
Abstract
Background
Haloacid dehalogenase (HAD)-like hydrolases represent the largest superfamily of phosphatases, which release inorganic phosphate from phosphate containing compounds, such as sugar phosphates. The HAD-like phosphatases with highly substrate specificity, which perform irreversible dephosphorylation, are always integrated into in vitro synthetic enzymatic biosystems as the last enzymatic step for the cost-efficient production of biochemicals. Therefore, identification and characterization of substrate specificity of HAD-like phosphatases are important for exploring their application.
Results
In this study, a hyperthermophilic HAD-like phosphatase from Archaeoglobus fulgidus (AfPase) was cloned, expressed, and characterized. AfPase was identified as a type I Mg2+-dependent HAD-like phosphatase with high optimal temperature and thermostability. Among the tested phosphate containing compounds, AfPase exhibited the highest catalytic activity on p-nitrophenyl phosphate, followed by dihydroxyacetone phosphate (DHAP). On the basis of the high catalytic activity of AfPase to generate 1,3-dihydroxyacetone (DHA) from DHAP, an in vitro synthetic enzymatic biosystem containing this phosphatase and other five enzymes was constructed for the biosynthesis of DHA from inexpensive maltodextrin in one pot. About 14 mM (1.26 g/L) DHA was produced from 10 g/L maltodextrin.
Conclusions
A hyperthermophilic HAD-like phosphatase from Archaeoglobus fulgidus was characterized carefully, and the success of an in vitro synthetic enzymatic biosystem containing this phosphatase provided a promising approach for DHA production from maltodextrin.

Similar content being viewed by others

Background
In vitro synthetic enzymatic biosystem is using several cascade enzymes to implement complicated biotransformation, emerging as a promising biomanufacturing platform for producing some products (such as inositol and rare sugars) better than microbial fermentation (Li et al. 2011; Meng et al. 2018; You et al. 2017). This platform exhibits several advantages, such as high product yields (Opgenorth et al. 2016; Zhu et al. 2014), fast reaction rate (Karim and Jewett 2016), easy process control (Kim and Zhang 2016; Opgenorth et al. 2017), tolerance of toxic compounds (Korman et al. 2017), biosystem robustness (Zhu and Zhang 2017), and implementation of non-natural biosynthesis (You et al. 2013). The development of in vitro enzymatic biosystems is composed of three parts: (i) pathway reconstruction; (ii) enzyme selection, engineering, and production; and (iii) process engineering (Zhang 2010). In principle, in vitro pathway design should focus on commercial feasibility: (i) the cost of products should be higher than that of substrates; (ii) the product cost is mainly attributed to the enzyme cost; (iii) in vitro biosystems should be cofactor free or cofactor balance; and (iv) the last step should be irreversible to maximize the product yield (You and Zhang 2017).
The haloacid dehalogenase-like hydrolase superfamily is a large group of enzymes, whose members are involved not only in the enzymatic cleavage by nucleophilic substitution of carbon–halogen bonds (C–halogen), but also in a variety of hydrolytic enzyme activities including phosphoglucomutase (CO–P hydrolysis and intramolecular phosphoryl transfer) reactions, ATPases (PO–P cleavage), dehalogenases (C–Cl cleavage), phosphonatase (C–P), and phosphatase (CO–P) (Lahiri et al. 2004; Meng et al. 2019). Haloacid dehalogenase-like phosphatases (HAD-like phosphatases) catalyze the irreversible dephosphorylation of phosphate containing compounds, leading to their widespread application in in vitro synthetic enzymatic biosystems for catalyzing the last step. For example, YqaB obtained from Escherichia coli was used for rare sugars production, including d-sorbose and d-psicose (Yang et al. 2015), or l-fructose and l-tagatose (Li et al. 2011). Dihydroxyacetone phosphate dephosphorylase from Corynebacterium glutamicum was introduced into engineered Escherichia coli to produce dihydroxyacetone (Jain et al. 2016). In addition, a hyperthermophilic inositol monophosphatase from Thermotoga maritima was also introduced into an in vitro enzymatic pathway for inositol synthesis (You et al. 2017), resulting in the decrease of enzyme production costs and prolonged reaction time. However, these phosphatases always recognize and catalyze a wide array of substrates and reactions (Huang et al. 2015; Kuznetsova et al. 2006, 2015). The characterization, especially the substrate specificity, of these phosphatases is important for applying them for the production of many kinds of biochemicals in high yields.
The use of thermostable enzymes from hyperthermophilic microorganisms is beneficial for in vitro synthetic enzymatic biosystems due to (i) an obvious decrease in enzyme purification costs, because the thermophilic enzymes can be overexpressed in mesophilic hosts (e.g., E. coli) and purified through simple heat precipitation, (ii) lower chances of microbial contamination odds, (iii) minimum side reactions catalyzed by contaminated mesophilic proteins, and (iv) the decrease of the aqueous solutions viscosity at high temperatures accompanied with enhanced mass transfer (Jaturapaktrarak et al. 2014; Ninh et al. 2015; Wang and Zhang 2009; You et al. 2017; Zhang et al. 2017). Therefore, gene mining, characterization, and application of thermophilic enzymes for in vitro synthetic enzymatic biosystems are crucial to improve the efficiency of this emerging biomanufacturing platform.
In this study, a hyperthermophilic HAD-like phosphatase was cloned from the genome of Archaeoglobus fulgidus (AfPase, GeneBank accession no. AF_0374), expressed in E. coli BL21 (DE3) and purified by Ni–NTA resin and simple heat precipitation. The enzymatic properties of the purified recombinant AfPase, including cation dependence, optimal pH and temperature, thermostability, and substrate specificity, were investigated carefully. Because this AfPase exhibited much higher activity in dephosphorylation of dihydroxyacetone phosphate than other phosphate containing compounds, such as glucose 1-phosphate (G1P), glucose 6-phosphate (G6P), fructose 6-phosphate (F6P), and glyceraldehyde 3-phosphate (GA3P), this phosphatase was introduced into an in vitro synthetic enzymatic biosystem to catalyze the last step for 1,3-dihydroxyacetone (DHA) production from maltodextrin.
