
Abstract
Background
Liriodendron chinense ranges widely in subtropical China and northern Vietnam; however, it inhabits several small, isolated populations and is now an endangered species due to its limited seed production. The objective of this study was to develop a set of nuclear SSR (simple sequence repeats) and multiple chloroplast genome markers for genetic studies in L. chinense and their characterization in diverse germplasm.
Results
We performed low-coverage whole genome sequencing of the L. chinense from four genotypes, assembled the chloroplast genome and identified nuclear SSR loci by searching in contigs for SSR motifs. Comparative analysis of the four chloroplast genomes of L. chinense revealed 45 SNPs, 17 indels, 49 polymorphic SSR loci, and five small inversions. Most chloroplast intraspecific polymorphisms were located in the interspaces of single-copy regions. In total, 6147 SSR markers were isolated from low-coverage whole genome sequences. The most common SSR motifs were dinucleotide (70.09%), followed by trinucleotide motifs (23.10%). The motif AG/TC (33.51%) was the most abundant, followed by TC/AG (25.53%). A set of 13 SSR primer combinations were tested for amplification and their ability to detect polymorphisms in a set of 109 L. chinense individuals, representing distinct varieties or germplasm. The number of alleles per locus ranged from 8 to 28 with an average of 21 alleles. The expected heterozygosity (He) varied from 0.19 to 0.93 and the observed heterozygosity (Ho) ranged from 0.11 to 0.79.
Conclusions
The genetic resources characterized and tested in this study provide a valuable tool to detect polymorphisms in L. chinense for future genetic studies and breeding programs.
Similar content being viewed by others


Background
Liriodendron chinense (Hemsl.) Sarg., one of the only two living Liriodendron species on the earth, is Asia’s native Liriodendron species, known as the Chinese tulip tree [1, 2]. Depending on fossil evidence, Liriodendron species were reported to become extinct in Europe due to large-scale glaciation and climate aridity during glacial phases, leaving a discontinuous distribution of L. chinense and its American relative, L. tulipifera [3]. L. chinense grows in central and southern China and locally in northern Vietnam. These tulip trees can grow to more than 40 m in height, and their large flowers superficially resemble tulips. The trees are therefore cultivated on other continents as ornamental trees [4]. In addition, L. chinense wood is hard but light and difficult to deform, making it useful for construction, ship building and furniture framing. The tree’s leaves and bark are used medicinally for dispersing cold and relieving cough [5].
Liriodendron chinense is a cross-pollinated plant; however, parthenogenesis exists, and gynoecium can develop without insemination, causing a low germination percentage in the natural environment. Seed reproduction of this species often requires artificial pollination, whereas seeds still have poor vitality [6]. The development and utilization of L. chinense germplasm resources began in the 1960s when crossbreeding of Asia and American tulip trees was successfully accomplished. The hybrids maintained parental advantages, such as peculiar leaf shape and long flowering phase; moreover, they performed even better in flower color, growth rate, insect resistance, etc. Excellent characteristics are inherited through asexual reproduction techniques, such as cuttage, grafting and tissue culture in modern L. chinense cultivation. Various cultivars flourished from different reproducing techniques, growing areas and genetic backgrounds. The original genetic resources that breed all kinds of cultivars may be missing due to long cultivating history, multiple market circulation and careless management. Comprehensive lineage cataloguing and genetic diversity investigation are required to supervise and protect a healthy development of tulip tree resources [7].
DNA sequences may be applied in species identification, molecular phylogeny, population genetics etc. For various kinds of DNA molecular markers, microsatellite sequences are thought to be sensitive in assessing the genetic diversity and structure of plant populations. This term refers to simple sequence repeats (SSR) markers, which are codominant, highly polymorphic, reproducible, reliable, and distributed throughout the genome. In the traditional methodology, microsatellite development involves an enriched library followed by gene cloning. Tedious lab work is time-consuming and still may not be able to produce even a small number of polymorphic loci [8, 9]. Next-generation sequencing provides a good alternative, in which the genome is fully screened literally, developing thousands of SSR candidates at one time [10, 11].
The chloroplast genome is conservatively inherited uniparentally mostly via maternal inheritance [12]. In general, with a size from 120 to 160 kb, the chloroplast genome is structurally highly conserved across land plants. The chloroplast genome in angiosperms has a circular structure of two copies of large inverted repeats (IR) separated by small (SSC) and large (LSC) single-copy regions [13, 14]. Chloroplast genome markers, such as single nucleotide polymorphisms (SNPs), indels, simple sequence repeats (SSRs), and small inversion, have been used for studying genetic and genome diversity and phylogenetic and systematic evolutionary analyses [15,16,17,18,19,20]. For example, using the whole plastid genome sequence data of wild extant Ginkgo populations revealed the deepest temporal footprint dating back to approximately 390,000 year ago [21]. Diversity and phylogenetic analyses using the chloroplast genome data revealed some selection characteristics in the chloroplast genome that Asian rice had been domesticated at least twice [22]. Phyloplastomic and network analyses clarified the taxonomic position of Pepper species (Capsicum spp.) [23]. Moreover, the genetic information in angiosperm chloroplasts is inherited maternally, making the chloroplast markers a good indicator of maternal ancestry. Intraspecific chloroplast sequence variation is used to investigate the population structure of L. chinense germplasm and is applied to guide molecular breeding.
