
Abstract
Natural organic matter (NOM) plays important roles in biological, chemical, and physical processes within the terrestrial and aquatic ecosystem. Despite its importance, a clear and exhaustive knowledge on NOM chemistry still lacks. Aiming to prove that advanced solid-state 13C nuclear magnetic resonance (NMR) techniques may contribute to fill such a gap, in this paper we reported relevant examples of its applicability to NOM components, such as biomass, deposition material, sediments, and kerogen samples. It is found that nonhydrolyzable organic carbons (NHC), chars, and polymethylene carbons are important in the investigated samples. The structure of each of the NHC fractions is similar to that of kerogens, highlighting the importance of selective preservation of NOM to the kerogen origin in the investigated aquatic ecosystems. Moreover, during the artificial maturation experiments of kerogen, the chemical and structural characteristics such as protonated aromatic, nonprotonated carbons, and aromatic cluster size play important roles in the origin and variation of nanoporosity during kerogen maturation.

NMR parameters of thermally stimulated kerogens
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Background
Terrigenous NOM generally includes a large fraction of biologically and chemically resistant (recalcitrant) compounds such as nonhydrolyzable organic matter, kerogen, and residual NOM, resulting from an enhanced degradation carried out by soil microbes [2, 19]. Despite its intrinsic recalcitrance, terrigenous NOM can also be recycled in the terrestrial environment. Ancient terrigenous NOM fractions also comprise kerogen or old soil NOM and humic substances, albeit to a lesser extent (<25%; [14]). Terrigenous NOM supplied by rivers undergoes intensive reworking in estuaries and deltas [9], with some estimates suggesting losses of two-thirds of the global fluvial input of particulate NOM (POM) in these environments [21]. Part of terrigenous NOM may be lost if leached (conversion to dissolved NOM), completely mineralized, exported, or buried. However, the controlling mechanisms for these processes are highly variable and are not yet well known [49].
Biomass residues (such as pollens and algae) are transformed via chemical, biological, and physical processes into more stable forms during humification processes. During humification, the number of aromatic and paraffinic carbons increases, whereas the level of alkyl C–O carbons decreases. Therefore, humification and degradation processes result in the loss of some of the characteristic signals of structurally identifiable materials such as carbohydrates and lead to the enhancement of signals from refractory structures, such as aromatic and alkyl compounds. It is well established that refractory biomacromolecules (such as lignin and sporopollenin biopolymers) are a recalcitrant and important constituent of NOM [3, 11].
Atmospheric particulate matter contains organic matter and inorganic materials and its sources can be either anthropogenic or natural. Its composition is closely related to issues such as direct and indirect climate modifications, air pollution, and reduced visibility. In the literature, most of the studies on the characterization of organic matter in atmospheric particulate matter have been conducted on the water soluble fraction using 1H NMR [5]. Note that even 2D 1H-13C HSQC and HMQC can only detect protonated carbons. Thus, these studies completely lose the nonprotonated structural information. In addition, the characterization of insoluble fractions is also examined. To some extent, solid-state NMR is essential for providing relatively complete structural information on organic matter in atmospheric organic matter. To the best of our knowledge, there are only a number of cases using solid-state NMR to study its organic matter. In almost all these studies, simple 13C cross polarization/magic angle spinning (CP/MAS) and spectral editing techniques have been used. The high-spinning speed multi-ramped amplitude cross polarization/magic angle spinning (multiCP/MAS) technique was applied for acquiring quantitative 13C NMR spectra [20]. This multiple-cross polarization (multiCP) technique provides a simple, robust way to obtain quantitative solid-state 13C NMR spectra of organic materials, with good signal-to-noise ratio. The spectral editing techniques (i.e., dipolar dephasing and a 13C chemical-shift-anisotropy filter) are employed to investigate the subspectra of the chemical groups. Therefore, the advanced solid-state techniques to the characterization are yet to be applied.
Various physical and chemical methods have been used, whereas advanced solid-state NMR techniques have not been applied yet, despite they are potentially capable to significantly improve the knowledge on black carbon (BC) and kerogen carbon (KC). Methods for BC quantification in soils and sediments have been extensively evaluated [13, 16, 32, 35, 42]. By contrast, less efforts have been directed at the quantification of KC in soils and sediments, and the current protocols are mainly restricted to the fields of organic–petrographic analyses and chemical treatments [6, 12, 22, 39, 40, 46]. Kerogen is the most abundant organic matter in the Earth’s crust [48]. Major input of kerogen to soils and sediments is from the natural weathering processes of sedimentary rocks and from coal since the latter, which is mainly composed of kerogen, is dispersed during mining operations. Kerogen typically has a three-dimensional structure with aromatic nuclei cross-linked by aliphatic chain-like bridges. These nuclei consist of aromatic sheets separated by voids with an average width of 30–40 nm. The aliphatic bridges contain linear or branched chains, and/or S- or O-containing functional groups [10]. Although most kerogens are distributed in deeply buried reservoirs [48], they can be present in significant amounts in soils and sediments. For example, (Ran et al. [40] found that Borden aquifer with a total OC content of 0.21 g kg−1 consisted of at least 186 g kg−1 of KC. Song et al. [46] reported 240–480 g kg−1 of KC out of total OC in one soil and three sediments. However, all existing KC quantification methods may suffer from operational shortcomings such as fine NOM particle loss, and incomplete amorphous organic carbon removal [13, 40, 46].