Materials and methods
Chemicals and strains
All chemicals were reagent grade and purchased from Sigma-Aldrich (St. Louis, MO, USA) or Sinopharm (Shanghai, China), unless otherwise noted. The dextrose equivalent of maltodextrin used in this study was 4.0–7.0. Escherichia coli Top10 was used for DNA manipulation and BL21 (DE3) containing a protein expression plasmid was used to produce the recombinant protein. Luria–Bertani (LB) medium was used for E. coli cell growth and recombinant protein expression supplemented with 100 μg/mL ampicillin or 50 μg/mL kanamycin. Oligonucleotides were synthesized by Genewiz (Suzhou, China).
Cloning, expression, and purification of AfPase
Plasmid pET20b-AfPase encoding phosphatase from a hyperthermophilic bacterium Archaeoglobus fulgidus was constructed based on the T7 promoter plasmid pET20b. The inserted DNA sequence of ORF AFULGI_RS01755 was amplified by PCR with a pair of primers IF (5′ gttta acttt aagaa ggaga tatac atatg atgcc ggata agaag ggcta c 3′) and IR (5′ gatct cagtg gtggt ggtgg tggtg aagct tcccc tccag agcct caacc atatc c 3′) using the genomic DNA of A. fulgidus as template, which was purchased from ATCC (Manassas, Virginia). A linear vector backbone was amplified based on pET20b (+) with a pair of primers VF (5′-ggata tggtt gaggc tctgg agggg aagct tcacc accac cacca ccact gagat c-3′) and the reverse primer VR (5′-gtagc ccttc ttatc cggca tcata tgtat atctc cttct taaag ttaaa c-3′). Plasmid pET20b-AfPase based on these two DNA fragments was obtained by Simple Cloning (You et al. 2012a, b).
For the overexpression of recombinant protein, the recombinant plasmid pET20b-AfPase containing the target sequence was transferred into E. coli BL21(DE3) to induce the protein expression. Two hundred milliliters of the LB culture containing 100 μg/mL of ampicillin in 1-L Erlenmeyer flasks was incubated in a rotary shaking machine under 220 rpm and 37 °C. When the OD600 reached 2, a final of 0.1 mM isopropyl-β-d- thiogalactopyranoside (IPTG) was added to the medium. Then, the culture was incubated at 37 °C for another 5 h. Cells were harvested by centrifugation, washed twice by 0.9% NaCl, and re-suspended in 50 mM HEPES buffer (pH 7.5) containing 50 mM NaCl. The cell pellets were lysed using a Scientz Sonic Dismembrator Model 500 (5-s pulse on and off, total 360 s, at 20% amplitude) in an ice bath. After centrifugation, the soluble his-tagged proteins in the supernatant were purified using a packed column of Ni-charged resin (Bio-Rad, Profinity IMAC Ni-Charged Resin). The endogenous E. coli proteins were washed away with a binding buffer (50 mM HEPES buffer, pH 7.5) containing 50 mM NaCl and 25 mM imidazole. The adsorbed recombinant proteins were eluted with 50 mM HEPES buffer (pH 7.5) containing 50 mM NaCl and 500 mM imidazole. Alternatively, heat treatment was used to purify the thermostable enzyme, that is, the cell lysate was treated in a water bath at 70, 80, and 90 °C for 20 min, respectively. After centrifugation at 12,000 rpm for 20 min, nearly, homogeneity recombinant proteins were obtained in the supernatant. The purity of the proteins was analyzed using sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). The protein concentration was measured by the Bio-Rad Bradford protein kit with bovine serum albumin as the reference.
Biochemical properties of AfPase
The effects of cations on the specific activity of AfPase were performed in 100 mM HEPES buffer (pH 7.0) containing 1 mM p-nitrophenyl phosphate (pNPP), 5 mM different ions, and appropriate concentration of AfPase at 85 °C for 3 min. The reaction was started by the addition of AfPase and stopped by ice bath. The product phosphate was determined by the mild-pH phosphate assay (Myung et al. 2010). One unit of enzyme activity was defined as the amount of enzyme that released 1 μmol of phosphate from pNPP per min.
The effects of pH on the specific activity of AfPase were performed in 100 mM Glycine–HCl buffer (pH 4.0–5.0), acetic acid–acetate sodium buffer (pH 5.0–6.0), Bis–Tris buffer (pH 6.0–7.0), HEPES buffer (pH 7.0–8.0) containing 1 mM pNPP, 5 mM MgCl2, and appropriate concentration of AfPase at 85 °C.
The effects of temperature on the specific activity of AfPase were performed in 100 mM HEPES buffer (pH 7.0) containing 1 mM pNPP, 5 mM MgCl2, appropriate concentration of AfPase at different temperatures from 40 to 100 °C.
For the thermostability of AfPase, enzymes (1 g/L) were preincubated at 80, 90, and 100 °C in 50 mM HEPES buffer (pH 7.0) for different time periods, respectively, followed by enzyme activity assay at 70 °C using pNPP as substrate. Thermal inactivation was determined by assuming a first-order inactivation reaction of logarithmic value of residual activity over time in hours.
Substrate specificity of AfPase
The enzyme–substrate specificity assays were performed at 60 °C in HEPES buffer (pH 7.0) containing 5 mM MgCl2 and different substrates. Appropriate concentration of AfPase was incubated with pNPP, glucose 1-phosphate (G1P), glucose 6-phosphate (G6P), glucosamine 6-phosphate (Glu6P), fructose 6-phosphate (F6P), tagatose 6-phosphate (T6P), psicose 6-phosphate (P6P), mannose 6-phosphate (M6P), fructose 1,6-diphosphate (FDP), glyceraldehyde 3-phosphate (GA3P), dihydroxyacetone phosphate (DHAP), and deoxyribose 5-phosphate (DR5P) at a final concentration of 10 mM, respectively. The released reaction product inorganic phosphate was measured by the mild-pH phosphate assay.