In our study, we performed low-coverage shotgun sequencing of the four genotypes of L. chinense using Next Generation Sequencing (NGS) technology. The study aimed to (1) assemble the chloroplast genome of L. chinense and identify the chloroplast genome markers, including chloroplast SSRs, indels, and SNPs; (2) develop the nuclear SSR loci by searching in contigs for SSR motifs, design candidate SSR PCR primers, and screen for fragment length polymorphisms among different L. chinense individuals.
Materials and methods
DNA extraction and high-throughput sequencing
Four genotypes of L. chinense were used in this study (Table 1). L. chinense were obtained from Monan, Songtao, Guizhou (GZST), Jiujiang, Jiangxi (JXLS), Liping, Guizhou (GZLP), and Shuining, Hunan (HNSN) of China, representing the geographical distribution of this species. Young leaves of L. chinense were picked for silica gel conservation. Total genomic DNA was extracted from dried leaves using the modified CTAB method [24] and further purified via the Wizard® Genomic DNA Purification Kit (A1120, Promega, USA). DNA (5 ng) amount was accurately calculated on a Qubit fluorometer and digested for library construction with the TruePrep DNA Library Prep Kit V2 for Illumina (TD502, Vazyme, Nanjing, China) in accordance with the manufacturer’s instructions. A library of 350 bp was selected for sequencing on the HiSeq 4000 platform of the Novogene genome sequencing company in Tianjin, China.
Chloroplast genome assembly and annotation
The software Trimmomatic was employed to filter low-quality reads from the raw data [25]. The remaining high-quality reads were assembled into contigs with SPAdes 3.6.1 [26]. Chloroplast genome contigs were selected from the SPAdes assembly by using BLAST search using the published Liriodendron chloroplast genome as a reference (GenBank accession number: KU170538). The selected contigs were second assembled with Sequencher 5.4.5 (Gene Codes, MI, USA). Ambiguous nucleotides or gaps in the chloroplast genome sequences were further confirmed by PCR amplification and Sanger sequencing with specific primers [27]. Finally, clean reads were remapped to the draft genome sequences, yielding the sequences. The chloroplast genome annotations were performed with Plann [28]. The chloroplast genome map was drawn using Genome Vx software [29].
Chloroplast genome marker development and validation
To develop the chloroplast genome markers and to show the intraspecific variations in L. chinense, the four sequenced L. chinense chloroplast genomes were aligned using MIFFT v7 [30] and then adjusted manually using Se-Al 2.0. [31]. The markers of single nucleotide substitutions (SNPs), indels, SSRs, and inversions in the L. chinense chloroplast genome were identified. SNPs were calculated using MEGA 6.0 software [32]. Variable SSRs, indels and inversions were identified in the chloroplast genomes of four L. chinense genotypes based on the aligned sequence matrix. Using the GZST genotype genome sequence as the standard reference, the size, location, and evolutionary direction of the chloroplast genome markers were counted.
Nuclear SSR marker development and primer design
The GZST genotype was used to develop nuclear SSR markers. We applied MISA software [33] to detect microsatellite repeats in assembled contigs. The search parameters were fixed six di-, five tri- and tetranucleotide repeats, respectively while the minimum product size was set to 100 bp. Primer pairs were designed for all candidate loci in Primer3 software [34]. Primer size was controlled between 18 and 22 bp with an optimal size of 20 bp. The minimum primer annealing temperature was set to 60 °C, and other settings were performed with default values.
Primer testing and polymorphism detection
First, forty-eight di- or trinucleotide repeats were tested for PCR primer universality in two tulip tree samples. When synthetizing the candidate SSR primer pairs, an 18 bp tail (5′-TGTAAAACGACGGCCAGT-3′) was added to the 5′ end of the forward primer to improve efficiency and lower cost, as described in MJ Blacket, C Robin, RT Good, SF Lee and AD Miller [35]. Each 10-μL PCR mixture contained 1 × PCR buffer (with Mg2+), 0.25 mmol/L each dNTP, 0.25 μmol/L each primer, 1.25 U of Taq polymerase, and 20-30 ng of DNA. The PCR program was 94 °C for 4 min, followed by 35 cycles of 30 s at 94 °C, 40 s at 55 °C, and 30 s at 72 °C, with a final step of 10 min at 72 °C. The PCR products were examined via electrophoresis in a 1% agarose gel containing ethidium bromide and were visualized using an ultraviolet transilluminator. Loci amplified to be strong bands in both samples were considered for further testing in the next step.