Kerogen is the sedimentary organic matter, which generates petroleum and natural gas. In this respect, many efforts have been devoted to its isolation and to the characterization of its chemical structure and evolution during sediment burial, aiming at a better prediction of oil and gas pools. With kerogen being a complex mixture of organic materials tightly mixed with minerals in sediments, its characterization represents a challenging goal. This notwithstanding, many progresses have been so far done and are due not only to the development of oil exploration via geological sample availability, but also to the huge increase in analytic capabilities during the last 40 years. However, despite the numerous advances made with respect to kerogen knowledge, important points are still far from being elucidated.
Solid-state NMR spectroscopy is commonly used to characterize complex humic substances [7, 18, 33, 38] including humic and fulvic acids [1, 17, 44, 47] and humin Preston and Newman [36, 37]. The primary technique used in these studies is the solid-state 13C CP-MAS technique. Our understanding of chemical structures of humic substances has markedly improved over decades of extensive investigations using the solid-state 13C CP-MAS spectroscopy [47, 51]. For example, it is well documented that marine humic acids generally contain less aromatics than lignin-derived terrigenous ones [27, 33]. Despite the knowledge gained by these NMR studies using 13C CP-MAS, the full potential of solid-state NMR for characterizing humic substances has yet to be realized [33]. 13C CP-MAS has drawbacks such as loss of structural information and poor quantification [8, 23, 30, 41, 45]. Moreover, due to their complex and heterogeneous nature, 13C CP-MAS or direct polarization magic angle spinning (DP-MAS) NMR spectra of NOM generally reveal broad, poorly resolved peaks pertaining to major functional groups. Therefore, ambiguous assignments can be caused by the overlapping chemical shifts of numerous different structural moieties [33]. Thus, other analytic methods have also been used in sediment NOM characterization as reviewed in the previous review [19, 33].
During the recent years, many advanced solid-state 13C NMR techniques have been developed, improved, and applied for characterizing complexity of NOM in algaes, plants, soils, sediments, and kerogens [23, 43]; Mao and Schmidt-Rohr [24–26], Mao et al. [27–29]. These advanced solid-state NMR techniques include 13C DP/MAS NMR, quantitative DP/MAS plus dipolar dephasing, 13C chemical-shift anisotropy filter (CSA), and CH, CH2 spectral editing [28]. They overcome the drawbacks of conventional CP/MAS techniques and enable us to obtain more detailed chemical and structural information on various types of NOM. The identification and quantification of specific functional groups such as CH2, CH3, aromatic CH, aromatic C–O, protonated aromatic C, nonprotonated aromatic C, carbonyl C, and carboxyl C can be achieved. The structural parameters such as aromaticity and aromatic cluster size can also be estimated by using spectral analysis and long-range 1H-13C recoupled dipolar dephasing [4, 28]. These advanced techniques provide reliable measurements and have proven to be powerful tools for the characterization of NOM [28]. In the following sections, we will discuss several case studies (algae organic matter, pollen organic matter, sediment organic matter, atmospheric deposition particles, and kerogens) to prove the applicability of advanced 1D solid-state in this research field. Moreover, solid-state NMR experiments were performed on a Bruker spectrometer operating at 400-MHz 1H and 100-MHz 13C frequencies in all reported cases.
Algal organic matter
The three algae (Spirulina, Seaweed, and Porphyra) samples were fractionated into the lipid (LP), lipid free (LF), alkaline nonhydrolyzable carbon (ANHC), and acid nonhydrolyzable carbon (NHC) to obtain the refractory component. The major steps for isolation of the algal fractions were described elsewhere [54]. Briefly, the bulk samples (OS) were extracted to separate lipids by using Soxhlet extraction with 2:1 CH2Cl2/CH3OH (v/v) for 24 h. The LP fractions were dried at 100 °C. Subsamples of the LF fractions were saponified for 1 h in 1 N KOH in 85:15 methanol/H2O (v/v). The ANHC fractions were hydrolyzed twice with 2 N trifluoroacetic acid (TFA) at 100 °C for 3 h. Subsequently, samples were hydrolyzed in 4 and 6 N TFA at 100 °C for 18 h. Finally, the residual hydrolyzable organic matter was removed with 6 M HCl at 110 °C for 24 h.
