
Abstract
Background
In acute respiratory distress syndrome (ARDS), uncontrolled production of activators of coagulation and proinflammatory mediators results in a shift from an adequate local innate immune response to hypercoagulability and inflammation. This study aimed to investigate whether the protease inhibitors antithrombin (AT) and alpha-1 protease inhibitor (A1PI) may attenuate an exaggerated pulmonary immune response.
Methods
Lung injury was induced either by single intranasal administration of lipopolysaccharide (LPS) (5 mg/kg) in BALB/c mice or by combination of an intravenous injection of LPS (10 mg/kg) with subsequent injurious ventilation using high tidal volumes (12–15 ml/kg) for 4 h in RccHan Wistar rats. Animals received either a single bolus of AT (250 IU/kg) or A1PI (60 mg/kg) alone or in combination, with or without intravenous low-dose heparin (100 U/kg). Control animals received saline. Additional controls received neither LPS, nor ventilation, nor treatment. Endpoints were local and systemic markers of coagulation, e.g., thrombin–antithrombin complexes (TATc), and inflammation, e.g., interleukin-6.
Results
Both lung injury models resulted in a pronounced immune response within the pulmonary compartment shown by elevated levels of markers of coagulation and inflammation. The two-hit lung injury model also induced profound systemic coagulopathy and inflammation. Monotherapy with AT or A1PI did not reduce pulmonary coagulopathy or inflammation in any lung injury model. Nor did combination therapy with AT and A1PI result in a decrease of coagulation or inflammatory parameters. AT markedly reduced systemic levels of TATc in the two-hit lung injury model. Systemic inflammation was not affected by the different interventions. Additional administration of heparin did not lead to macroscopic bleeding incidences.
Conclusions
In two different murine models of acute lung injury, neither single therapy with AT or A1PI nor combination of both agents attenuates the pronounced pulmonary coagulation or inflammatory response.
Similar content being viewed by others


Background
ARDS is a heterogeneous syndrome characterized by local production of coagulation proteases and proinflammatory mediators causing a shift from a potentially adequate innate immune response to harmful coagulation and inflammation [1, 2]. In ARDS patients, the degree of pulmonary hypercoagulability and inflammation is clearly linked to clinical outcome [3,4,5].
Antithrombin (AT) and alpha-1 protease inhibitor (A1PI) are serine protease inhibitors playing a pivotal role in maintaining equilibrium between protective and harmful coagulation and inflammation [6,7,8]. AT contributes to a balanced coagulation response by inhibiting the activity of thrombin, a coagulation factor facilitating the conversion of fibrinogen to fibrin [9]. Preclinical studies also suggest an anti-inflammatory effect of AT by downregulating the inflammatory response and preventing premature neutrophil activation [10, 11]. A1PI is an acute-phase protein exerting anti-inflammatory effects by blocking proteolytic enzymes released by activated neutrophils into the pulmonary compartment, acting as an “anti-protease screen” [7]. Previously, both AT and A1PI were administered in an indirect ARDS model in sheep leading to attenuation of microvascular protein permeability within the lungs and prevention of a decrease in arterial oxygenation. None of these effects were observed when only one intervention was administered [12].
Studies in ARDS patients with anticoagulants, such as protein C, heparin, or AT, did not result in improved outcome despite inhibiting an exaggerated coagulation response [13,14,15,16,17]. As coagulation and inflammation are closely interrelated and act as “brothers-in-arms,” treatment strategies addressing both host-protective responses may be more successful in restoring a balanced immune response than one intervention alone. We hypothesized that combination therapy of AT or A1PI exerts a beneficial synergistic effect by decreasing pulmonary coagulopathy and inflammation, as well as improving endothelial dysfunction compared to AT or A1PI therapy alone. Therefore, we investigated the effect of combination therapy on pulmonary coagulation and inflammation in two models of acute lung injury. Heparin was added in an extra group receiving combination therapy to assess a potential increased bleeding risk.
