fungus Pestalotiopsis microspora. Microbiol Res. 2017;203:29–39.
Farkas V, Betina V. The intracellular level of ATP during the photoinduced sporulation of Trichoderma viride. Folia Microbiol. 1977;22:438.
Farkas V, Sulova Z, Lehotsky J. Effect of light on the concentration of adenine nucleotides in Trichoderma viride. Folia Microbiol. 1985;131:317–20.
Gresik M, Kolarova N, Farkas V. Membrane potential, ATP, and cyclic AMP changes induced by light in Trichoderma viride. Exp Mycol. 1988;12:295–301.
Nemcovic Farkas V. Stimulation of conidiation by derivatives of cAMP in Trichoderma viride. Folia Microbiol. 1998;43:399–402.
Sestak S, Farkas V. Metabolic regulation of endoglucanase synthesis in Trichoderma reesei: participation of cyclic AMP and glucose-6-phosphate. Can J Microbiol. 1993;39(3):342–7.
Schuster A, Tisch D, Seidl-Seiboth V, Kubicek CP, Schmoll M. Roles of protein kinase A and adenylate cyclase in light-modulated cellulase regulation in Trichoderma reesei. Appl Environ Microbiol. 2012;78(7):2168–78.
Nogueira KM, Costa Mdo N, de Paula RG, Mendonca-Natividade FC, Ricci-Azevedo R, Silva RN. Evidence of cAMP involvement in cellobiohydrolase expression and secretion by Trichoderma reesei in presence of the inducer sophorose. BMC Microbiol. 2015;15:195.
Schmoll M, Schuster A, do Nascimento Silva R, Kubicek CP. The G-alpha protein GNA3 of Hypocrea jecorina (anamorph Trichoderma reesei) regulates cellulase gene expression in the presence of light. Eukaryot Cell. 2009;8(3):410–20.
Seibel C, Gremel G, Silva RD, Schuster A, Kubicek CP, Schmoll M. Light-dependent roles of the G-protein subunit GNA1 of Hypocrea jecorina (anamorph Trichoderma reesei). BMC Biol. 2009;7(1):58.
Tisch D, Kubicek CP, Schmoll M. New insights into the mechanism of light modulated signaling by heterotrimeric G-proteins: ENVOY acts on gna1 and gna3 and adjusts cAMP levels in Trichoderma reesei (Hypocrea jecorina). Fungal Genet Biol. 2011;48(6):631–40.
Tisch D, Schuster A, Schmoll M. Crossroads between light response and nutrient signalling: ENV1 and PhLP1 act as mutual regulatory pair in Trichoderma reesei. BMC Genomics. 2014;15:425.
Schmoll M. Light, stress, sex and carbon—the photoreceptor ENVOY as a central checkpoint in the physiology of Trichoderma reesei. Fungal Biol. 2018;122(6):479–86.
Schmoll M, Franchi L, Kubicek CP. Envoy, a PAS/LOV domain protein of Hypocrea jecorina (Anamorph Trichoderma reesei), modulates cellulase gene transcription in response to light. Eukaryot Cell. 2005;4(12):1998–2007.
Tisch D, Kubicek CP, Schmoll M. The phosducin-like protein PhLP1 impacts regulation of glycoside hydrolases and light response in Trichoderma reesei. BMC Genomics. 2011;12:613.
Liu R, Chen L, Jiang Y, Zou G, Zhou Z. A novel transcription factor specifically regulates GH11 xylanase genes in Trichoderma reesei. Biotechnol Biofuels. 2017;10:194.
Monroy AA, Stappler E, Schuster A, Sulyok M, Schmoll M. A CRE1—regulated cluster is responsible for light dependent production of dihydrotrichotetronin in Trichoderma reesei. PLoS ONE. 2017;12:e0182530.
Derntl C, Rassinger A, Srebotnik E, Mach RL, Mach-Aigner AR. Identification of the main regulator responsible for synthesis of the typical yellow pigment produced by Trichoderma reesei. Appl Environ Microbiol. 2016;82(20):6247–57.
Marie-Nelly H, Marbouty M, Cournac A, Flot JF, Liti G, Parodi DP, et al. High-quality genome (re)assembly using chromosomal contact data. Nat Commun. 2014;5:5695.
Schmoll M, Kubicek CP. ooc1, a unique gene expressed only during growth of Hypocrea jecorina (anamorph: Trichoderma reesei) on cellulose. Curr Genet. 2005;48(2):126–33.
Atanasova L, Knox BP, Kubicek CP, Druzhinina IS, Baker SE. The polyketide synthase gene pks4 of Trichoderma reesei provides pigmentation and stress resistance. Eukaryot Cell. 2013;12(11):1499–508.