The multiCP/MAS spectra were measured at a spinning speed of 14 kHz, where spinning sidebands are fairly small (<3%) and have little overlap with center bands. The 90° pulse lengths were 4.3 μs for 1H and 4 μs for 13C. To achieve dead time-free detection, which is indispensable for spectra with broad lines, all spectra were recorded with a Hahn echo generated by an exorcycled 180° pulse applied one rotation period (t r) after the end of cross polarization. The 1H decoupling field strength was |γB 1|/2π = 65 kHz during the period of 2 t r = 0.14 ms duration before the Hahn echo, and ca. 55 kHz during signal detection. The ramp for CP was implemented with 11 steps of 0.1 ms duration and a 1% amplitude increment (90–100%). The duration of the repolarization period (t z) in multiCP was 0.7 s. Corresponding multiCP spectra of nonprotonated C and mobile segments were selected by recoupled dipolar dephasing (multiCP/DD) with a dipolar dephasing time of 68 µs. The 13C spectra of three commercial algae and their LF and NHC fractions are shown in Fig. 1. Thin lines and thick lines represent quantitative 13C NMR spectra of all C (multiCP/MAS) and of nonprotonated or mobile C (multiCP/MAS with recoupled dipolar dephasing), respectively. The relative percentages of different C functional groups are listed in Table 1 [55]. In general, the three bulk commercial algae samples contain high alkyl C–O (31.7–43.1% of all C) and alkyl C (17.7–24.7%), but low aromatic C (5.91–7.89%). After organic solvent extraction, the (CH2)n percentages decrease in the LF fractions relative to the corresponding bulk samples for Spirulina and Seaweed (Table 1). But for Porphyra, the (CH2)n signal varies slightly and is quite similar to that of the OS, likely due to the lower content of lipid in Porphyra. The NHC fractions of three commercial algae generally contain much lower alkyl C–O (8.72–12.6% of all C) than their corresponding OS (31.7–43.1%) and LF samples (28.9–43.4%), indicating that the majority of polysaccharides and carbohydrates were removed during alkaline and acid hydrolysis. In addition, the aromatic C including aromatic C–C, aromatic C–H and aromatic C–O, and ketone/aldehyde C are much more abundant in the NHC fractions than in corresponding OS and LF samples. The signal at 27.5–31.8 ppm is the most intense in the NHC fraction of Porphyra, suggesting that its NHC fraction contains the highest polymethylene carbon among the three commercial algae.

MultiCP 13C spectra (thin lines) and multiCP 13C spectra after recoupled dipolar dephasing (thick lines, 2 s recycle delay) of the bulk algal samples (OS) and related fractions (LF and NHC)
Pollen organic matter
The two pollens (Nelumbo nucifera and Brassica campestris L.) were sequentially fractionated into the lipid (LP), lipid free (LF), residue obtained after successive TFA hydrolysis (TFAR), residue obtained after saponification (SR), and acid nonhydrolyzable carbon (NHC) to obtain the refractory component, as described elsewhere [53]. In brief, the lyophilized OS samples were extracted to separate lipids via Soxhlet extraction with CH2Cl2:CH3OH (2:1, v/v) for 24 h. The LP fractions were dried at 60 °C. Subsamples of the LF fractions were hydrolyzed twice with 2 N TFA at 100 °C for 3 h. Subsequently, samples were hydrolyzed in 4 and 6 N TFA at 100 °C for 18 h. The residue obtained after successive TFA hydrolysis (TFAR) fractions was saponified by refluxing for 1 h in 5% KOH in 2-methoxy ethanol/H2O (88/12, v/v). The residue obtained after saponification (SR) fractions was treated by 6 N HCl at 110 °C for 24 h. The remaining residues made up the final NHC fraction. The cross polarization (CP) experiments included 13C cross polarization/total sideband suppression (CP/TOSS), CP/TOSS plus dipolar dephasing (CP/TOSS/DD), and 13C chemical-shift-anisotropy (CSA) filter. The 13C solid NMR spectra of the investigated bulk pollens and their fraction samples represent 13C CP/TOSS NMR spectra of all C (Additional file 1: Figure S1). A pair of spectral editing techniques, dipolar dephasing, and 13C CSA were employed to study the structures of the bulk pollens and their fractions in more detail [53].
The bulk lotus and rape pollen samples contain the largest amounts of alkyl C–O (53.9 and 47.7%, respectively) (Additional file 1: Table S1). The relative proportions of alkyl C–O for TFAR, SR, and NHC fractions (11.6–19.1%) decrease greatly compared to those for LF fractions (44.7–52.4%). After successive TFA and 6 N HCl treatments, alkyl C increases to a greater extent than does aromatic C. The TFAR, SR, and NHC fractions display the highest relative amount of alkyl carbon (44.1–53.2%), which includes polymethylene-based functionalities. The NHC fractions mainly consist of alkyl carbon (44.1–53.2%), aromatic moieties (110–165 ppm) (20.5–25.8%), alkyl C–O (11.6–12.6%), and O–CH3/NCH (8.82–10.4%). The NHC fractions have extremely high poly(methylene) carbon percent, up to 21.0%. NMR spectroscopy indicates that the structure of the NHC fractions in pollen samples is similar to that of sporopollenin, which principally contains aliphatic, aromatic, ether, and carbonyl and carboxylic groups in varying degrees [1, 15, 31, 50].