Methods
Animals
The experiments were conducted under protocols approved by the Animal Care and Use Committee of the Academic Medical Center at the University of Amsterdam (LEICA132-AB and -AD). Animals were used in compliance with the Institutional Standards for Use of Laboratory Animals and were handled 1 week before experiments to diminish stress activation. They were housed in a specific-pathogen-free facility on a 12/12-h light/dark cycle. Standard laboratory chow and water were available ad libitum. Single-hit lung injury model: Experiments were performed in 44 male specific-pathogen-free BALB/c mice (Charles River, The Netherlands), aged 8–12 weeks and with a mean ± SD body weight of 23.4 ± 1 .2 g. Two-hit lung injury model: Experiments were performed in 45 male specific-pathogen-free RccHan Wistar rats (Envigo, The Netherlands) with a mean ± SD body weight of 322 ± 19 g.
Study groups
Animals were randomized into five different intervention groups: (1) antithrombin (AT; 250 IU/kg; Anbinex™, kind gift from Grifols), (2) alpha-1 protease inhibitor (A1PI; 60 mg/kg; Prolastin™, kind gift from Grifols), (3) AT + A1PI, (4) AT + A1PI + heparin (100 U/kg; LEO Pharma, Lier, Belgium), and (5) sodium chloride 0.9% (saline; Braun, Melsungen, Germany), referred to as the “hit” group (N = 8). Control animals received neither LPS, nor ventilation, nor treatment (N = 4). Administered dosages of intervention drugs were based on previous preclinical research [18,19,20].
Study design
Single-hit lung injury model
At baseline, mice were weighed and labeled. At time point T = 0, mice were sedated using isoflurane 2–4%, followed by intranasal (i.n.) inoculation of 5 mg/kg LPS (Escherichia coli, serotype: 0127:B8, Sigma Aldrich, St. Louis, MO, USA) diluted in 50 μl NaCl 0.9% or 50 μl NaCl 0.9% (control). Appropriate dosages of LPS to achieve an adequate lung injury model were assessed in a prior pilot trial (data not shown). One hour after i.n. inoculation (T = 1), mice received the intervention drugs intraperitoneally (i.p.). Five hours after administration of the intervention (T = 6), mice were anesthetized using a bolus of 9.5 μl KDA per gram body weight i.p. (KDA 1.26 ml 100 mg/ml ketamine (Anesketin, EuroVetAnimal Health B.V., Bladel, The Netherlands) + 0.2 ml 0.5 mg/ml dexmedetomidine (Pfizer Animal Health B.V., Capelle a/d Ijssel, The Netherlands) + 1 ml 0.5 mg/ml atropine (Pharmachemie, Haarlem, The Netherlands) in 5 ml 0.9% NaCl) and sacrificed by exsanguination from the carotid artery (Fig. 1a). Whole blood was drawn using a heparin-coated syringe and subsequently centrifuged for 10 min at 4000 rpm at 4 °C (Eppendorf, microcentrifuge). Plasma was obtained and stored at − 80 °C for further analyses. Following exsanguination, the sternum of the mouse was opened surgically and lungs with bronchi and trachea were resected. To obtain bronchoalveolar lavage fluid (BALF), the right main bronchus was clipped and a 1-ml syringe was connected to the trachea. The left lung was lavaged three times with 0.5 ml normal saline. BALF was centrifuged for 10 min at 2000 rpm at 4 °C (Eppendorf, microcentrifuge); the upper lobe of the right lobe was snap frozen in liquid nitrogen and stored at − 80 °C for further analyses.