Second, polymorphism examination of selected repeats in previous steps was performed in eight L. chinense from eight different populations. Each 10-μL PCR mixture contained 1 × PCR buffer (with Mg2+), 0.25 mmol/L each dNTP, 0.25 μmol/L each primer, 0.25 μmol/L 18 bp tail primer modified by fluorescence (FAM (blue), HEX (green), and ROX (red)) including different colors, 1.25 U of Taq polymerase, and 20-30 ng of DNA. The amplification program was the same as above. The ABI 3730xl DNA Analyzer (Applied Biosystems, Foster, CA, USA) was used to analyze the amplified PCR fragments with the GeneScan 500 LIZ size standard (Applied Biosystems).
Finally, marker primers screened out by the first two steps were validated in all 109 samples from different populations. The amplification mixture, amplification program, and analysis of PCR fragments were the same as the second step. Genotyping data were identified, and errors were corrected by GeneMapper software version 4.0 (Applied Biosystems, Thermo Fisher, USA). Genetic analyses for polymorphic loci were performed using ATetra version1.2 [36] to calculate such parameters as the number of alleles, effective number of alleles, expected heterozygosity, observed heterozygosity and Shannon’s information index.
Result
Chloroplast genome assembly and genome features
Using the Illumina HiSeq 4000 system, total DNA from four genotypes of L. chinense were sequenced to produce 13,142,198—18,356,014 paired-end raw reads (150 bp average read length) per genotype. We obtained four chloroplast genome sequences of L. chinense with coverage of 1035–1506 X after de nova assembly. The chloroplast genome sequences were deposited in GenBank (Table 1).
The whole chloroplast genome sequences of the four genotypes of L. chinense ranged from 159,428 to 159,890 bp in length (Table 1 and Fig. 1). The chloroplast genome of L. chinense displayed the typical circular quadripartite structure, consisting of a pair of inverted repeat (IR) regions (26,325–26,333 bp) separated by a larger single copy (LSC) region (87,765–88,240 bp), and small single copy (SSC) region (18,997–19,029 bp). The overall GC contents were 37.8% in the LSC region, 43.2% in the IR regions, 34.3% in the SSC region, and 39.2% in the entire chloroplast genome.

Chloroplast genome map of L. chinense. Genes drawn outside of the circle are transcribed clockwise, while those inside are counterclockwise. Small single copy (SSC), large single copy (LSC), and inverted repeats (IRa, IRb) are indicated
Gene content and arrangement were identical in four genotypes of L. chinense chloroplast genomes. The L. chinense chloroplast genome contains 113 different genes, including 79 protein-coding genes, 30 tRNA genes and 4 rRNA genes. Ten protein-coding genes (atpF, ndhA, ndhB, petB, petD, rpl2, rpl16, rpoC1, rps12 and rps16) and six tRNA genes (trnA-UGC, trnG-UCC, trnI-GAU, trnK-UUU, trnL-UAA, trnV-UAC) had a single intron, while two protein-coding genes (ycf3, and clpP) contained two introns. matK was located within the intron of trnK-UUU in the L. chinense chloroplast genomes.
Chloroplast genome marker development
There were 45 SNPs in the four genotypes of the L. chinense chloroplast genome, including 26 SNPs in the intergenic regions, 3 in the intronic regions, and 16 SNPs in the coding sequences (Table 2). All detected SNPs were located in the single copy region. More than two SNPs were detected in each of three positions (trnK-rps16, psbE-petL, and ycf1). Ycf1, which had the highest number of SNPs of all the positions examined, contained four SNPs. The C to G and A to T SNPs had the lowest frequencies among the six direction types of SNPs. We designed primer pairs for amplification of all the SNPs (Additional file 1: Table S1).
We identified 17 indels among the four genotypes of the L. chinense chloroplast genome (Table 3, Additional file 2: Table S2). All indels were found in noncoding regions. Most indels were located in the single copy regions. The size of the indels ranged from 2 to 458 bp. The largest indel (458 bp), in ndhC-trnV, was a deletion in chloroplast genome of the GZST and JXLS genotypes. The other two larger indels (> 100 bp) were found in the ndhC-trnV and petN-psbM regions.
Forty-nine SSR loci showed polymorphism after in silico comparative analysis among the four genotypes of the L. chinense chloroplast genome (Additional file 1: Table S1). All polymorphic SSR loci were located in noncoding regions. Thirty-seven regions harbored SSRs; the trnG intron had the highest number of SSRs (three), followed by trnH-psbA, rps16-trnQ, atpF-atpH, atpH-atpI, rps2-rpoC2, psbM-trnD, trnE-trnT, ycf4-cemA, and rpl32-trnL, all of which had two SSRs. Mononucleotide motifs were the most abundant type of repeat (95.92%). Furthermore, almost all SSR loci were composed of A or T, which contributed to the bias in base composition (A/T; both 60.8%) in the chloroplast genomes of L. chinense. We designed primer pairs for amplification of all the SSRs (Additional file 3: Table S3).