Based on the 13C NMR spectra of the classes reported by Nelson and Baldock [34], the constituents of the bulk pollen samples and their fractions are largely aliphatic components (including sporopollenin), carbohydrates, protein, and lignin as estimated by the four listed biochemical classes (Additional file 1: Table S2). Carbohydrate and aliphatic components account for 63.3–68.0 and 12.5–15.4% of the total organic carbon in the bulk pollen samples, respectively, followed by protein and lignin ranging from 9.34 to 14.9% and from 5.83 to 8.67% of the OC content, respectively. The aliphatic components of LF fractions range from 10.8 to 11.7%, which are lower than in their OS samples. In NHC fractions, alkyl C and aromatic C (especially aromatic C–C) were more visible because of the removal of carbohydrates. The NHC fractions are composed of aliphatic components (52.1–58.2%), lignin (20.5–25.9%), carbohydrate (11.5–13.4%), and protein (7.96–10.4%), consistent with the previous studies that sporopollenin is not a uniform macromolecule but rather a series of closely related biopolymers with different chemical groups occurring in varying amounts [15, 50].
Sediment organic matter
Three surface sediment samples (B1, A01, and A04) were sequentially fractionated into the demineralized (DM), lipid (LP), lipid free (LF), and acid nonhydrolyzable carbon (NHC) to obtain the refractory component, as described elsewhere [52]. Briefly, to remove paramagnetic material and enrich the relative OC content in samples, the bulk samples (OS) were tested with 1 N HCl/10% HF to obtain the DM fraction. The free lipids (LP) were extracted repeatedly from an aliquot of DM with a 2:1 CH2Cl2/CH3OH (v/v) using soxhlet extraction for 24 h, and then dried at 50 °C. The solid residue (lipid-free fraction, LF) was dried at 50 °C. The LF fractions were hydrolyzed twice with 2 N TFA at 100 °C for 3 h. Subsequently, samples were hydrolyzed in 4 and 6 N TFA at 100 °C for 18 h. Finally, the residual hydrolyzable organic matter was removed with 6 M HCl at 110 °C for 24 h.
The 13C solid NMR spectra in the demineralized carbon (DM), LF, and NHC fractions for the three sediments collected from the Pearl River Delta represent 13C CP/TOSS NMR spectra of all C. In addition, the spectra resulting from two spectral editing, dipolar dephasing, and CSA are shown in Additional file 1: Figure S2, and described elsewhere [52]. The 13C CP/TOSS data show that the alkyl carbon components account for 25.6–32.4% of the total organic carbon in the DM fractions (Table 2). The LP fractions mainly consist of alkyl carbon. As a result, alkyl carbon contents of the LF fractions decrease to 22.0–27.3% relative to the DM fractions. The LF fractions have higher aromatic C contents (38.8–39.2%) than DM fractions (32.8–35.9%) do.
Based on the 13C NMR spectra, the contribution of four biomacromolecule classes (lignin, protein, carbohydrate, and aliphatic component) in sediments and their isolated fractions is estimated according to Nelson and Baldock (Table 2). As the sediments were reported to contain char or black carbon (BC), the char end member is added. The solid-state 13C CP/MAS NMR spectra of char are cited from one previous investigation [34]. The result shows that aliphatic components and carbohydrate account for 22.7–34.8 and 21.3–25.5% of the total OC contents in the DM fractions, followed by char and lignin ranging from 15.6 to 24.1% and from 13.7 to 16.8% of the total OC contents, respectively (Table 3). After organic solvent extraction, the aliphatic components of LF fractions range from 17.9 to 25.6%, which are lower than those of their DM fractions. The NHC fractions (normalized to OC contents) are composed of char (30.8–40.9%), aliphatic component (25.0–33.9%), lignin (14.0–17.4%), carbohydrate (11.2–11.9%), and protein (6.71–8.27%). The NHC fractions are enriched in aromatic moieties due to the removal of carbohydrates and proteins.
Atmospheric deposition particles
Atmospheric dry and wet deposition particles were seasonally collected with several stainless steel buckets (i.d. 50 ± 0.5 cm, height 50 cm, flat bottom) in the Guangzhou Institute of Geochemistry (DHS) in Guangzhou, an urban area of Pearl River Delta (PRD), South China from Apr. 2010 to Jul. 2011. Bulk dry and wet deposition samples were collected by using 25 L plastic buckets and were carried back to the laboratory. The samples were centrifuged by using a high-speed refrigerated centrifuge (Changsha, China) to collect the suspended particulate materials (SPM). Then the separated atmospheric SPM were analyzed by advanced solid-state 13C NMR techniques combined with elemental analysis and Rock-Eval pyrolysis for their chemical and structural characterization. In order to calculate the deposition fluxes, the rainfall amount records at each sampling site were periodically obtained from the reservoir administration offices or from the local meteorological departments.
The results show that the deposition fluxes of SPM range from 47.58 to 65.07 mg/d/m2 (data not shown), and are higher in spring and fall than in summer and winter. The total organic carbon (TOC) of SPM ranges from 13.15 to 17.09% in DHS.