Lung injury models. a Single-hit lung injury model. b Two-hit lung injury model. Abbreviations: FiO2, fraction of inspired oxygen; i.n., intranasal; i.p., intraperitoneal; i.v., intravenous; LPS, lipopolysaccharide; T time point in hours; VT, tidal volume
Two-hit lung injury model
Intravenous (i.v.) injection of LPS to mimic sepsis-induced ARDS was regarded as the first hit to the lungs, followed by high tidal volume mechanical ventilation for ventilator-induced lung injury (VILI) as the second pulmonary hit. At baseline (T = 0), rats were sedated using isoflurane 2–4%, followed by an i.v. injection of 10 mg/kg LPS (Escherichia coli, serotype: 0127:B8, Sigma Aldrich, St. Louis, MO, USA) diluted in 1 ml NaCl into the penile vein. Dosage of LPS to achieve an adequate sepsis-induced ARDS model was assessed in a pilot trial (data not shown). Simultaneously, rats received one dosage of 0.03 mg/kg buprenorphine (Temgesic®; Indivior UK Limited, Slough, UK) subcutaneously. Rats were placed back into their cage. Two hours after LPS injection (T = 2), rats were sedated by i.p. injection of 85 mg/kg pentobarbital (Euthasaol® 20%; AST Farma B.V., Oudewater, The Netherlands) and intervention drugs were administered intravenously via the tail vein. Tracheotomy was performed, and a polyethylene catheter was inserted into the carotid artery for hemodynamic monitoring and sampling for blood gas analysis (T = 2, T = 4, and T = 6). Anesthesia was maintained by continuous intravenous infusion of pentobarbital at a rate of 15 mg/kg/h, and fluid resuscitation was performed by infusing Ringer’s lactate (Baxter; Utrecht, The Netherlands) at a rate of 8 ml/kg/h to maintain a mean arterial pressure of above 60 mmHg. In case of hypotension, a maximum of three boluses of 10 ml/kg were administered. Body temperature was maintained between 36.5 and 37.5°C using a heat pad. Rats were subjected to a mechanical ventilation in a volume-controlled mode with tidal volumes between 12 and 15 ml/kg and a positive end-expiratory pressure (PEEP) of 3.4 mbar (Babylog® 8000, Dräger, Germany). The inspired oxygen fraction (Fi02) was set at 30%, the respiratory rate at 60 breaths per minute with an inspiratory-to-expiratory ratio of 1:1.5. Rats were mechanically ventilated for 4 h and sacrificed by exsanguination from the carotid artery after a bolus of pentobarbital (T = 6) (Fig. 1b). Arterial blood was drawn using a heparin-coated syringe and subsequently centrifuged for 15 min at 1500 rcf at 4 °C (Eppendorf, Centrifuge 5804 R). Plasma was obtained and stored at − 80 °C for further analyses. Following exsanguination, the sternum of the rat was opened surgically and lungs with bronchi and trachea were resected. For BALF, the right main bronchus was clipped and the left lung was lavaged three times with 2 ml sterile saline. BALF was centrifuged for 15 min at 1500 rcf at 4 °C and stored at − 80 °C for further analyses. The middle and accessory lobe were snap frozen in nitrogen and stored at − 80 °C until processing to lung homogenate. Plasma, BALF, and lung homogenate samples were analyzed using a coding system; hence, the outcome assessor was blinded for which treatment each animal received.
Coagulation and fibrinolysis
Single-hit lung injury model
Thrombin–antithrombin complex (TATc) levels were determined as a marker for coagulation and assessed in BALF and plasma using a mouse-specific ELISA kit (Abcam, Cambridge, The UK) according to the manufacturer’s instructions, detection range 0.031–8 ng/ml.
Two-hit lung injury model
Markers for coagulation and fibrinolysis, e.g., thrombin–antithrombin complexes (TATc) and plasminogen activator inhibitor (PAI)-1, were analyzed in BALF and plasma using rat-specific ELISA kits (Nordic Biosite AB, Täby, Sweden) according to the manufacturer’s instructions, detection range: TATc 15.625–1000 pg/ml, PAI-1 0.313–20 ng/ml.
Inflammatory response and endothelial injury
Single-hit lung injury model
Inflammatory cytokines and endothelial markers, e.g., tumor necrosis factor (TNF)-α, interleukin (IL)-1β, were assessed in BALF, and TNFα, IL-1β, IL-6, and keratinocyte-derived chemokine (KC) were assessed in plasma using the multi-analyte bead assay Luminex (Bio-Techne, R&D systems, Minneapolis, MN, USA) according to the manufacturer’s instructions. Detection limits for each biomarker are TNFα 1.13, IL-1β 100.55, KC 24.02, and IL-6 18.84. Total protein levels in BALF were assessed using Bradford Protein Assay Kit (OZ Biosciences, Marseille, France) according to official instructions with bovine serum albumin (BSA) as standard and a detection limit of 50 μg/ml.
Two-hit lung injury model
Inflammatory cytokines, e.g., interleukin (IL)-6 and cytokine-induced neutrophil chemoattractant (CINC)-3, were assessed in BALF and plasma using rat-specific ELISA kits (R&D systems; Minneapolis, MN, USA) according to the manufacturer’s instructions. Detection limits for each biomarker are the following: IL-6 125–8000 pg/ml and CINC-3 31.2–2000 pg/ml. Myeloperoxidase (MPO) activity was measured in lung homogenate and plasma using a rat-specific ELISA kit according to the manufacturer’s instructions (Hycult Biotech; Uden, The Netherlands). Total protein levels in BALF were determined by the bovine serum albumin (BSA) Lowry method.