Chauhan N, Inglis D, Roman E, Pla J, Li D, Calera JA, et al. Candida albicans response regulator gene SSK1 regulates a subset of genes whose functions are associated with cell wall biosynthesis and adaptation to oxidative stress. Eukaryot Cell. 2003;2(5):1018–24.
Metz B, de Vries RP, Polak S, Seidl V, Seiboth B. The Hypocrea jecorina (syn. Trichoderma reesei) lxr1 gene encodes a D-mannitol dehydrogenase and is not involved in L-arabinose catabolism. FEBS Lett. 2009;583(8):1309–13.
Richard P, Putkonen M, Vaananen R, Londesborough J, Penttila M. The missing link in the fungal L-arabinose catabolic pathway, identification of the L-xylulose reductase gene. Biochemistry. 2002;41(20):6432–7.
Cai P, Wang B, Ji J, Jiang Y, Wan L, Tian C, et al. The putative cellodextrin transporter-like protein CLP1 is involved in cellulase induction in Neurospora crassa. J Biol Chem. 2015;290(2):788–96.
Lehmann L, Ronnest NP, Jorgensen CI, Olsson L, Stocks SM, Jorgensen HS, et al. Linking hydrolysis performance to Trichoderma reesei cellulolytic enzyme profile. Biotechnol Bioeng. 2016;113(5):1001–10.
Glass NL, Schmoll M, Cate JH, Coradetti S. Plant cell wall deconstruction by ascomycete fungi. Annu Rev Microbiol. 2013;67:477–98.
Brown SA, Kowalska E, Dallmann R. (Re)inventing the circadian feedback loop. Dev Cell. 2012;22(3):477–87.
Ivanova C, Ramoni J, Aouam T, Frischmann A, Seiboth B, Baker SE, et al. Genome sequencing and transcriptome analysis of Trichoderma reesei QM9978 strain reveals a distal chromosome translocation to be responsible for loss of vib1 expression and loss of cellulase induction. Biotechnol Biofuels. 2017;10:209.
Hitzenhammer E, Büschl C, Sulyok M, Schuhmacher R, Kluger B, Wischnitzki E, et al. YPR2 is a regulator of light modulated carbon and secondary metabolism in Trichoderma reesei. BMC Genomics. 2019;20(1):211.
De Souza CP, Hashmi SB, Osmani AH, Andrews P, Ringelberg CS, Dunlap JC, et al. Functional analysis of the Aspergillus nidulans kinome. PLoS ONE. 2013;8(3):e58008.
Dattenböck C, Tisch D, Schuster A, Monroy AA, Hinterdobler W, Schmoll M. Gene regulation associated with sexual development and female fertility in different isolates of Trichoderma reesei. Fungal Biol Biotechnol. 2018;5:9.
Ivey FD, Kays AM, Borkovich KA. Shared and independent roles for a Galpha(i) protein and adenylyl cyclase in regulating development and stress responses in Neurospora crassa. Eukaryot Cell. 2002;1(4):634–42.
Poole CF. Instrumental thin-layer chromatography. Amsterdam: Elsevier; 2015. p. 652.
Spangenberg B, Poole CF, Weins C. Quantitative thin-layer chromatography—a practical survey. Berlin Heidelberg: Springer-Verlag; 2011. p. 388.
Bazafkan H, Dattenböck C, Böhmdorfer S, Tisch D, Stappler E, Schmoll M. Mating type dependent partner sensing as mediated by VEL1 in Trichoderma reesei. Mol Microbiol. 2015;96(6):1103–18.
Hogan DA, Muhlschlegel FA. Candida albicans developmental regulation: adenylyl cyclase as a coincidence detector of parallel signals. Curr Opin Microbiol. 2011;14(6):682–6.
Dong W, Qu Y, Gao P. Regulation of cellulase synthesis in mycelial fungi: participation of ATP and cyclic-AMP. Biotechnol Lett. 1995;17:593–8.
Farkas V, Gresik M, Kolarova N, Sulova Z, Sestak S. Biochemical and physiological changes during photo-induced conidiation and derepression of cellulase synthesis in Trichoderma. In: Kubicek CP, Eveleigh DE, Esterbauer W, Steiner W, Kubicek-Pranz EM, editors. Trichoderma reesei cellulase: biochemistry, genetics, physiology, and application. Cambridge: Graham House; 1990. p. 139–55.