Moreover, a total of five small inversions were uncovered based on the sequence alignment of the four chloroplast genomes (Table 4, Additional file 4: Table S4). Of which these inversions, three were located in the LSC region and two were in the SSC region. All inversions were accompanied by a pair of inverted repeats immediately flanking the inversion. The inversions were from 3 to 23 bp and the franking repeats were from 17 to 29 bp in length. The two small inversions from petA-psbJ and the inversion from trnH-psbA only occurred in the GZLP genotype. The small inversions in rpl32-trnL, and ccsA-ycf1 occurred in the GZLP and HNSN genotypes.
Nuclear microsatellite marker development
The paired end reads of the GZST genotype were qualitatively assessed and assembled with SPAdes 3.6.1. There were 161,179 contigs assembled and the contig length ranged from 150 bp to 113,929 bp.
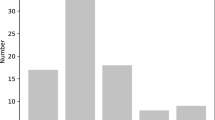
A total of 9155 SSRs were discovered using MISA from the assembled contigs. These SSRs included 6417 di-, 2115 tri-, 312 tetra-, 219 penta- and 92 hexanucleotide repeats, which corresponded to 70.09%, 23.10%, 3.41%, 2.39%, and 1.00% of total SSRs, respectively (Fig. 2a). According to the distribution of microsatellites, SSR frequency and density varied with motif length, as motif length increased (from mono- to hexanucleotide repeats). Dinucleotide repeats are more common than the higher order motif, which is in agreement with previous research examining other wood plants. The ten most frequent motif types in the L. chinense genome (Fig. 2b) were five dinucleotides (GA/CT, TC/AG, TG/AC, AT/TA, GT/CA, AT/TA), and five trinucleotides (TTC/AAG, GAA/CTT, CAT/GTA, TTA/AAT, TCT/AGA).

Distribution and type of SSRs in L. chinense. a Number of different SSRs types. b Number of identified SSR motifs in different repeat class types
Primer design and evaluation
Primer3 was used to generate primer pairs targeting these SSR regions (Additional file 5: Table S5). In total, there were 5339 SSR-designed primers. We randomly selected 48 SSRs for initial validation in eight individuals. After screening in 2% agarose electrophoresis, 18 of the 48 primer pairs produced clear, unique amplification products of the expected size. Of 18 SSRs, 13 loci had polymorphic amplifications, and 5 loci were monomorphic. These polymorphic loci serve as candidate markers in the following analysis.
Genetic diversity and relationships among genotypes
To evaluate the genetic diversity of L. chinense, 109 individuals were collected and analyzed using 13 primer pairs selected. Based on 13 polymorphic primer pairs, the number of alleles (A) per locus ranged from 8 to 28, with an average of 21. The expected heterozygosity (He) varied from 0.1919 to 0.9344 and the observed heterozygosity (Ho) ranged from 0.1101 to 0.7944 (Table 5).
Discussion
Nuclear SSR markers developed by NGS
Recent developments in sequencing technologies and bioinformatic analysis have provided an unprecedented opportunity to discover SSR markers of high quality and effective cost/time in nonmodel organisms about which genomic information was lacking. Moreover, this approach is also rapid and more cost-effective than traditional SSR development methods and Sanger sequencing [37, 38]. In this study, we randomly obtained partition nuclear genome sequences, and this approach was sufficient for the development of 9155 SSR markers for L. chinense. The density of SSRs was 1 per 7.04 kb in the L. chinense chloroplast genome, while in the most plant genomes, every 6.8 kb has one SSR. This density was less than the density in coffee (1/2.16 kb), and Amorphophallus (1/3.63 kb) [39, 40], but it was higher than that of Arabidopsis (1/14 kb) [41]. The variable frequency of genic and genomic SSRs may reflect a difference in their distribution in coding sequences compared to the entire genome. In the L. tulipifera EST data, the average frequency of SSR was 1 per 8.5 kb, which was less frequent than the genomic data [42].
Among the selected genomic SSRs, dinucleotide repeats were the most abundant (70.09%), followed by trinucleotide motifs (23.10%). One of the 10 most abundant motif types in jujube was tetranucleotide motifs (ATTT/AAAT) which was not found in L. tulipifera [43]. The motif length frequency differences between genomic and genic SSRs are most likely due to selection pressure on genic SSRs which reduces the fixation of mutations leading to frameshifts. Among dinucleotide repeats, AG/CT was most frequently observed (33.51%), followed by AG/TC (25.53%) which is in agreement with Xu et al. [42], who reported that AG/CT was the most frequent genic dinucleotide (57.4%) in L. chinense.
Several studies used expressed sequence tags (ESTs) developing SSR markers [1, 6, 42]. Compared with this study, those SSRs had the lower variable, for example, the average effective number of alleles was 3.95 to 5.93 [38, 44]. Using the low- coverage whole genome sequencing method, we quickly obtained a number of nuclear SSRs with low costs.