Statistical analyses
A sample size of eight animals per group was needed, using a power of 0.8 with a chance of 0.904 (effect size 1.85) with double signification level of 0.05 to detect a difference in IL-6 levels between intervention groups and the control group. All data are expressed as median (interquartile range (IQR)) according to data distribution. The saline control and healthy control groups were compared using the Mann–Whitney U test. Comparisons between the intervention groups and the saline-treated control group were performed using Kruskal–Wallis test followed by uncorrected Dunn’s test for multiple comparisons (if p < 0.05). p < 0.05 is considered statistically significant. Statistical analyses were performed with SPSS (IBM SPSS Statistics, version 24.0, Chicago, IL, USA), and scatter plots were created using GraphPad Prism version 7.0a (GraphPad Software; San Diego, CA).
Results
Lung injury was induced in both models evidenced by increased levels of protein in the BALF indicative of acute lung injury as well as coagulation and inflammatory parameters in the hit group compared to the control group (Figs. 2, 3, 4, 5, and 6). Notably, the two-hit lung injury model elicited a far more vigorous pulmonary coagulation and inflammatory response than the single-hit model.

Marker for acute lung injury. Single-hit lung injury model: a protein count in BALF. Two-hit lung injury model: b protein count in BALF. Abbreviations: BALF, bronchoalveolar lavage fluid; AT, antithrombin; A1PI, alpha-1 protease inhibitor; Hep, heparin

Markers of pulmonary coagulation. Single-hit lung injury model: a TATc levels in BALF. Two-hit lung injury model: b TATc levels in BALF. c PAI-1 levels in BALF. Abbreviations: BALF, bronchoalveolar lavage fluid; AT, antithrombin; A1PI, alpha-1 protease inhibitor; Hep, heparin; TATc, thrombin–antithrombin complexes; PAI-1, plasminogen activator inhibitor 1

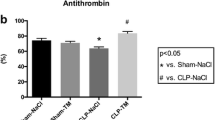
Markers of systemic coagulation. Single-hit lung injury model: a TATc levels in plasma. Two-hit lung injury model: b TATc levels in plasma. c PAI-1 levels in plasma. *p < 0.05. Abbreviations: AT, antithrombin; A1PI, alpha-1 protease inhibitor; Hep, heparin; TATc, thrombin–antithrombin complexes; PAI-1, plasminogen activator inhibitor 1

Markers of pulmonary inflammation. Single-hit lung injury model: a TNFα levels in BALF. b IL-1β levels in BALF. Two-hit lung injury model: c IL-6 levels in BALF. d CINC-3 levels in BALF. e MPO levels in lung homogenate. Abbreviations: AT, antithrombin; A1PI, alpha-1 protease inhibitor; BALF, bronchoalveolar lavage fluid; CINC, cytokine-induced neutrophil chemoattractant; Hep, heparin; IL, interleukin; MPO, myeloperoxidase; TNF, tumor necrosis factor

Markers of systemic inflammation. Single-hit lung injury model: a IL-6 levels in plasma. b KC levels in plasma. c TNFα levels in plasma. Two-hit lung injury model: d IL-6 levels in plasma. e CINC-3 levels in plasma. f MPO levels in plasma. Abbreviations: AT, antithrombin; A1PI, alpha-1 protease inhibitor; CINC, cytokine-induced neutrophil chemoattractant; Hep, heparin; IL, interleukin; KC, keratinocyte chemoattractant; MPO, myeloperoxidase; TNF, tumor necrosis factor
No abnormal macroscopic bleeding was observed in any of the animals. Intraperitoneal administration of AT and A1PI resulted in increased levels of the agents within the pulmonary compartment (data not shown). In the single-hit lung injury model, all animals survived the experiment. In the two-hit lung injury model, all animals survived i.v. injection of LPS and the following 2 h without medical intervention. The two-hit lung injury model yielded a mortality of 2.5% (1 out of 40). It concerned one animal from the A1PI group with the terminal endpoint being prolonged hypotension (MAP < 55 mmHg) unresponsive to fluid resuscitation followed by severe lactate acidosis after 3 h of mechanical ventilation. The experiment was repeated to achieve a similar number of animals per group.