Schuster A, Kubicek CP, Friedl MA, Druzhinina IS, Schmoll M. Impact of light on Hypocrea jecorina and the multiple cellular roles of ENVOY in this process. BMC Genomics. 2007;8(1):449.
Xiong Y, Sun J, Glass NL. VIB1, a link between glucose signaling and carbon catabolite repression, is essential for plant cell wall degradation by Neurospora crassa. PLoS Genet. 2014;10(8):e1004500.
Seidl V, Seibel C, Kubicek CP, Schmoll M. Sexual development in the industrial workhorse Trichoderma reesei. Proc Natl Acad Sci USA. 2009;106(33):13909–14.
Mandels M, Andreotti R. Problems and challenges in the cellulose to cellulase fermentation. Proc Biochem. 1978;13:6–13.
Priebe S, Kreisel C, Horn F, Guthke R, Linde J. FungiFun2: a comprehensive online resource for systematic analysis of gene lists from fungal species. Bioinformatics. 2015;31(3):445–6.
Coppe A, Danieli GA, Bortoluzzi S. REEF: searching REgionally Enriched Features in genomes. BMC Bioinform. 2006;7:453.
Schuster A, Bruno KS, Collett JR, Baker SE, Seiboth B, Kubicek CP, et al. A versatile toolkit for high throughput functional genomics with Trichoderma reesei. Biotechnol Biofuels. 2012;5(1):1.
Steiger MG, Vitikainen M, Uskonen P, Brunner K, Adam G, Pakula T, et al. Transformation system for Hypocrea jecorina (Trichoderma reesei) that favors homologous integration and employs reusable bidirectionally selectable markers. Appl Environ Microbiol. 2011;77(1):114–21.
Schmoll M. Sexual development in Trichoderma—scrutinizing the aspired phenomenon. In: Mukherjee PK, Horwitz BA, Singh US, Mukherjee M, Schmoll M, editors. Trichoderma: Biology and Applications. Wallingford: CAB International; 2013. p. 67–86.
Malachova A, Sulyok M, Beltran E, Berthiller F, Krska R. Optimization and validation of a quantitative liquid chromatography-tandem mass spectrometric method covering 295 bacterial and fungal metabolites including all regulated mycotoxins in four model food matrices. J Chromatogr A. 2014;1362:145–56.
Lee D, Lee JH, Cai XF, Shin JC, Lee K, Hong YS, et al. Fungal metabolites, sorbicillinoid polyketides and their effects on the activation of peroxisome proliferator-activated receptor gamma. J Antibiot (Tokyo). 2005;58(10):615–20.
Acknowledgements
We acknowledge NMR Center and MS Center, Faculty of Chemistry, University of Vienna for measuring NMR and MS spectra, respectively. We want to thank Sabrina Beier, Michael Sulyok, Kaja S. Liepert and Bianca Mikulasek for technical assistance.
Funding
Work of WH was supported by the NFB (Science Fund of Lower Austria; grant LC16-04 to MS). Work of DT and AS was supported by the Austrian Science Fund (FWF; grant P22511 to MS). Work of HB was supported by the Austrian Science Fund (FWF, grants P24350 and P26935).
Author information
Authors and Affiliations
Contributions
DT and AS performed experimental work for transcriptome analysis and contributed to bioinformatic analyses. WH contributed to secondary metabolite analysis and to drafting of the manuscript. EÖ extracted and measured secondary metabolites. HB contributed construction and analysis of the deletion strain. StB contributed to secondary metabolite analysis and quantification. WH and StB supervised work of EÖ. JS extracted trichodimerol. LB performed NMR structure elucidation. MS conceived of the study, supervised work of DT, AS, and WH, contributed to bioinformatic analyses and wrote the final version of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1.
Target genes of PKAc1 during growth on cellulose in light and darkness.
Additional file 2.
Functional category analysis of genes with altered transcript levels in Δpkac1 and the subset of genes with overlapping regulation to photoreceptor targets.
Additional file 3.
Genes with altered transcript levels in Δpkac1 and regulation in an induction specific manner.
Additional file 4.
Comparative analysis of genes with altered transcript levels in Δpkac1 and genes regulated by photoreceptors, CRE1 and by light upon growth on cellulose.
Additional file 5: Figure S1.
Biological replicates of high performance thin layer chromatography (HPTLC) analysis of wildtype and pkac1 mutants under asexual and sexual conditions.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Hinterdobler, W., Schuster, A., Tisch, D. et al. The role of PKAc1 in gene regulation and trichodimerol production in Trichoderma reesei. Fungal Biol Biotechnol 6, 12 (2019). https://doi.org/10.1186/s40694-019-0075-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40694-019-0075-8