Application of nuclear SSR markers
Nuclear microsatellite, with a mutation rate ranging from 10−6 to 10−2 [45], are highly polymorphic in comparison with other marker systems, which have been widely used in many living organisms including plant, insects, birds, humans and animals for different kinds of basic genetics research. There were many applications of nuclear SSR markers in plants, such as, genetic diversity and phylogenetic relationships, population and evolutionary studies, cultivar identification and marker-assisted selection, genome mapping [46].
SSR markers often are powerful system for revealing interspecific and/or intraspecific phylogenetic relationships. Several applications show nuclear SSRs have led to a better understanding of close relationships between species. Relationships among eight Actinidia species were resolved with SSRs [47]. The fragment length polymorphism of SSRs among the Cucumis accessions made it possible to distinguish three main groups [48]. The genetic diversity for germplasm collections have been assessed by SSR markers, such as apple [49], eggplant [50], walnut [51]. Belgj et al. examined the pattern of genetic variability and genetic relationships of wild olive populations in the north-western Mediterranean and indicated a degree of admixture in all the populations [52]. Evaluation of genetic diversity, genome mapping and phylogenetic relationships has resulted in information of the history process and will provide important information for breeding programs.
Chloroplast genome variation in L. chinense
The chloroplast genomes of plants are a valuable resource for developing molecular markers to study intraspecies and interspecies ecolution [15, 53]. Chloroplast genomic sequences are highly conserved within species; however, nucleotide substitutions, SSRs, indels, and other microstructure mutations within these sequences can be used to elucidate the genetic diversity and guide molecular breeding [54, 55]. However, few studies have used whole chloroplast genome data to examine intraspecific diversity. In this study, we assembled chloroplast genome sequences of four accession samples from wild L. chinense germplasm using low-coverage NGS data.
Comparative analysis of the four chloroplast genomes of L. chinense revealed 45 SNPs, 17 indels, 49 polymorphic SSR loci, and five small inversions. The abundant genetic diversity could be applied to phylogenetic analysis and development of molecular markers to verify the genetic diversity of L. chinense. Chloroplast genome sequence diversity in L. chinense is relatively high compared to that reported for other plant species including Scutellaria baicalensis (25 SNPs, 19 indels, two individuals) [56], Brachypodium distachyon (298 SNPs, 53 individuals) [20], Jacobaea vulgaris (32 SNPs, 17 individuals) [57] and Dioscorea polystachya (141 SNPs, 43 indels, 24 polymorphic SSRs, six individuals) [54].
Previously, trnH-psbA, trnL-F, rbcL, and matK genes were used in evolutionary studies, and which were reported to be variation hotspots [58, 59]. Of these genes, matK and rbcL were the core DNA barcodes in plants [60]. More studies have been shown that the mutation hotspot regions in the chloroplast genome are concentrated in noncoding regions. For example, Dong et al. identified 23 highly variable chloroplast markers (4 coding regions, 2 introns, and 17 intergenic spacers) that were used to resolve phylogenies and for DNA barcoding of closely related flowering plant species [61]. Regions of particularly high variability in L. chinense included the LSC intergenic spacer regions trnK-rps16 (five polymorphisms) and rps16-trnQ (five polymorphisms) followed by atpH-atpI, petA-psbJ, trnG intron and ycf1. trnK-rps16 and rps16-trnQ have been identified as variable and underutilized regions of the angiosperm chloroplast genome suitable for intraspecific phylogenetic studies [61, 62]. Ycf1 is the second longest gene and the most rapidly evolving chloroplast gene [63], the function of which is essential for plant viability and encodes Tic214, a vital component of the Arabidopsis TIC complex [64]. There were two highly variable regions (ycf1a and ycf1b) in the SSC of the ycf1 gene [61, 63].
The polymorphisms found in this study can be used to elucidate evolutionary history such as promoting practical applications for breeding new cultivars of L. chinense. Furthermore, chloroplast polymorphism markers will be useful in testing maternal inheritance of the chloroplast genome, in identifying genotype differentiation and even in developing breeding programs.
Conclusion
In this study, we obtained four chloroplast genomes of L. chinense from four genotypes, and identified SNPs indels, SSRs and small inversions in L. chinense by comparative analyses of chloroplast genomes. We also developed nuclear SSRs by low-coverage whole genome sequencing. These newly developed chloroplast genome resources and SSR markers will become useful tools for molecular genetics, genotype identification, genetic mapping, and molecular breeding of Chinese tulip tree.
Availability of data and materials
The newly sequenced plastomes have been submitted to GenBank with Accession Numbers MK887904–MK887907.
Abbreviations
- SSR:
-
Simple sequence repeat
- SNP:
-
Single nucleotide polymorphisms
- LSC:
-
Large single copy
- SSC:
-
Small single copy
- IR:
-
Inverted repeat
References
Yang AH, Zhang JJ, Tian H, Yao XH. Characterization of 39 novel EST-SSR markers for Liriodendron tulipifera and cross-species amplification in L. chinense (Magnoliaceae). Am J Bot. 2012;99(11):e460–4.