The NMR data indicate a similar distribution of dominated nonpolar alkyl C and alkyl C–O in all the deposited SPM, followed by aromatic, and aromatic C–O, COO, NC=O, ketone, quinone, and aldehyde carbon (Duan et al. unpublished data). Relative high contents of COO are observed in the suburb area during summer and fall seasons. Aromaticity is in the range of 11.04–18.4%, whereas polymethylene carbon varies from 5.00 to 5.97%. The percentages of nonprotonated aromatic carbon accounting for aromatic carbon range from 5.5 to 14.81%. The above results are further confirmed by the data of the Rock-Eval pyrolysis and the five-component model, as used in the above sediment section. The Rock-Eval shows that the S2 concentrations range from 43.67 to 61.34 mg HC/g and the hydrogen index (HI) values are in the range of 332–380 mg HC/g TOC, suggesting the presence of high aliphatic components (Table 4). Moreover, the S1 and S2 values are positively related to aliphatic carbon. The carbohydrates accounting for 32.47–47.95% of the total carbon are the main components in the atmospheric bulk deposition, followed by aliphatic components (19.88–27.33%), proteins (12.65–17.87%), lignin (16.68–26.86%), and BC (0.00–1.61%) (spectra not shown). In general, the chemical compounds of alkyl groups are more abundant than those of aromatic groups.
Bulk kerogen and thermally stimulated kerogen
Kerogen was isolated from the Maoming shale and sequentially treated at different temperatures (250, 300, 350, 400, 450, and 500 °C) to understand changes in chemical, structural, and nanoporous characteristics during artificial maturation. Quantitative 13C direct polarization magic angle spinning (DP/MAS) NMR experiments were performed at a spinning speed of 14 kHz. The 90° 13C pulse length was 4 μs. Nonprotonated carbons and mobile carbon fractions were quantified using a combination of the DP/MAS technique with a recoupled dipolar dephasing (DP/MAS/DD).
Advanced solid-state 13C NMR techniques (DP/MAS NMR, DP/MAS-DD, and CSA filter) were employed and combined with Rock–Eval pyrolysis to characterize the kerogen sample, whereas the nanoporosity and surface properties were elucidated using N2 and CO2 adsorption techniques. The results show that the H/C and O/C atomic ratios decrease, but vitrinite reflectance (R o) and maturation (T max) values increase with the increasing temperature, which are consistent with the declining trends for S2, S3, HI, and oxygen index (OI) parameters derived from the Rock-Eval pyrolysis (data not shown), suggesting that the dehydration, decarboxylation, and condensation predominately occurred throughout the artificial thermal process.
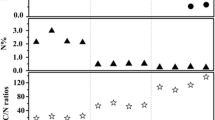
The aliphatic and carbonyl carbons of the kerogen samples significantly decrease, but the aromatic nonprotonated and protonated carbons significantly increase when the temperature increases (Fig. 2). Aromaticity (F aro) is an important parameter for the kerogen structure characterization. The aromaticity ranges from 47.4 to 94.7% for the investigated kerogens (Table 5). As the simulation temperature increases, Faro (%) increases slightly from 47.4% at 25 °C to 54.9% at 300 °C, and then sharply increases from 82.7% at 400 °C to 94.7% at 500 °C. Moreover, F aro (%) is highly correlated with Ro (R 2 = 0.93, p < 0.01) and T max (R 2 = 0.73, p < 0.01). In addition, the nonpolar nonprotonated aromatic carbon (F naro) is highly correlated with R o (%) (Fig. 2). The above statistics analysis demonstrates that F aro (%) and F naro (%) could be used as appropriate proxies for the kerogen maturity. Furthermore, the average aromatic cluster size can be obtained by the following equation: \(n_{\text{C}} \, \ge \, 6/\chi_{{_{\text{edge}} }}^{{^{ 2} }} \, \ge \, 6/\chi_{\text{edge, max}}^{2}\), which gives a lower limit to the number of carbons of an aromatic cluster in the kerogen [28]. The minimum aromatic cluster size ranges from 10 carbons at 350 °C to 38 carbons at 500 °C. In addition, the nanopore volumes (V o, d-co2) increase from 28.5 at 25 °C to 80.3 μL/g at 450 °C but significantly decrease to 78.9 μL/g on the kerogen at 500 °C. It is found that aromaticity and aromatic cluster size are tightly related to the nanoporous volumes for the thermally matured kerogen samples. In conclusion, the advanced NMR techniques are very useful for investigating the structure and maturation degrees of the thermally simulated kerogen samples.

13C NMR DP/MAS (thin lines a–g), DP with dipolar dephasing (solid lines, a–g, 20 s recycle delay), and CSA filtered spectra (h–n, 1 s recycle delay) for thermally treated kerogens
Conclusions
NMR techniques have facilitated our ability to identify the key NOM composition, structure, transformation, and fates, and their changes as a function of environmental factors. Terrestrial environments are important sites of organic carbon cycling. In this manuscript, several case studies have been reported to prove the applicability of NMR tools to study the structures and compositions of NOM. This study characterized the isolated NHC from biomass, atmospheric deposition, and sediments in the Pearl River Delta by using advanced NMR analytical technique and Rock-Eval pyrolysis technique. It is feasible that biochemical macromolecules can be effectively removed, and condensed NOM (NHC and BC) can be effectively isolated from the investigated samples with the multiple step hydrolysis procedure. The NHC fractions are important or even dominant components of the NOM in the investigated samples. Systematic characterization of NHC shows that it is originated from polymethylene C (algaenan, sporopollenin, cutin, and cuticle), and/or bitumen, kerogen, aged terrigenous NOM (lignin), and black carbon. Moreover, during the artificial maturation experiments of kerogens, as their chemical composition and structure are altered, their nanoporosity is changed to some extent. The chemical and structural characteristics such as aromatic protonated, nonprotonated carbons, and aromatic cluster size play important roles in the development of nanoporosity during kerogen maturation. In summary, the structure of each of the NHC fractions is similar to that of kerogen, highlighting the importance of selective preservation of NOM to the kerogen origin in the investigated aquatic ecosystems. The advanced NMR techniques are very useful to investigate the compositions, constituents, and structures for the biomass, sediment NOM, atmospheric SPM, and thermally matured kerogen samples.