The kinetic parameters of AfPase
The Michaelis–Menten kinetic parameters of purified AfPase against DHAP were determined at 60 °C and 37 °C. 0.1 g/L AfPase was incubated at 60 °C with DHAP at various concentrations from 1 to 20 mM for 3 min. 1 g/L AfPase was incubated at 37 °C with DHAP at various concentrations from 5 to 80 mM for 5 min. The amounts of the catalytic product phosphate were detected and plotted against substrate concentrations to calculate the initial reaction rates. Kinetic constants (the Michaelis constant Km and the turnover number kcat) were estimated using the Michaelis–Menten equation with the GraphPad Prism 5.01 software (San Diego, CA, USA) by employing nonlinear regression.
The application of AfPase on DHA production
DHA was produced from maltodextrin by an in vitro synthetic enzymatic biosystem containing alpha-glucan phosphorylase (αGP), phosphoglucomutase (PGM), glucose 6-phosphate isomerase (PGI), fructose 6-phosphate aldolase (FSA), triosephosphate isomerase (TIM), and AfPase. Plasmid pET28a-EcoαGP, pET20b-CtPGM, pET20b-CtPGI, pET20b-TtcTIM, and pET20b-StIA were used for the preparation of corresponding recombinant αGP from E. coli, PGM from Clostridium thermocellum, PGI from Clostridium thermocellum, TIM from Thermus thermophiles, and isoamylase (IA) from Sulfolobus tokodaii as described elsewhere (Myung et al. 2011, 2014; Wang and Zhang 2010; Zhou et al. 2016). Plasmid pET28a-EcoFSA was prepared for the overexpression of FSA from E. coli and was constructed based on the T7 promoter plasmid pET28a using Simple Cloning described above. The FSA gene was amplified by PCR with a pair of primers of FSA-IF (5′-cggcc tggtg ccgcg cggca gccat atgga actgt atctg gatac ttcag-3′) and FSA-IR (5′-cagtg gtggt ggtgg tggtg ctcga gaatc gacgt tctgc caaac gctcc-3′) based on the genomic DNA of E. coli. The linear vector backbones were amplified based on pET28a (+) with a pair of primers FSA-VF (5′-ggagc gtttg gcaga acgtc gattc tcgag cacca ccacc accac cactg-3′) and the reverse primer FSA-VR (5′-ctgaa gtatc cagat acagt tccat atggc tgccg cgcgg cacca ggccg-3′). These plasmids were transformed into E. coli BL21(DE3). Each recombinant E. coli strain was cultivated in a 5-L fermenter (T&J Bio-engineering Co., LTD, Shanghai, China) for enzyme expression as described elsewhere (Meng et al. 2019). His-tagged αGP and FSA were purified by a packed column of Ni-charged resin; PGI and PGM were purified by RAC adsorption/desorption; and the others were purified by simple heat treatment at 70 °C for 20 min.
The maltodextrin was fist pretreated by mixing 1 U/mL of IA with 100 g/L maltodextrin (DE 4–7) in 5 mM acetate buffer (pH 5.5) containing 0.5 mM MgCl2. After incubation at 80 °C overnight, the IA-treated maltodextrin was obtained. One-pot biosynthesis of DHA was performed in 100 mM HEPES buffer containing 10 g/L IA-treated maltodextrin, 10 mM phosphate, 5 mM MgCl2, 0.18 g/L (1 U/mL) αGP, 0.05 g/L (1 U/mL) PGM, 2.52 mg/L (1 U/mL) PGI, 0.17 g/L (1 U/mL) FSA, 6.7 mg/L (1 U/mL) TIM, and 10 g/L (1.6 U/mL) AfPase at 37 °C. DHA was determined by HPLC equipped with Bio-Rad HPX87H column with 5 mM H2SO4 as a mobile phase (0.6 mL/min) and a refractive index detector at 60 °C.
Results and discussion
Sequence analysis of AfPase
In the genome of Archaeoglobus fulgidus DSM 4304, an opening reading frame (AFULGI_RS01755) encoding a 265 amino-acid protein was annotated as a probable p-nitrophenyl phosphatase (AfPase). The crystal structure of AfPase (PDB ID: 4jdp) shows the typical α/β core domain catalytic scaffold of HAD members (Fig. 1a), with the active site formed by four loops containing highly conserved motifs I–IV (Fig. 1b), indicating that AfPase is a member of HAD-like superfamily. Moreover, the deduced amino-acid sequence of this phosphatase has modest identities with three identified p-nitrophenyl phosphatase from Saccharomyces cerevisiae (24.2%, NCBI accession CAB56540.1) (Kaneko et al. 1989), Bacillus stearothermophilus (30.6%, NCBI accession AAM29189.1) (Shen et al. 2014), and Thermoplasma acidophilum (13.6%, NCBI accession WP_110641440.1) (Ekaterina et al. 2010). Despite the identities of the whole protein sequence of AfPase with other HAD phosphatases are relatively low, the typical α/β core domains including motif I–IV and a mobile cap domain are highly conserved (Additional file 1: Figure S1). Motif I is AfPase active site, which has conserved amino-acid sequence of DxDx (where x is any amino acid) among the p-nitrophenyl phosphatases and is used to mediate phosphoryl or carbon group transfer in all HAD members (Collet et al. 1998). The Thr147 and Lys184 of the AfPase are also as conservative as other p-nitrophenyl phosphatases, coinciding with amino-acid sequence of motif II and motif III of HAD-like phosphatases, respectively. Motif II possesses a hydrogen bond donor (Thr or Ser) to assist in substrate binding, and motif III contributes a positively charged group (Arg or Lys) to orient the electrophile and/or nucleophile (Zhang et al. 2004). The amino-acid sequence between positions 208 to 212 of AfPase traces a five-residue hairpin turn [(G/S)(D/S)x2–6(D/N)], which is consistent with motif IV of HAD-like phosphatases and functions in metal binding (Cho and Yan 2001; Zhang et al. 2004). The cap domain contains a stringently conserved Gly, and the rest of the cap domain sequence was highly variable among subfamilies (Lahiri et al. 2004). Based on these information, we could deduce that the cap domain of AfPase is around position 106, locating between motif I and motif II (Additional file 1: Figure S1). The results of amino-acid sequence analysis implied that the AfPase was an HAD-like phosphatase.