Coagulation and fibrinolysis
Both lung injury models induced pulmonary coagulopathy as shown by elevated TATc levels in BALF (p < 0.05 hit versus control). Fibrinolysis, as depicted by PAI-1 levels, was not affected within the pulmonary compartment in the two-hit lung injury model (p = 0.55 hit versus control). None of the interventions exerted a significant effect on pulmonary coagulopathy in the models studied (Fig. 3).
Systemic coagulation was not enhanced in the single-hit lung injury model. In the two-hit lung injury model, systemic hypercoagulability and reduced fibrinolysis were present in all animals as proven by increased plasma levels of TATc and PAI-1 (p < 0.01 hit versus control, for both). Administration of AT significantly reduced systemic TATc levels (p < 0.05 hit versus AT) (Fig. 4).
Inflammation and endothelial injury
In the single-hit lung injury model, i.n. LPS inoculation resulted in pulmonary inflammation shown by a marked increase of acute inflammatory markers TNFα and IL-1β in BALF (p < 0.01 hit versus control). Likewise, the two-hit lung injury model led to an acute inflammatory response within the lungs as shown by markedly elevated levels of IL-6, CINC-3, and MPO (p < 0.01 hit versus control, for all). None of the interventions affected the pulmonary inflammatory response (Fig. 5).
Both lung injury models also resulted in systemic inflammation as shown by markedly increased IL-6 and KC levels in plasma (p < 0.01 versus control) in the single-hit lung injury model and elevated levels of IL-6, CINC-3, and MPO in plasma in the two-hit lung injury model (p < 0.01 hit versus control, for all). The interventions did not affect systemic inflammation (Fig. 6).
Physiological parameters in the two-hit lung injury model
All animals showed a decrease in mean arterial pressure throughout the 4 h of mechanical ventilation, and 24% (11 out of 46) animals required additional fluid resuscitation. Those animals were evenly distributed between all groups, with only the AT group not having animals requiring fluid resuscitation. All animals showed a respiratory alkalosis mainly due to high tidal volume MV. Lactate levels were elevated in all animals confirming a model of septic shock. Inspiratory pressures ranged between 16 and 23 cmH2O, and a FiO2 of 30% was sufficient in all animals (Table 1).
Discussion
The main results of this study are that neither single therapy with AT or A1PI nor combination therapy of both agents attenuates pulmonary coagulopathy or inflammation in two different murine models of acute lung injury.
Combination therapy of AT and A1PI does not exert a synergistic beneficial effect on pulmonary coagulation, nor on pulmonary inflammation. The only previous study investigating the combined effect of AT and A1PI showed that combination therapy prevents pathophysiological changes within the lungs [12]. The rationale of a synergistic effect of combination therapy comprises of two parts: AT is thought to reduce binding of neutrophils to the endothelium [21] leading to a decrease in neutrophil activation [22]. This in turn should result in lower levels of reactive oxygen species (ROS) from neutrophilic granules [23], known to inactivate A1PI [24, 25]. Both protease inhibitors were assumed to protect each other’s functionality and to exert a synergistic effect in their anticoagulant and anti-inflammatory capacity. Nonetheless, this present study does not support these assumptions.
Interestingly, this present experiment did not find a beneficial effect of AT on pulmonary coagulopathy and thereby contradicts previous studies administering the same dosage of AT [18, 19]. Differences in the applied animal models may attribute to the different outcomes as discussed below. This study confirms previous research [18, 19] that AT does not affect pulmonary or systemic inflammatory parameters in in vivo models [26].
In the Kybersept trial [27], an interaction between prophylactic heparin and AT was found. Therefore, we also studied the effect of adding heparin to the combination therapy. Co-administration of heparin did not result in an increased bleeding risk.
The current study assessed the effect of A1PI on pulmonary coagulation and inflammation for the first time and did not find an effect of A1PI on coagulopathy, nor on inflammation. An explanation may be the time frame of the experiment. A1PI is an acute-phase protein synthesized by the liver and seems to play a dual role within the inflammatory response [7]. A preclinical study sacrificing animals 2 h after A1PI administration found increased markers of inflammation [28], whereas most studies sacrificing animals at a later time point detected decreased inflammatory cytokines [20, 29, 30]. Hence, one may hypothesize that A1PI does exert its anti-inflammatory effects after the acute phase of lung injury but not in the early phase, as in our model.