Yang Y, Xu M, Luo Q, Wang J, Li H. De novo transcriptome analysis of Liriodendron chinense petals and leaves by Illumina sequencing. Gene. 2014;534(2):155–62.
He S, Hao R. Study on the natural population dynamics and the endangering habitat of Liriodendron chinense in China. Acta Phytoecologica Sinica. 1999;23(1):87–95.
Li K, Chen L, Feng Y, Yao J, Li B, Xu M, Li H. High genetic diversity but limited gene flow among remnant and fragmented natural populations of Liriodendron chinense Sarg. Biochem Syst Ecol. 2014;54:230–6.
Moon MK, Oh HM, Kwon BM, Baek NI, Kim SH, Kim JS, Kim DK. Farnesyl protein transferase and tumor cell growth inhibitory activities of lipiferolide isolated from Liriodendron tulipifera. Arch Pharm Res. 2007;30(3):299–302.
Zhang X, Carlson A, Tian Z, Staton M, Schlarbaum SE, Carlson JE, Liang H. Genetic characterization of Liriodendron seed orchards with EST-SSR markers. J Plant Sci Mol Breeding. 2015. https://doi.org/10.7243/2050-2389-4-1.
Li B, Li Y, Cai Q, Lin F, Meng Q, Zheng Y. The complete chloroplast genome of a Tertiary relict species Liriodendron chinense (Magnoliaceae). Conserv Genet Resour. 2016;8(3):279–81.
Tian HL, Chen XQ, Wang JX, Xue JH, Wen J, Mitchell G, Zhou SL. Development and characterization of microsatellite loci for lotus (Nelumbo nucifera). Conserv Genet. 2008;9(5):1385–8.
Wang J, Xia T, Zhang J, Zhou S. Isolation and characterization of fourteen microsatellites from a tree peony (Paeonia suffruticosa). Conserv Genet. 2009;10(4):1029–31.
Yang T, Fang L, Zhang X, Hu J, Bao S, Hao J, Li L, He Y, Jiang J, Wang F, et al. High-throughput development of SSR markers from pea (Pisum sativum) based on next generation sequencing of a purified chinese commercial variety. PLOS ONE. 2015;10(10):e0139775.
Qi W, Lin F, Liu Y, Huang B, Cheng J, Zhang W, Zhao H. High-throughput development of simple sequence repeat markers for genetic diversity research in Crambe abyssinica. BMC Plant Biol. 2016;16(1):139.
Kuroiwa T, Ogawa K, Nakamura S. Mechanisms of maternal inheritance—preferential destruction of chloroplast nuclei. Jpn J Genet. 1984;59(6):633–4.
Rogalski M, do Nascimento Vieira L, Fraga HP, Guerra MP. Plastid genomics in horticultural species: importance and applications for plant population genetics, evolution, and biotechnology. Front Plant Sci. 2015;6:586.
Lima MS, Woods LC, Cartwright MW, Smith DR. The (in)complete organelle genome: exploring the use and non-use of available technologies for characterizing mitochondrial and plastid chromosomes. Mol Ecol Resour. 2016;16:1279.
Dong W, Liu H, Xu C, Zuo Y, Chen Z, Zhou S. A chloroplast genomic strategy for designing taxon specific DNA mini-barcodes: a case study on ginsengs. BMC Genet. 2014;15(1):138.
Xu C, Dong W, Li W, Lu Y, Xie X, Jin X, Shi J, He K, Suo Z. Comparative analysis of six lagerstroemia complete chloroplast genomes. Front Plant Sci. 2017;8(15):15.
Li W, Liu Y, Yang Y, Xie X, Lu Y, Yang Z, Jin X, Dong W, Suo Z. Interspecific chloroplast genome sequence diversity and genomic resources in Diospyros. BMC Plant Biol. 2018;18(1):210.
Morris GP, Grabowski PP, Borevitz JO. Genomic diversity in switchgrass (Panicum virgatum): from the continental scale to a dune landscape. Mol Ecol. 2011;20(23):4938–52.
Huang DI, Hefer CA, Kolosova N, Douglas CJ, Cronk QCB. Whole plastome sequencing reveals deep plastid divergence and cytonuclear discordance between closely related balsam poplars, Populus balsamifera and P. trichocarpa (Salicaceae). New Phytol. 2014;204:693.
Sancho R, Cantalapiedra CP, Lopez-Alvarez D, Gordon SP, Vogel JP, Catalan P, Contreras-Moreira B. Comparative plastome genomics and phylogenomics of Brachypodium: flowering time signatures, introgression and recombination in recently diverged ecotypes. New Phytol. 2018;218(4):1631–44.
Hohmann N, Wolf EM, Rigault P, Zhou W, Kiefer M, Zhao Y, Fu C-X, Koch MA. Ginkgo biloba’s footprint of dynamic Pleistocene history dates back only 390,000 years ago. BMC Genomics. 2018;19(1):299.