Crystal structure of probable p-nitrophenyl phosphatase from Archaeoglobus fulgidus (AfPase, PDB 4jdp). Cartoon representations of the structure of the fold with close-up of different active site configurations. Beta strands are colored dark gray, while alpha-helices are colored light gray. a Side view of AfPase. It shows the Rossmann-like fold of the HAD superfamily and reveals the typical spatial orientations of the conserved residues involved in catalysis. b Close-up active site view of AfPase with motif I–IV signatures. Motif I: conserved amino-acid sequence of DxDx, Motif II: conserved amino-acid of Thr or Ser, Motif III: conserved amino acid of Arg or Lys, Motif IV: conserved amino-acid sequence of (G/S)(D/S)x2–6(D/N)
Heterologous expression and purification of recombinant AfPase
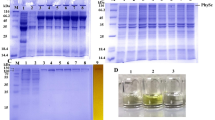
The expression plasmid, pET20b-AfPase, was constructed for the production of his-tagged AfPase. The AfPase was expressed efficiently in BL21(DE3) (Fig. 2, line T and line S). After expression, the enzymes were purified by heat treatment at 70, 80, and 90 °C for 20 min, and were also purified by a packed column of nickel resin (Fig. 2, line 70H, 80H, 90H, and Ni–NTA), respectively. The purified AfPase appeared to be homogeneous with a molecular mass of 30 kDa (Fig. 2), which was close to the estimated value of 29.2 kDa from the deduced amino-acid sequence. The purification information is shown in Table 1. Although the purity of AfPase purified by a packed column of Ni-charged resin was the highest, the loss of protein in the purification process was higher, leading to the low recovery yield of activity. Compromising the purity and activity yield, heat treatment was the best method for AfPase purification, especially at 80 °C for 20 min. Then, the purified enzymes after heat treatment at 80 °C for 20 min were used for the following experiments.

SDS-PAGE analysis of recombinant enzyme expression in E. coli BL21 (DE3) and purification of AfPase. M, protein marker; T, total cell lysate of recombinant E. coli cells; S, the supernatant of cell lysate; 70H, purified AfPase after heat treatment of cell supernatant at 70 °C for 20 min; 80H, purified AfPase after heat treatment of cell supernatant at 80 °C for 20 min; 90H, purified AfPase after heat treatment of cell supernatant at 90 °C for 20 min; Ni–NTA, purified AfPase by Ni–NTA resin
Biochemical properties of AfPase
The carboxylate residues on loop 4 (motif IV) and their positions on the loop were related to the metal ion activation among HAD family members (Zhang et al. 2004), indicating that AfPase was metal-dependent enzymes. Therefore, the effects of metal ions, including Mn2+, Mg2+, Ca2+, Cu2+, Zn2+, Co2+, Fe3+, and Ni2+ on AfPase activity towards p-nitrophenyl phosphate were examined (Fig. 3). The activity of AfPase without any metal ion was set as 100%, and the highest AfPase activity, which is about 32-fold higher than that without the addition of metal ions, was observed in the presence of 5 mM Mg2+. Ni2+, Mn2+, Co2+, and Zn2+ can increase the AfPase activity by 12, 3.8, 3.4, and 1.5-folds, respectively. Cu2+ and Ca2+ had no significant influences on its activities, and Fe3+ caused obvious salt precipitates and decreased the activity by 34%. The results suggested that AfPase was an Mg2+-dependent phosphatase.

Effects of cations on the specific activity of AfPase. The reactions were performed in 100 mM HEPES buffer (pH 7.0) containing 1 mM p-nitrophenyl phosphate, 5 mM different ions, and appropriate concentration of phosphatase at 85 °C
The effects of pH on AfPase activity towards p-nitrophenyl phosphate were analyzed using four different buffer systems (Fig. 4). The recombinant protein exhibited relatively high activity around pH 4.5–5.0 in glycine–HCl buffer. The shape and maximum of the pH velocity curves, however, varied somewhat with the nature of the buffer ion present. Given that the optimum growth temperature of Archaeoglobus fulgidus DSM 4304 is 85 °C (Klenk et al. 1997), the temperature dependence of the enzymatic activity was analyzed between 40 and 100 °C (Fig. 5a). AfPase had the highest activity at 100 °C, which was the highest value of all p-nitrophenyl phosphatases of the superfamily characterized to our best knowledge. Furthermore, the enzyme exerted a good thermostability (Fig. 5b). The half-life of 1 g/L AfPase was 8.7, 3.5, and 0.16 h at 80, 90, and 100 °C, respectively. In contrast, the optimal temperature of thermophilic p-nitrophenyl phosphatases from Bacillus stearothermophilus (BsPase) was 55 °C (Guo et al. 2014). The higher temperature optimum of activity and thermostability of AfPase than BsPase might due to aromatic–aromatic interaction and electrostatic interaction. The aromatic–aromatic interaction was an important factor to maintain the stability of enzymes formed by aromatic amino acids (Tyr, Phe, and Try) (Burley and Petsko, 1985). The analysis of amino-acid residue (Additional file 1: Table S1) showed that the percentage of aromatic amino acid (Tyr, Phe, Try) of AfPase (10.7%) was higher than BsPase (8.2%), indicating that there might be more aromatic–aromatic interactions in AfPase than that of BsPase. Electrostatic interaction formed by charged amino acid (Glu, Asp, Arg, and Lys) also had positive effect on improving the thermal stability of the enzyme (Yip et al. 1995). The percentage of charged amino acid of AfPase (30.3%) was also higher than that of BsPase (23.5%), indicating that there might be more electrostatic interactions in AfPase. The extremely high-temperature optimum of activity and thermostability of AfPase showed the great potential in in vitro synthetic enzymatic biosystems, which always need thermophilic enzymes (You and Zhang 2017).