This animal study has several strong points. First of all, this study investigated the effect of the interventions in two clinically relevant and validated models mimicking ARDS [31,32,33,34]. Secondly, rigorous methods such as randomization of animals, blinding of groups before analysis of outcomes, and a power calculation were used. Importantly, all interventions were administered as a treatment. Many previous preclinical studies chose to give AT or A1PI as a pre-treatment, hence before induction of lung injury—which is clinically less relevant [29, 30, 35, 36]. With regard to drug dosage, a dosage of 250 IU/kg has been proven to attenuate enhanced coagulation, but did not have a beneficial effect on inflammation in rat models [18, 19]. With regard to A1PI, the dosage used proved to be effective in previous murine models [20, 29]. It may be hypothesized that different etiologies of lung injury require different dosages and that higher dosages of AT or A1PI may exert a more pronounced effect.
One strength of this present study is actually also one of the major limitations. It is important to realize that both models are simulating lung injury within the acute phase, e.g., within the first 6 h after the induction of lung injury. Comparable studies are scarce, and most studies using lung injury models sacrificed animals after 16 up to 75 h focusing on a beneficial effect of therapy at a later time point [29, 30]. Hence, this study gives insight in the effects of drugs studied within the acute phase of lung injury, but does not allow to draw any conclusions about the effects at a later time point of lung injury.
In conclusion, in this present study, neither single therapy with AT or A1PI nor combination therapy exerts beneficial effects on pulmonary coagulation or inflammation in two murine acute lung injury models. AT markedly decreased systemic coagulopathy. Adding heparin did not result in aggravation of the anticoagulant effect leading to abnormal macroscopic bleeding. Further research is warranted to gain insights about the effects of AT and A1PI on coagulation and inflammation after the acute phase of lung injury.
Abbreviations
- A1PI:
-
Alpha-1 protease inhibitor
- ARDS:
-
Acute respiratory distress syndrome
- AT:
-
Antithrombin
- BALF:
-
Bronchoalveolar lavage fluid
- FiO2 :
-
Inspired oxygen fraction
- i.n.:
-
Intranasal
- i.p.:
-
Intraperitoneal
- i.v.:
-
Intravenous
- IL:
-
Interleukin
- KDA:
-
Ketamine, dexmedetomidine, atropine
- LPS:
-
Lipopolysaccharide
- MPO:
-
Myeloperoxidase
- NaCl:
-
Sodium chloride
- PEEP:
-
Positive end-expiratory pressure
- SD:
-
Standard deviation
- TNF:
-
Tumor necrosis factor
- VILI:
-
Ventilator-induced lung injury
References
Matthay MA, Zemans RL (2011) The acute respiratory distress syndrome: pathogenesis and treatment. Annu Rev Pathol 6:147–163
Tuinman PR, Dixon B, Levi M, Juffermans NP, Schultz MJ (2012) Nebulized anticoagulants for acute lung injury - a systematic review of preclinical and clinical investigations. Crit Care 16(2):R70
Afshari A, Wetterslev J, Brok J, Moller AM (2008) Antithrombin III for critically ill patients. Cochrane Database Syst Rev 3 CD005370
Ware LB, Fang X, Matthay MA (2003) Protein C and thrombomodulin in human acute lung injury. Am J Physiol Lung Cell Mol Physiol 285(3):L514–L521
Ware LB, Matthay MA, Parsons PE, Thompson BT, Januzzi JL, Eisner MD et al (2007) Pathogenetic and prognostic significance of altered coagulation and fibrinolysis in acute lung injury/acute respiratory distress syndrome. Crit Care Med 35(8):1821–1828
Chambers RC, Scotton CJ (2012) Coagulation cascade proteinases in lung injury and fibrosis. Proc Am Thorac Soc 9(3):96–101
Ehlers MR (2014) Immune-modulating effects of alpha-1 antitrypsin. Biol Chem 395(10):1187–1193
Janciauskiene SM, Bals R, Koczulla R, Vogelmeier C, Kohnlein T, Welte T (2011) The discovery of alpha1-antitrypsin and its role in health and disease. Respir Med 105(8):1129–1139