Cheng L, Nam J, Chu SH, Rungnapa P, Min MH, Cao Y, Yoo JM, Kang JS, Kim KW, Park YJ. Signatures of differential selection in chloroplast genome between japonica and indica. Rice. 2019;12(1):65.
Magdy M, Ou L, Yu H, Chen R, Zhou Y, Hassan H, Feng B, Taitano N, van der Knaap E, Zou X, et al. Pan-plastome approach empowers the assessment of genetic variation in cultivated Capsicum species. Horticult Res. 2019;6(1):108.
Li J, Wang S, Jing Y, Wang L, Zhou S. A modified CTAB protocol for plant DNA extraction. Chin Bull Bot. 2013;48(1):72–8.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics. 2014;30(15):2114–20.
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19(5):455–77.
Dong W, Xu C, Cheng T, Lin K, Zhou S. Sequencing angiosperm plastid genomes made easy: a complete set of universal primers and a case study on the phylogeny of Saxifragales. Genome Biol Evol. 2013;5(5):989–97.
Huang DI, Cronk QCB. Plann: a command-line application for annotating plastome sequences. Appl Plant Sci. 2015;3(8):1500026.
Conant GC, Wolfe KH. GenomeVx: simple web-based creation of editable circular chromosome maps. Bioinformatics. 2008;24(6):861–2.
Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30(4):772–80.
Se-Al: sequence alignment editor. version 2.0.
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30(12):2725–9.
Beier S, Thiel T, Munch T, Scholz U, Mascher M. MISA-web: a web server for microsatellite prediction. Bioinformatics. 2017;33(16):2583–5.
Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, Rozen SG. Primer3—new capabilities and interfaces. Nucleic Acids Res. 2012;40(15):e115–e115.
Blacket MJ, Robin C, Good RT, Lee SF, Miller AD. Universal primers for fluorescent labelling of PCR fragments–an efficient and cost-effective approach to genotyping by fluorescence. Mol Ecol Resour. 2012;12(3):456–63.
Puyvelde VAN. A VANG, Triest L: atetra, a new software program to analyse tetraploid microsatellite data: comparison with tetra and tetrasat. Mol Ecol Resour. 2010;10(2):331–4.
Xu M, Li HG, Zhang B. Fifteen polymorphic simple sequence repeat markers from expressed sequence tags of Liriodendron tulipifera. Mol Ecol Notes. 2006;6(3):728–30.
Yao X, Zhang J, Ye Q, Huang H. Characterization of 14 novel microsatellite loci in the endangered Liriodendron chinense (Magnoliaceae) and cross-species amplification in closely related taxa. Conserv Genet. 2008;9(2):483–5.
Zheng X, Pan C, Diao Y, You Y, Yang C, Hu Z. Development of microsatellite markers by transcriptome sequencing in two species of Amorphophallus (Araceae). BMC Genomics. 2013;14(1):490.
Aggarwal RK, Hendre PS, Varshney RK, Bhat PR, Krishnakumar V, Singh L. Identification, characterization and utilization of EST-derived genic microsatellite markers for genome analyses of coffee and related species. Theor Appl Genet. 2007;114(2):359.
Cardle L, Ramsay L, Milbourne D, Macaulay M, Marshall D, Waugh R. Computational and experimental characterization of physically clustered simple sequence repeats in plants. Genetics. 2000;156(2):847–54.
Xu M, Sun Y, Li H. EST-SSRs development and paternity analysis for Liriodendron spp. New Forest. 2010;40(3):361–82.
Fu PC, Zhang YZ, Ya HY, Gao QB. Characterization of SSR genomic abundance and identification of SSR markers for population genetics in Chinese jujube (Ziziphus jujuba Mill.). PeerJ. 2016;4:e1735.
Zhang H, Li H, Xu M, Feng Y. Identification of Liriodendron tulipifera, Liriodendron chinense and hybrid Liriodendron using species-specific SSR markers. Scientia Silvae Sinicae. 2010;46(1):36–9.
Schlotterer C. Evolutionary dynamics of microsatellite DNA. Chromosoma. 2000;109(6):365–71.
Wang ML, Barkley NA, Jenkins TM: Microsatellite Markers in Plants and Insects. Part I: Applications of Biotechnology. Genes, genomes and genomics 2009, v. 3(no. 1):pp. 54-67-2009 v.2003 no.2001.
Korkovelos AE, Mavromatis AG, Huang WG, Hagidimitriou M, Giakoundis A, Goulas CK. Effectiveness of SSR molecular markers in evaluating the phylogenetic relationships among eight Actinidia species. Sci Hortic. 2008;116(3):305–10.
Garcia-Mas J, Monforte AJ, Arús P. Phylogenetic relationships among Cucumis species based on the ribosomal internal transcribed spacer sequence and microsatellite markers. Plant Syst Evol. 2004;248(1):191–203.