Effects of pH on the specific activity of AfPase. The reactions were performed in 100 mM glycine–HCl buffer (pH 4.0–5.0), acetic acid–acetate sodium buffer (pH 5.0–6.0), Bis–Tris buffer (pH 6.0–7.0), HEPES buffer (pH 7.0–8.0) containing 1 mM p-nitrophenyl phosphate, 5 mM MgCl2, and appropriate concentration of phosphatase at 85 °C

Effects of temperature on the specific activity (a) and thermostability (b) of AfPase. The reactions were performed in 100 mM HEPES buffer (pH 7.0) containing 1 mM p-nitrophenyl phosphate, 5 mM MgCl2, and appropriate concentration of phosphatase at different temperatures (a). The enzyme (1 g/L) was preincubated at different temperatures in 50 mM HEPES buffer (pH 7.0). The residual activity was measured at 70 °C using p-nitrophenyl phosphate as substrate. Thermal inactivation is plotted on logarithmic scale to demonstrate first-order kinetics (b)
The activity promiscuity of AfPase
HAD-like phosphatases showed catalytic efficiency and affinity to a wide range of phosphorylated metabolites and this phenomenon was called “promiscuity” (Huang et al. 2015; Kuznetsova et al. 2006, 2015). However, the promiscuity of them is unpredictable from sequence similarity even at subfamily level (Jojima et al. 2012). Therefore, basic enzymatic characterization might be the most reliable method to obtain accurate substrate spectrum of them. In this study, the substrate specificity of recombinant AfPase was analyzed towards a range of phosphate metabolites, including pNPP, glucose 1-phosphate, glucose 6-phosphate, glucosamine 6-phosphate, fructose 6-phosphate, tagatose 6-phosphate, psicose 6-phosphate, mannose 6-phosphate, fructose 1,6-diphosphate, glyceraldehyde 3-phosphate (GA3P), dihydroxyacetone phosphate (DHAP), and deoxyribose 5-phosphate (DR5P) (Table 2). AfPase decomposed pNPP with the specific activity of 3.36 U/mg at 60 °C, yielding a yellow product p-nitrophenol and phosphate. Except for pNPP, AfPase had the highest activity towards DHAP, followed by GA3P and DR5P, and AfPase had low or no activity to other sugar phosphates. The Km values of AfPase against DHAP were 4.72 mM and 10.47 mM at 60 °C and 37 °C, respectively, while kcat were 0.64/s and 0.16/s, respectively (Table 3). The kcat/Km value of AfPase against DHAP at 60 °C was about 10 times of that at 37 °C. These results indicated that AfPase is a potential DHAP phosphatase that can be used for 1,3-dihydroxyacetone (DHA) production by the dephosphorylation of DHAP in the last step.
The application of AfPase for 1,3-dihydroxyacetone production
DHA is a valuable chemical with a wide range of applications in the cosmetics, food, and pharmaceutical industries (Wang et al. 2001). In addition, it is also used as a versatile building block for the synthesis of fine chemicals (Pagliaro et al. 2010), such as heterocyclic compounds (e.g., imidazole, furan), triglyceride, and so on. On the basis of the substrate specificity of AfPase, an in vitro synthetic enzymatic biosystem for DHA production from maltodextrin was constructed (Fig. 6). Six steps of reaction were occurred sequentially in this biosystem: (i) maltodextrin is phosphorylated by αGP in the presence of Pi, yielding G1P; (ii) G1P is converted to G6P under the catalysis of PGM; (iii) G6P is isomerized to F6P catalyzed by PGI; (iv) FSA cleaves F6P into GA3P and the final product—DHA; (v) GA3P is isomerized to DHAP by TIM; (vi) DHAP is dephosphorylated into DHA by AfPase, yielding a by-product Pi, which can be used in Step (i). The standard Gibbs energy changes (ΔG′°) of reactions 1–6 were calculated to be +2.8, − 7.4, + 2.5, + 15.6, − 5.5, and − 12.7 kJ/mol at pH 7.0 and ionic strength of 0.1 M (http://equilibrator.weizmann.ac.il/), respectively (Additional file 1: Table S2). The overall Gibbs energy change was − 4.7 kJ/mol, indicating that reactions can occur spontaneously, and achieve high product yield. All enzymes for DHA synthesis were expressed in E. coli BL21 (DE3) and purified as described above (Additional file 1: Figure S2). The specific activities of αGP, PGM, PGI, FSA, TIM, and AfPase were 5.6, 20, 396, 6, 150, and 0.3 U/mg at 37 °C, respectively (Additional file 1: Table S2).

Scheme of pathway for the conversion of maltodextrin to 1,3-dihydroxyacetone (DHA) by in vitro synthetic enzymatic biosystem. The enzymes are alpha-glucan phosphorylase (αGP), phosphoglucomutase (PGM), phosphoglucose isomerase (PGI), fructose 6-phosphate aldolase (FSA), triosephosphate isomerase (TIM), and phosphatase (Pase)
DHA was synthesized from 10 g/L IA-treated maltodextrin (DE 4–7) (Fig. 7). When the AfPase was absent, DHA was only produced by the cleavage of F6P. ΔG′° of reaction 4 (F6P = GA3P + DHA, ΔG′° =+15.6 kJ/mol) indicates that it is an extremely thermodynamically unfavorable reaction. Although reaction 5 (GA3P = DHAP) can push the reaction 4 forward, ΔG′° (− 5.5 kJ/mol) of reaction 5 is much lower than reaction 4. Therefore, only about 2 mM DHA was produced after 4 h and no longer increased (Fig. 7, red line). When AfPase was integrated into the reaction system, DHA was produced not only by F6P cleavage, but also by the dephosphorylation of DHAP. The driving force of reaction 5 plus reaction 6 (DHAP = DHA + Pi) was capable of pushing the whole reactions to produce more DHA. After incubation 24 h at 37 °C, about 14 mM DHA was produced (Fig. 7, black line), much higher than the biosystem without adding AfPase, indicating the success of the construction of this biosystem.