KA B. Antithrombin deficiency UpToDate2016 [updated April 28, 2016. Available from: www.uptodate.com/contents/antithrombin-deficiency?source=search_result&search=antithrombin&selectedTitle=1~150
Kaneider NC, Forster E, Mosheimer B, Sturn DH, Wiedermann CJ (2003) Syndecan-4-dependent signaling in the inhibition of endotoxin-induced endothelial adherence of neutrophils by antithrombin. Thromb Haemost 90(6):1150–1157
Oelschlager C, Romisch J, Staubitz A, Stauss H, Leithauser B, Tillmanns H et al (2002) Antithrombin III inhibits nuclear factor kappaB activation in human monocytes and vascular endothelial cells. Blood 99(11):4015–4020
Redens TB, Leach WJ, Bogdanoff DA, Emerson TE, Jr. Synergistic protection from lung damage by combining antithrombin-III and alpha 1-proteinase inhibitor in the E. coli endotoxemic sheep pulmonary dysfunction model. Circ Shock 1988;26(1):15–26
Cornet AD, Groeneveld AB, Hofstra JJ, Vlaar AP, Tuinman PR, van Lingen A et al (2014) Recombinant human activated protein C in the treatment of acute respiratory distress syndrome: a randomized clinical trial. PLoS One 9(3):e90983
Cornet AD, Hofstra JJ, Vlaar AP, Tuinman PR, Levi M, Girbes AR et al (2013) Activated protein C attenuates pulmonary coagulopathy in patients with acute respiratory distress syndrome. J Thromb Haemost 11(5):894–901
Juschten J, Tuinman PR, Juffermans NP, Dixon B, Levi M, Schultz MJ (2017) Nebulized anticoagulants in lung injury in critically ill patients-an updated systematic review of preclinical and clinical studies. Ann Transl Med 5(22):444
Schultz MJ, Haitsma JJ, Zhang H, Slutsky AS (2006) Pulmonary coagulopathy as a new target in therapeutic studies of acute lung injury or pneumonia--a review. Crit Care Med 34(3):871–877
Bernard GR, Vincent JL, Laterre PF, LaRosa SP, Dhainaut JF, Lopez-Rodriguez A et al (2001) Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med 344(10):699–709
Aslami H, Haitsma JJ, Hofstra JJ, Florquin S, Dos Santos C, Streutker C et al (2012) Plasma-derived human antithrombin attenuates ventilator-induced coagulopathy but not inflammation in a Streptococcus pneumoniae pneumonia model in rats. J Thromb Haemost 10(3):399–410
Choi G, Vlaar AP, Schouten M, Van't Veer C, van der Poll T, Levi M et al (2007) Natural anticoagulants limit lipopolysaccharide-induced pulmonary coagulation but not inflammation. Eur Respir J 30(3):423–428
Gao W, Zhao J, Kim H, Xu S, Chen M, Bai X et al (2014) alpha1-antitrypsin inhibits ischemia reperfusion-induced lung injury by reducing inflammatory response and cell death. J Heart Lung Transplant 33(3):309–315
Kaneider NC, Reinisch CM, Dunzendorfer S, Romisch J, Wiedermann CJ (2002) Syndecan-4 mediates antithrombin-induced chemotaxis of human peripheral blood lymphocytes and monocytes. J Cell Sci 115(Pt 1):227–236
Uchiba M, Okajima K, Murakami K, Okabe H, Takatsuki K (1996) Attenuation of endotoxin-induced pulmonary vascular injury by antithrombin III. Am J Phys 270(6 Pt 1):L921–L930
Grommes J, Soehnlein O (2011) Contribution of neutrophils to acute lung injury. Mol Med 17(3–4):293–307
Cochrane CG (1988) Alpha-1-proteinase inhibitor in inflammatory states of humans and laboratory animals. Am J Med 84(6A):75–79
Wallaert B, Aerts C, Gressier B, Gosset P, Voisin C (1993) Oxidative inactivation of alpha 1-proteinase inhibitor by alveolar epithelial type II cells. J Appl Physiol 75(6):2376–2382
Choi G, Hofstra JJ, Roelofs JJ, Rijneveld AW, Bresser P, van der Zee JS et al (2008) Antithrombin inhibits bronchoalveolar activation of coagulation and limits lung injury during Streptococcus pneumoniae pneumonia in rats. Crit Care Med 36(1):204–210
Warren BL, Eid A, Singer P, Pillay SS, Carl P, Novak I et al (2001) Caring for the critically ill patient. High-dose antithrombin III in severe sepsis: a randomized controlled trial. Jama 286(15):1869–1878
Subramaniyam D, Steele C, Kohnlein T, Welte T, Grip O, Matalon S et al (2010) Effects of alpha 1-antitrypsin on endotoxin-induced lung inflammation in vivo. Inflamm Res 59(7):571–578