Abbreviations
- ANHC:
-
alkaline nonhydrolyzable carbon
- BC:
-
black carbon
- 13C CSA:
-
13C chemical shift anisotropy filter
- 13C CP/MAS:
-
13C cross polarization/magic angle spinning
- 13C CP/TOSS:
-
13C cross polarization/total sideband suppression
- 13C DP/MAS:
-
13C direct polarization/magic angle spinning
- 13C DP/MAS-DD:
-
13C cross polarization/magic angle dipolar dephasing
- Multi CP:
-
high-spinning speed multi-ramped amplitude cross polarization
- DHS:
-
Guangzhou Institute of Geochemistry
- DM:
-
demineralized carbon
- Faro :
-
aromaticity
- HMQC:
-
proton-detected heteronuclear multiple quantum coherence
- HSQC:
-
proton-detected heteronuclear single quantum coherence
- KC:
-
kerogen carbon
- LP:
-
lipid
- LF:
-
lipid free
- NHC:
-
acid nonhydrolyzable carbon
- NMR:
-
nuclear magnetic resonance
- NOM:
-
natural organic matter
- OS:
-
bulk sample
- POM:
-
particulate NOM
- PRD:
-
Pearl River Delta
- SR:
-
residue obtained after saponification
- TFA:
-
trifluoroacetic acid
- TFAR:
-
residue obtained after successive TFA hydrolysis
- TOC:
-
total organic carbon
- Vo :
-
nanoporosity
References
Ahlers F, Bubert H, Steuernage S, Wiermann R. The nature of oxygen in sporopollenin from the pollen of Typha angustifolia L. Zeitschrift für Naturforschung C. 2000;55(3–4):129–36.
Bergamaschi BA, Tsamakis E, Keil RG, Eglinton TI, Montlucon DB, Hedges JI. The effect of grain size and surface area on organic matter, lignin and carbohydrate concentration, and molecular compositions in Peru Margin sediments. Geochim Cosmochim Acta. 1997;61:1247–60.
Blokker P, Schouten S, van den Ende H, de Leeuw JW, Hatcher PG, Sinninghe Damsté JS. Chemical structure of algaenans from the fresh water algae Tetraedron minimum, Scenedesmus communis and Pediastrum boryanum. Org Geochem. 1998;29:1453–68.
Brewer CE, Schmidt-Rohr K, Satrio JA, Brown RC. Characterization of biochar from fast pyrolysis and gasification systems. Environ Prog Sustain Energy. 2009;28:386–96.
Chalbot MCG, Kavouras IG. Nuclear magnetic resonance spectroscopy for determining the functional content of organic aerosols: a review. Environ Pollut. 2014;191:232–49.
Cornelissen G, Gustafsson O, Bucheli TD, Jonker MTO, Koelmans AA, Van Noort PCM. Extensive sorption of organic compounds to black carbon, coal, and kerogen in sediments and soils: mechanisms and consequences for distribution, bioaccumulation, and biodegradation. Environ Sci Technol. 2005;39:6881–95.
Conte P, Piccolo A. Conformational arrangement of dissolved humic substances. Influence of solution composition on association of humic molecules. Environ Sci Technol. 1999;33:1682–90.
Duer MJ. Introduction to solid-state NMR spectroscopy. Oxford: Blackwell; 2004.
Eglinton TI, Repeta DJ. Organic matter in the contemporary ocean. In: Elderfield H, editor. The oceans and marine geochemistry. London: Elsevier Pergamon; 2004.
Engel MH, Macko SA. Organic geochemistry: principle and application. New York: Plenum Press; 1993.
Gelin F, Volkman JK, Largeau C, Derenne S, Damsté JS, De Leeuw JW. Distribution of aliphatic, nonhydrolyzable biopolymers in marine microalgae. Org Geochem. 1999;30(2):147–59.
Ge´linas Y, Prentice KM, Baldock JA, Hedges JI. An improved thermal oxidation method for the quantification of soot/graphitic black carbon in sediments and soils. Environ Sci Technol. 2001;35(17):e3519–25.
Ge´linas Y, Baldock JA, Hedges JI. Organic carbon composition of marine sediments: effect of oxygen exposure on oil generation potential. Science. 2001;294:e145–8.