Production of DHA from maltodextrin. The reaction was performed in 100 mM HEPES buffer (pH 7.0) containing 10 g/L isoamylase treated maltodextrin, 5 mM MgCl2, 10 mM Pi, 0.18 g/L (1 U/mL) EcoαGP, 0.05 g/L (1 U/mL) CtPGM, 2.52 mg/L (1 U/mL) CtPGI, 0.17 g/L (1 U/mL) EcoFSA, 6.7 mg/L (1 U/mL) TtcTIM, and in the absence (close circle) and presence (close square) of 10 g/L (1.6 U/mL) AfPase at 37 °C
Even though the cell-based DHA production by the glycerol fermentation of Gluconobacter oxydans, which is the main method for DHA production at present, is already very successful (Bories et al. 1991; Li et al. 2010), we believe that our in vitro enzymatic route is highly promising despite the current product yield of DHA was lower than theoretical yield. The major advantages of our biomanufacturing platform are the high purity of the product (no by-products), the fast optimization possibilities (no need to consider culture conditions or regulatory cell mechanisms, etc.), and cofactor free (no need to add NADH or NADPH regeneration system). To achieve high product yield of this in vitro synthetic enzymatic biosystem, some shortcomings need to be overcome: (i) the low reaction temperature (at 37 °C) limited the activity of thermophilic AfPase, leading to a prolonged reaction time. The low reaction temperature is owing to the instability of FSA at high temperatures, because we have not find any thermophilic FSA yet, except for mesophilic FSA from E. coli. The engineering of current EcoFSA by directed evolution using high-throughput screening method (Alvizo et al. 2014; Zhou et al. 2018) and gene mining for thermostable FSA could be performed in the future to obtain thermal-stable FSA; (ii) the instability of DHAP (Myung et al. 2010) may also be a reason for the low yield of DHA. In this system, if DHAP is not phosphorylated by AfPase to generate DHA in time, the accumulated DHAP would degrade spontaneously. In the future, the enzyme complex containing TIM and AfPase can be constructed to make DHAP produced by TIM transfer to AfPase quickly (You et al. 2012a), mitigating the degradation of DHAP; and (iii) although the AfPase has very low activity towards G1P, G6P, and F6P, it still has a considerable activity to GA3P (Table 2), which lead to a waste of substrate and further to decrease the yield of DHA. The fundamental solution to this problem is to engineer AfPase to obtain a highly specific DHAP phosphatase by protein engineering method.
Conclusion
In this study, a probable p-nitrophenyl phosphatase was cloned from Archaeoglobus fulgidus genome, expressed in E. coli BL21(DE3), purified by simple heat treatment, and identified as a type I Mg2+-dependent HAD-like phosphatase. It exhibited relatively high activity around pH 4.5–5.0 in glycine–HCl buffer and had high optimal temperature (about 100 °C) and thermostability. The substrate specificity analysis revealed that AfPase was a potential DHAP phosphatase which could be used for DHA production by the dephosphorylation of DHAP. Therefore, an AfPase-contained in vitro synthetic enzymatic biosystem for DHA biosynthesis from maltodextrin was constructed, providing a promising approach for DHA production. The gene mining and characterization of more phosphatases will be of importance for the production of fine chemicals by in vitro synthetic enzymatic biosystems.
Availability of data and materials
The data sets supporting the conclusions of this article are included in the main manuscript. The authors promise to provide any missing data on request.
Abbreviations
- DHAP:
-
dihydroxyacetone phosphate
- DHA:
-
1,3-dihydroxyacetone
- G1P:
-
glucose 1-phosphate
- G6P:
-
glucose 6-phosphate
- F6P:
-
fructose 6-phosphate
- GA3P:
-
glyceraldehyde 3-phosphate
- pNPP:
-
p-nitrophenyl phosphate
- Glu6P:
-
glucosamine 6-phosphate
- T6P:
-
tagatose 6-phosphate
- P6P:
-
psicose 6-phosphate
- M6P:
-
mannose 6-phosphate
- FDP:
-
fructose 1,6-diphosphate
- DR5P:
-
deoxyribose 5-phosphate
- AfPase:
-
phosphatase from Archaeoglobus fulgidus
- EcoαGP:
-
alpha-glucan phosphorylase from Escherichia coli
- CtPGM:
-
phosphoglucomutase from Clostridium thermocellum
- CtPGI:
-
phosphoglucose isomerase from C. thermocellum
- EcoFSA:
-
fructose 6-phosphate aldolase from E. coli
- TtcTIM:
-
triosephosphate isomerase from Thermus thermophilus
- IA:
-
isoamylase
References
Alvizo O, Nguyen LJ, Savile CK, Bresson JA, Lakhapatri SL, Solis EO, Fox RJ, Broering JM, Benoit MR, Zimmerman SA, Novick SJ, Liang J, Lalonde JJ (2014) Directed evolution of an ultrastable carbonic anhydrase for highly efficient carbon capture from flue gas. Proc Natl Acad Sci USA 111:16436–16441
Bories A, Claret C, Soucaille P (1991) Kinetic-study and optimization of the production of dihydroxyacetone from glycerol using Gluconobacter oxydans. Process Biochem 26:243–248
Burley SK, Petsko GA (1985) Aromatic-aromatic interaction: a mechanism of protein structure stabilization. Science 229:23–28
Cho H, Yan D (2001) BeF3− acts as a phosphate analog in proteins phosphorylated onaspartate: structure of a BeF3− complex with phosphoserinephosphatase. Proc Natl Acad Sci USA 98:8525–8530
Collet JF, Stroobant V, Pirard M, Delpierre G, Van Schaftingen E (1998) A new class of phosphotransferases phosphorylated on an aspartate residue in an amino-terminal DXDX(T/V) motif. J Biol Chem 273:14107–14112