Lassois L, Denancé C, Ravon E, Guyader A, Guisnel R, Hibrand-Saint-Oyant L, Poncet C, Lasserre-Zuber P, Feugey L, Durel C-E. Genetic diversity, population structure, parentage analysis, and construction of core collections in the french apple germplasm based on SSR markers. Plant Mol Biol Rep. 2016;34(4):827–44.
Liu J, Yang Y, Zhou X, Bao S, Zhuang Y. Genetic diversity and population structure of worldwide eggplant (Solanum melongena L.) germplasm using SSR markers. Genet Resour Crop Evol. 2018;65(6):1663–70.
Bernard A, Barreneche T, Lheureux F, Dirlewanger E. Analysis of genetic diversity and structure in a worldwide walnut (Juglans regia L.) germplasm using SSR markers. PLOS ONE. 2018;13(11):e0208021.
Belaj A, Muñoz-Diez C, Baldoni L, Porceddu A, Barranco D, Satovic Z. Genetic diversity and population structure of wild olives from the north-western Mediterranean assessed by SSR markers. Ann Bot. 2007;100(3):449–58.
Curci PL, De Paola D, Sonnante G. Development of chloroplast genomic resources for Cynara. Mol Ecol Resour. 2016;16(2):562–73.
Cao J, Jiang D, Zhao Z, Yuan S, Zhang Y, Zhang T, Zhong W, Yuan Q, Huang L. Development of chloroplast genomic resources in Chinese Yam (Dioscorea polystachya). Biomed Res Int. 2018;2018:6293847.
Dong W, Xu C, Li W, Xie X, Lu Y, Liu Y, Jin X, Suo Z. Phylogenetic resolution in Juglans based on complete chloroplast genomes and nuclear DNA sequences. Front Plant Sci. 2017;8:1148.
Jiang D, Zhao Z, Zhang T, Zhong W, Liu C, Yuan Q, Huang L. The chloroplast genome sequence of Scutellaria baicalensis provides insight into intraspecific and interspecific chloroplast genome diversity in scutellaria. Genes. 2017;8(9):227.
Doorduin L, Gravendeel B, Lammers Y, Ariyurek Y, Chin AWT, Vrieling K. The complete chloroplast genome of 17 individuals of pest species Jacobaea vulgaris: sNPs, microsatellites and barcoding markers for population and phylogenetic studies. DNA Res. 2011;18(2):93–105.
Pang X, Liu C, Shi L, Liu R, Liang D, Li H, Cherny SS, Chen S. Utility of the trnH-psbA intergenic spacer region and its combinations as plant DNA barcodes: a meta-analysis. PLoS ONE. 2012;7(11):e48833.
Saarela JM, Sokoloff PC, Gillespie LJ, Consaul LL, Bull RD. DNA barcoding the Canadian Arctic flora: core plastid barcodes (rbcL + matK) for 490 vascular plant species. PLoS ONE. 2013;8(10):e77982.
Hollingsworth PM, Graham SW, Little DP. Choosing and using a plant DNA barcode. PLoS ONE. 2011;6(5):e19254.
Dong W, Liu J, Yu J, Wang L, Zhou S. Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PLoS ONE. 2012;7(4):e35071.
Shaw J, Lickey EB, Schilling EE, Small RL. Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: the tortoise and the hare III. Am J Bot. 2007;94(3):275–88.
Dong W, Xu C, Li C, Sun J, Zuo Y, Shi S, Cheng T, Guo J, Zhou S. ycf1, the most promising plastid DNA barcode of land plants. Sci Rep. 2015;5:8348.
Kikuchi S, Bedard J, Hirano M, Hirabayashi Y, Oishi M, Imai M, Takase M, Ide T, Nakai M. Uncovering the protein translocon at the chloroplast inner envelope membrane. Science. 2013;339(6119):571–4.
Acknowledgements
None.
Funding
This work was supported by the National Natural Science Foundation of China (31570657) and National Forest and Grass Germplasm Resources Bank Program (2016–2020).
Author information
Authors and Affiliations
Contributions
BL and YZ designed the experiment; BL, FL and PH, and WG collected samples and performed the experiment; BL analyzed the data and wrote the manuscript; All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1.
Primers of SNP markers in the L. chinense chloroplast genome.
Additional file 2: Table S2.
Primers of Indels markers in the L. chinense chloroplast genome.
Additional file 3: Table S3.
SSRs identified from in silico comparative analysis of the chloroplast genomes of four L. chinense genotypes.
Additional file 4: Table S4.
Primers of small inversions.
Additional file 5: Table S5.
Nuclear SSRs identified in this study.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, B., Lin, F., Huang, P. et al. Development of nuclear SSR and chloroplast genome markers in diverse Liriodendron chinense germplasm based on low-coverage whole genome sequencing. Biol Res 53, 21 (2020). https://doi.org/10.1186/s40659-020-00289-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40659-020-00289-0