Goni MA, Yunker MB, Macdonald RW, Eglinton TI. The supply and preservation of ancient and modem components of organic carbon in the Canadian Beaufort Shelf of the Arctic Ocean. Mar Chem. 2005;93:53–73.
Guilford WJ, Schneider DM, Labovitz J, Opella SJ. High resolution solid state 13C NMR spectroscopy of sporopollenins from different plant taxa. Plant Physiol. 1988;86(1):134–6.
Gustafsson O, Haghseta K, Chan F, McFarlane A, Gschwend PM. Quantification of the dilute sedimentary soot phase: implications for PAH speciation and bioavailability. Environ Sci Technol. 1997;31(1):e203–9.
Hatcher PG, Lerch HE III, Bates AL, Verheyen TV. Solid-state 13C nuclear magnetic resonance studies of coalified gymnosperm xylem tissue from Australian brown coals. Org Geochem. 1989;14:145–55.
Hayes CE, Edelstein WA, Schenck JF, Mueller OM, Eash M. An efficient, highly homogeneous radiofrequency coil for whole-body NMR imaging at 1.5 T. J Magn Reson. 1985;63(3):622–8.
Hedges JI, Oades JM. Comparative organic geochemistries of soils and marine sediments. Org Geochem. 1997;27:319–61.
Johnson RL, Schmidt-Rohr K. Quantitative solid-state 13C NMR with signal enhancement by multiple cross polarization. J Magn Reson. 2014;239:44–9.
Keil RG, Mayer LM, Quay PD, Richey JE, Hedges JI. Loss of organic matter from riverine particles in deltas. Geochim Cosmochim Acta. 1997;61:1507–11.
Kleineidam S, Rügner H, Ligouis B, Grathwohl P. Organic matter facies and equilibrium sorption of phenanthrene. Environ Sci Technol. 1999;33(10):1637–44.
Mao JD, Hu WG, Schmidt-Rohr K, Davies G, Ghabbour EA, Xing BS. Quantitative characterization of humic substances by solid-state carbon-13 nuclear magnetic resonance. Soil Sci Soc Am. 2000;64:873–84.
Mao JD, Schmidt-Rohr K. Accurate quantification of aromaticity and nonprotonated aromatic carbon fraction in natural organic matter by 13C solid-state nuclear magnetic resonance. Environ Sci Technol. 2004;38:2680–4.
Mao JD, Schmidt-Rohr K. Separation of aromatic-carbon 13 C NMR signals from di-oxygenated alkyl bands by a chemical-shift-anisotropy filter. Solid State Nucl Magn Reson. 2004;26:36–45.
Mao JD, Schmidt-Rohr K. Methylene spectral editing in solid-state 13C NMR by three-spin coherence selection. J Magn Reson. 2005;176:1–6.
Mao JD, Cory RM, McKnight DM, Schmidt-Rohr K. Characterization of a nitrogen-rich fulvic acid and its precursor algae from solid state NMR. Org Geochem. 2007;38:1277–92.
Mao JD, Fang X, Lan Y, Schimmelmann A, Mastalerz M, Xu L, Schmidt-Rohr K. Chemical and nanometer-scale structure of kerogen and its change during thermal maturation investigated by advanced solid-state 13C NMR spectroscopy. Geochim Cosmochim Acta. 2010;74:2110–27.
Mao JD, Fang X, Schmidt-Rohr K, Carmo AM, Hundal LS, Thompson ML. Molecular-scale heterogeneity of humic acid in particle-size fractions of two Iowa soils. Geoderma. 2007;140:17–29.
Mehring M, Weber H, Müller W, Wegner G. High resolution 13 C NMR in undoped cis-and trans-polyacetylene. Solid State Commun. 1983;45(12):1079–82.
Meuter-Gerhards A, Schwerdtfeger C, Steuernagel S, Wilmesmeier S, Wiermann R. Studies on sporopollenin structure during pollen development. Zeitschrift für Naturforschung C. 1995;50(7–8):487–92.
Middelburg JJ, Nieuwenhuize J, Breugel P. Black carbon in marine sediments. Mar Chem. 1999;65(4):e245–52.
Mopper K, Stubbins A, Ritchie JD, Bialk HM, Hatcher PG. Advanced instrumental approaches for characterization of marine dissolved organic matter: extraction techniques, mass spectrometry, and nuclear magnetic resonance spectroscopy. Chem Rev. 2007;107:419–42.
Nelson PN, Baldock JA. Estimating the molecular composition of a diverse range of natural organic materials from solid-state 13C NMR and elemental analyses. Biogeochemistry. 2005;72:1–34.
Nguyen TH, Brown R, Ball WP. An evaluation of thermal resistance as a measure of black carbon content in diesel soot, wood char, and sediment. Org Geochem. 2004;35(3):217–34.
Preston CM, Newman RH. Demonstration of spatial heterogeneity in the organic matter of de-ashed humin samples by solid-state 13C CPMAS NMR. Can J Soil Sci. 1992;72(1):13–9.
Preston C, Newman R. A long-term effect of N fertilization on the 13C CPMAS NMR of de-ashed soil humin in a second-growth Douglas-fir stand of coastal British Columbia. Geoderma. 1995;68:229–41.