Ekaterina K, Michael P, Sanders SA, Jeffrey R, Alexei S, Arrowsmith CH, Edwards AM, Yakunin AF (2010) Enzyme genomics: application of general enzymatic screens to discover new enzymes. FEMS Microbiol Rev 29:263–279
Guo Z, Wang F, Shen T, Huang J, Wang Y, Ji C (2014) Crystal structure of thermostable p-nitrophenylphosphatase from Bacillus stearothermophilus (Bs-TpNPPase). Protein Pept Lett 21:483–489
Huang H, Pandya C, Liu C, Al-Obaidi NF, Wang M, Zheng L, Keating ST, Aono M, Love JD, Evans B, Seidel RD, Hillerich BS, Garforth SJ, Almo SC, Mariano P, Dunaway-Mariano D, Allen KN, Farelli JD (2015) Panoramic view of a superfamily of phosphatases through substrate profiling. Proc Natl Acad Sci USA 112:E1974–E1983
Jain VK, Tear CJ, Lim CY (2016) Dihydroxyacetone production in an engineered Escherichia coli through expression of Corynebacterium glutamicum dihydroxyacetone phosphate dephosphorylase. Enzyme Microb Technol 86:39–44
Jaturapaktrarak C, Napathorn SC, Cheng M, Okano K, Ohtake H, Honda K (2014) In vitro conversion of glycerol to lactate with thermophilic enzymes. Bioresour Bioprocess 1:1–8
Jojima T, Igari T, Gunji W, Suda M, Inui M, Yukawa H (2012) Identification of a HAD superfamily phosphatase, HdpA, involved in 1,3-dihydroxyacetone production during sugar catabolism in Corynebacterium glutamicum. FEBS Lett 586:4228–4232
Kaneko Y, Toh-E A, Banno I, Oshima Y (1989) Molecular characterization of a specific p-nitrophenylphosphatase gene, PHO13, and its mapping by chromosome fragmentation in Saccbaromyces cerevisiae. Mol Gen Genet 220:133–139
Karim AS, Jewett MC (2016) A cell-free framework for rapid biosynthetic pathway prototyping and enzyme discovery. Metab Eng 36:116–126
Kim JE, Zhang YH (2016) Biosynthesis of D-xylulose 5-phosphate from d-xylose and polyphosphate through a minimized two-enzyme cascade. Biotechnol Bioeng 113:275–282
Klenk HP, Clayton RA, Tomb JF, White O, Nelson KE, Ketchum KA, Dodson RJ, Gwinn M, Hickey EK, Peterson JD, Richardson DL, Kerlavage AR, Graham DE, Kyrpides NC, Fleischmann RD, Quackenbush J, Lee NH, Sutton GG, Gill S, Kirkness EF, Dougherty BA, McKenney K, Adams MD, Loftus B, Peterson S, Reich CI, McNeil LK, Badger JH, Glodek A, Zhou L, Overbeek R, Gocayne JD, Weidman JF, McDonald L, Utterback T, Cotton MD, Spriggs T, Artiach P, Kaine BP, Sykes SM, Sadow PW, D’Andrea KP, Bowman C, Fujii C, Garland SA, Mason TM, Olsen GJ, Fraser CM, Smith HO, Woese CR, Venter JC (1997) The complete genome sequence of the hyperthermophilic, sulphate-reducing archaeon Archaeoglobus fulgidus. Nature 390:364–370
Korman TP, Opgenorth PH, Bowie JU (2017) Asynthetic biochemistry platform for cell free production of monoterpenes from glucose. Nat Commun 8:15526
Kuznetsova E, Proudfoot M, Gonzalez CF, Brown G, Omelchenko MV, Borozan I, Carmel L, Wolf YI, Mori H, Savchenko AV, Arrowsmith CH, Koonin EV, Edwards AM, Yakunin AF (2006) Genome-wide analysis of substrate specificities of the Escherichia coli haloacid dehalogenase-like phosphatase family. J Biol Chem 281:36149–36161
Kuznetsova E, Nocek B, Brown G, Makarova KS, Flick R, Wolf YI, Khusnutdinova A, Evdokimova E, Jin K, Tan K, Hanson AD, Hasnain G, Zallot R, de Crecy-Lagard V, Babu M, Savchenko A, Joachimiak A, Edwards AM, Koonin EV, Yakunin AF (2015) Functional diversity of haloacid dehalogenase superfamily phosphatases from Saccharomyces cerevisiae: biochemical, structural and evolutionary insights. J Biol Chem 290:18678–18698
Lahiri SD, Zhang GF, Dai JY, Dunaway-Mariano D, Allen KN (2004) Analysis of the substrate specificity loop of the HAD superfamily cap domain. Biochemistry 43:2812–2820
Li MH, Wu J, Liu X, Lin JP, Wei DZ, Chen H (2010) Enhanced production of dihydroxyacetone from glycerol by overexpression of glycerol dehydrogenase in an alcohol dehydrogenase-deficient mutant of Gluconobacter oxydans. Bioresour Technol 101:8294–8299
Li Z, Cai L, Qi Q, Styslinger TJ, Zhao G, Wang PG (2011) Synthesis of rare sugars with l-fuculose-1-phosphate aldolase (FucA) from Thermus thermophilus HB8. Bioorg Med Chem Lett 21:5084–5087
Meng DD, Wei XL, Zhang Y-HPJ, Zhu Z, You C, Ma YH (2018) Stoichiometric conversion of cellulosic biomass by in vitro synthetic enzymatic biosystems for biomanufacturing. ACS Catal 8:9550–9559
Meng DD, Liang AL, Wei XL, You C (2019) Enzymatic characterization of a thermostable phosphatase from Thermomicrobium roseum and its application for biosynthesis of fructose from maltodextrin. Appl Microbiol Biotechnol. https://doi.org/10.1007/s00253-019-09917-6
Myung S, Wang YR, Zhang Y-HP (2010) Fructose-1,6-bisphosphatase from a hyper-thermophilic bacterium Thermotoga maritima: characterization, metabolite stability, and its implications. Process Biochem 45:1882–1887
Myung S, Zhang XZ, Zhang YH (2011) Ultra-stable phosphoglucose isomerase through immobilization of cellulose-binding module-tagged thermophilic enzyme on low-cost high-capacity cellulosic adsorbent. Biotechnol Prog 27:969–975
Myung S, Rollin J, You C, Sun F, Chandrayan S, Adams MW, Zhang YH (2014) In vitro metabolic engineering of hydrogen production at theoretical yield from sucrose. Metab Eng 24:70–77