Jonigk D, Al-Omari M, Maegel L, Muller M, Izykowski N, Hong J et al (2013) Anti-inflammatory and immunomodulatory properties of alpha1-antitrypsin without inhibition of elastase. Proc Natl Acad Sci U S A 110(37):15007–15012
Pott GB, Beard KS, Bryan CL, Merrick DT, Shapiro L (2013) Alpha-1 antitrypsin reduces severity of pseudomonas pneumonia in mice and inhibits epithelial barrier disruption and pseudomonas invasion of respiratory epithelial cells. Front Public Health 1:19
Matute-Bello G, Frevert CW, Martin TR (2008) Animal models of acute lung injury. Am J Physiol Lung Cell Mol Physiol. 295(3):L379–L399
Rojas M, Woods CR, Mora AL, Xu J, Brigham KL (2005) Endotoxin-induced lung injury in mice: structural, functional, and biochemical responses. Am J Physiol Lung Cell Mol Physiol. 288(2):L333–L341
Tuinman PR, Muller MC, Jongsma G, Hegeman MA, Juffermans NP (2013) High-dose acetylsalicylic acid is superior to low-dose as well as to clopidogrel in preventing lipopolysaccharide-induced lung injury in mice. Shock 40(4):334–338
Yang CL, Chen CH, Tsai PS, Wang TY, Huang CJ (2011) Protective effects of dexmedetomidine-ketamine combination against ventilator-induced lung injury in endotoxemia rats. J Surg Res 167(2):e273–e281
Hofstra JJ, Vlaar AP, Cornet AD, Dixon B, Roelofs JJ, Choi G et al (2010) Nebulized anticoagulants limit pulmonary coagulopathy, but not inflammation, in a model of experimental lung injury. J Aerosol Med Pulm Drug Deliv 23(2):105–111
Uchiba M, Okajima K (1997) Antithrombin III (AT III) prevents LPS-induced pulmonary vascular injury: novel biological activity of AT III. Semin Thromb Hemost 25(6):583–590
Acknowledgements
We would like to thank Grifols for their support in all stages of the study, and especially Bettina Dreger for expertise. Special gratitude we would like to express to Dr. Nina Hauck-Weber, Anita Tuip-de Boer, Rafaela Kerindongo and Jacoline Buchner for their help and expertise on various techniques in the laboratory.
Funding
This study has been supported by an investigator led research grant from Grifols with regards to finances and tested treatment agents, as described above. This article did not receive sponsorship for publication.
Availability of data and materials
Please contact author for data requests.
About this supplement
This article has been published as part of Intensive Care Medicine Experimental Volume 7 Supplement 1 2019: Proceedings from the Third International Symposium on Acute Pulmonary Injury and Translational Research (INSPIRES III). The full contents of the supplement are available at https://icm-experimental.springeropen.com/articles/supplements/volume-7-supplement-1.
Author information
Authors and Affiliations
Contributions
JJ, NPJ, ARJG, MJS, and PRT developed the study design. JJ, SAI, and MAW conducted the experiments, run the assays, and collected the data. JJ, SAI, ARJG, NPJ, MJS, and PRT analyzed and interpreted the data. JJ, NPJ, MJS, ARJG, and PRT wrote the manuscript. All authors reviewed the manuscript and approved the final submitted version.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The experiments were conducted under protocols approved by the Animal Care and Use Committee of the Academic Medical Center at the University of Amsterdam (LEICA132-AB and -AD).
Consent for publication
Not applicable.
Competing interests
This study has been supported by an investigator-led research grant from Grifols. This funding facilitated conductance of this study, but did not influence the study design, collection of the data, analysis, nor interpretation or writing of the article. We declare that the tested treatment agents AT and A1PI were a kind gift from Grifols.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Juschten, J., Ingelse, S.A., Maas, M.A.W. et al. Antithrombin plus alpha-1 protease inhibitor does not affect coagulation and inflammation in two murine models of acute lung injury. ICMx 7 (Suppl 1), 36 (2019). https://doi.org/10.1186/s40635-019-0240-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40635-019-0240-7