Preston CM. Applications of NMR to soil organic matter analysis: history and prospects. Soil Sci. 1996;161:144–66.
Ran Y, Huang W, Rao PSC, Sheng G, Fu J. The role of condensed organic matter in the nonlinear sorption of hydrophobic organic contaminants by a peat and sediments. J Environ Qual. 2002;31:1953–62.
Ran Y, Xiao B, Huang W, Peng P, Liu D, Fu J, Sheng G. Kerogen in aquifer material and its strong sorption for nonionic organic pollutants. J Environ Qual. 2003;32:1701.
Schmidt-Rohr K, Spiess HW. Multidimensional solid-state NMR and polymers. Hoboken: Elsevier; 1994.
Schmidt MWI, Skjemstad J, Czimczik CI, Glaser B, Prentice KM, Gelinas Y, Kuhlbusch TAJ. Comparative analysis of black carbon in soils. Global Biogeochem Cycles. 2001;15(1):163–7.
Schmidt-Rohr K, Mao JD. Efficient CH-group selection and identification in 13C solid-state NMR by dipolar DEPT and 1H chemical-shift filtering. J Am Chem Soc. 2002;124:13938–48.
Smernik RJ, Olk D, Mahieu N. Quantitative solid-state 13C NMR spectroscopy of organic matter fractions in lowland rice soils. Eur J Soil Sci. 2004;55:367–79.
Smernik RJ, Kookana RS, Skjemstad JO. NMR characterization of 13C-benzene sorbed to natural and prepared charcoals. Environ Sci Technol. 2006;40:1764–9.
Song JZ, Peng PA, Huang WL. Black carbon and kerogen in soils and sediments. 1. Quantification and characterization. Environ Sci Technol. 2002;36:3960–7.
Thorn KA. Structural characteristics of the IHSS Suwannee River fulvic and humic acids determined by solution state C-13 NMR spectroscopy. Sci Total Environ. 1987;62:175–83.
Tissot BP, Welte DH. Petroleum Formation and Occurrence. New York: Springer; 1984.
Wassmann P, Bauerfeind E, Fortier M, Fukuchi M, Hargrave B, Moran B, Noji T, Nothig EM, Olli K, Peinert R, Sasaki H, Shevchenko V. Particulate organic carbon flux to the Arctic Ocean sea floor. Organic Carbon Cycle in the Arctic Ocean; 2004. p. 101–138.
Wilmesmeier S, Steuernagel S, Wiermann R. Comparative FTIR and 13C CP/MAS NMR spectroscopic investigations on sporopollenin of different systematic origins. Zeitschrift für Naturforschung C. 1993;48(9–10):697–701.
Wilson MA. NMR techniques and applications in geochemistry and soil chemistry. Ocford: Pergamon Press; 1987.
Zhang D, Duan D, Huang Y, Xiong Y, Yang Y, Ran Y. Role of structure, accessibility and microporosity on sorption of phenanthrene and nonylphenol by sediments and their fractions. Environ Pollut. 2016. doi:10.1016/j.envpol.2016.05.052.
Zhang D, Duan D, Huang Y, Yang Y, Ran Y. Novel phenanthrene sorption mechanism by two pollens and their fractions. Environ Sci Technol. 2016;50(14):7305–14.
Zhang D, Ran C, Yang Y, Ran Y. Biosorption of phenanthrene by pure algae and field-collected planktons and their fractions. Chemosphere. 2013;93:61–8.
Zhang D, Ran Y, Cao X, Mao J, Cui J, Schmidt-Rohr K. Biosorption of nonylphenol by pure algae, field-collected planktons and their fractions. Environ Pollut. 2015;198:61–9.
Authors’ contributions
DNZ and DDD have collected the biomass, sediment, atmospheric samples, and done the fractionated extraction and NMR spectra, and written the manuscript, and YDH and YY have collected the sediment and kerogen samples and the NMR spectra. YR has designed the investigation and written the manuscript. All authors read and approved the final manuscript.
Acknowledgements
The authors thank Dr. Jingdong Mao in Old Dominion University, USA for the analytic measurements of the samples by using advanced NMR techniques. This is a contribution from GIGCAS, No. IS-2337.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
Supplemental Data and Tables attached in the Additional file 1. Other data and materials could be requested from the corresponding author.
Consent for publication
The authors agreed the publication of the manuscript in this journal.
Ethics approval and consent to participate
This manuscript is an original paper, and has not published in other journals. The authors agreed to keep the copyright rule.
Funding
This investigation was supported by a key joint project of the National Natural Foundation of China (NNSFC) the Guangdong Province Natural Science Foundation (U1201235), and a general project of the NNSFC (41473103).
Author information
Authors and Affiliations
Corresponding author
Additional file
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zhang, D., Duan, D., Huang, Y. et al. Composition and structure of natural organic matter through advanced nuclear magnetic resonance techniques. Chem. Biol. Technol. Agric. 4, 8 (2017). https://doi.org/10.1186/s40538-017-0086-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40538-017-0086-8