Ninh PH, Honda K, Sakai T, Okano K, Ohtake H (2015) Assembly and multiple gene expression of thermophilic enzymes in Escherichia coli for in vitro metabolic engineering. Biotechnol Bioeng 112:189–196
Opgenorth PH, Korman TP, Bowie JU (2016) A synthetic biochemistry module for production of bio-based chemicals from glucose. Nat Chem Biol 12:393–395
Opgenorth PH, Korman TP, Iancu L, Bowie JU (2017) A molecular rheostat maintains ATP levels to drive a synthetic biochemistry system. Nat Chem Biol 13:938–942
Pagliaro M, Ciriminna R, Kimura H, Rossi M, Pina CD (2010) From glycerol to value-added products. Angew Chem Int Ed 46:4434–4440
Shen T, Guo Z, Ji C (2014) Structure of a His170Tyr mutant of thermostable pNPPase from Geobacillus stearothermophilus. Acta Crystallogr F Struct Biol Commun 70:697–702
Wang Y, Zhang YP (2009) Overexpression and simple purification of the Thermotoga maritima 6-phosphogluconate dehydrogenase in Escherichia coli and its application for NADPH regeneration. Microb Cell Fact 8:1–11
Wang Y, Zhang YH (2010) A highly active phosphoglucomutase from Clostridium thermocellum: cloning, purification, characterization and enhanced thermostability. J Appl Microbiol 108:39–46
Wang ZX, Zhuge J, Fang H, Prior BA (2001) Glycerol production by microbial fermentation: a review. Biotechnol Adv 19:201–223
Yang J, Zhu Y, Li J, Men Y, Sun Y, Ma Y (2015) Biosynthesis of rare ketoses through constructing a recombination pathway in an engineered Corynebacterium glutamicum. Biotechnol Bioeng 112:168–180
Yip K, Stillman TJ, Britton KL, Artymiuk PJ, Baker PJ, Sedelnikova SE, Engel PC, Pasquo A, Chiaraluce R, Consalvi V (1995) The structure of Pyrococcus furiosus glutamate dehydrogenase reveals a key role for ion-pair networks in maintaining enzyme stability at extreme temperatures. Structure 3:1147–1158
You C, Zhang YHP (2017) Biomanufacturing by in vitro biosystems containing complex enzyme mixtures. Process Biochem 52:106–114
You C, Myung S, Zhang YHP (2012a) Facilitated substrate channeling in a self-assembled trifunctional enzyme complex. Angew Chem Int Ed Engl 51:8787–8790
You C, Zhang XZ, Zhang YH (2012b) Simple cloning via direct transformation of PCR product (DNA Multimer) to Escherichia coli and Bacillus subtilis. Appl environ microbiol 78:1593–1595
You C, Chen HG, Myung S, Sathitsuksanoh N, Ma H, Zhang XZ, Li JY, Zhang YHP (2013) Enzymatic transformation of nonfood biomass to starch. Proc Natl Acad Sci USA 110:7182–7187
You C, Shi T, Li YJ, Han PP, Zhou XG, Zhang YHP (2017) An in vitro synthetic biology platform for the industrial biomanufacturing of myo-inositol from starch. Biotechnol Bioeng 114:1855–1864
Zhang YH (2010) Production of biocommodities and bioelectricity by cell-free synthetic enzymatic pathway biotransformations: challenges and opportunities. Biotechnol Bioeng 105:663–677
Zhang GF, Morais MC, Dai JY, Zhang WH, Dunaway-Mariano D, Allen KN (2004) Investigation of metal ion binding in phosphonoacetaldehyde hydrolase identifies sequence markers for metal-activated enzymes of the HAD enzyme superfamily. Biochemistry 43:4990–4997
Zhang YP, Sun J, Ma Y (2017) Biomanufacturing: history and perspective. J Ind Microbiol Biotechnol 44:773–784
Zhou W, You C, Ma H, Ma Y, Zhang YH (2016) One-pot biosynthesis of high-concentration alpha-glucose 1-phosphate from starch by sequential addition of three hyperthermophilic enzymes. J Agric Food Chem 64:1777–1783
Zhou W, Huang R, Zhu ZG, Zhang YHPJ (2018) Coevolution of both thermostability and activity of polyphosphate glucokinase from Thermobifida fusca YX. Appl Environ Microbiol 84:e01224–e01318
Zhu Z, Zhang YP (2017) In vitro metabolic engineering of bioelectricity generation by the complete oxidation of glucose. Metab Eng 39:110–116
Zhu Z, Kin Tam T, Sun F, You C, Percival Zhang YH (2014) A high-energy-density sugar biobattery based on a synthetic enzymatic pathway. Nat Commun 5:3026–3034
Acknowledgements
WW, ZML, and CY acknowledge the State Key Laboratory of Bioreactor Engineering and Shanghai Committee of Science and Technology, the Fundamental Research Funds for the Central Universities for financial support. CY acknowledges National Natural Science Foundation of China and the 1000-youth talent program of China for financial support.
Funding
This work was funded by the Open Funding Project of the State Key Laboratory of Bioreactor Engineering and Shanghai Committee of Science and Technology, the Fundamental Research Funds for the Central Universities (22221818014), National Natural Science Foundation of China (Grant No. 21778073), and the 1000-youth talent program of China.
Author information
Authors and Affiliations
Contributions
WW and QZL performed the experiments and analyzed the data. WW and CY conceived and designed the experiments. WW wrote the paper. DDM, CY, and ZML revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1.
Additional Figures and Tables.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Wang, W., Meng, D., Li, Q. et al. Characterization of a hyperthermophilic phosphatase from Archaeoglobus fulgidus and its application in in vitro synthetic enzymatic biosystem. Bioresour. Bioprocess. 6, 22 (2019). https://doi.org/10.1186/s40643-019-0257-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40643-019-0257-5