
Abstract
Leucine-rich repeat kinase 2 (LRRK2) is the most common causative gene for autosomal dominant Parkinson’s disease (PD) and is also known to be a susceptibility gene for sporadic PD. Although clinical symptoms with LRRK2 mutations are similar to those in sporadic PD, their pathologies are heterogeneous and include nigral degeneration with abnormal inclusions containing alpha-synuclein, tau, TAR DNA-binding protein 43, and ubiquitin, or pure nigral degeneration with no protein aggregation pathologies. We discovered two families harboring heterozygous and homozygous c.4332 G > A; p.R1441H in LRRK2 with consanguinity, sharing a common founder. They lived in the city of Makurazaki, located in a rural area of the southern region, the Kagoshima prefecture, in Kyushu, Japan. All patients presented late-onset parkinsonism without apparent cognitive decline and demonstrated a good response to levodopa. We obtained three autopsied cases that all presented with isolated nigral degeneration with no alpha-synuclein or other protein inclusions. This is the first report of neuropathological findings in patients with LRRK2 p.R1441H mutations that includes both homozygous and heterozygous mutations. Our findings in this study suggest that isolated nigral degeneration is the primary pathology in patients with LRRK2 p.R1441H mutations, and that protein aggregation of alpha-synuclein or tau might be secondary changes.
Similar content being viewed by others

Introduction
Parkinson’s disease (PD) is the most common neurodegenerative movement disorder. Pathologies include neuronal loss and gliosis in the substantia nigra pars compacta (SNpc), locus coeruleus (LC), and dorsal motor nucleus of the vagus nerve, as well as the appearance of Lewy pathologies [9]. Lewy pathologies are the pathological hallmark of PD, and their major component is alpha-synuclein, encoded by synuclein alpha (SNCA) [43]. Many genetic factors for PD (PARK from 1 to 23) have been detected in the past two decades [7]. LRRK2 was originally mapped as a candidate region by linkage analysis from a large family living in the Sagamihara region of the Kanagawa prefecture in Japan [13]. Following its discovery, two groups concurrently reported the mutations p.R1441G, p.Y1699C, and p.R1441C in LRRK2 from patients in England, Spain, Germany, and the United States [33, 54]. LRRK2 is located on 12q12 (MIM#609007) and includes 51 exons, and encodes a large protein (2527 amino acids) that belongs to the ROCO protein family and includes seven domains: armadillo, ankyrin, leucine-rich repeat (LRR), Ras in complex proteins (Roc), C-terminal of Roc (COR), kinase, and WD40 [26]. Currently, seven missense mutations (p.N1437H, p.R1441C/G/H, p.Y1699C, p.G2019S, and p.I2020T) are considered pathogenic variants. LRRK2 p.G2019S is the most common mutation, responsible for 36% of familial PD in North African Arab-Berbers, and 28% of familial PD in Ashkenazi Jews [21]. LRRK2 p.G2385R is also known as a risk variant for the onset of PD in Asian countries such as Japan, Taiwan, and Singapore [8, 14, 46].
To date, heterogeneous brain pathology has been reported in 55 autopsies of patients harboring LRRK2 mutations [40]. Even in the same mutation, for instance LRRK2 p.G2019S, pathologies have been reported with and without alpha-synuclein-positive Lewy bodies or Lewy neurites, Alzheimer’s disease (AD) pathology, or tau-immunopositive glial tangle pathologies [17, 36, 38]. Neuropathology in LRRK2 p.I2020T mutations from the original Sagamihara family also revealed heterogeneous pathologies, including pure degeneration of the SNpc without any inclusions, Lewy bodies, or multiple system atrophy (MSA) pathology [19]. It is still unclear why LRRK2 mutations have such varied pathological and clinical manifestations.
In the present study, we detected two families (families A and B) that harbored c.4332 G > A, p.R1441H mutations in LRRK2, including ten PD patients presenting slowly progressing, late-onset parkinsonism. The two families had consanguinity. Our genetic analysis of seven PD patients indicated five homozygote and two heterozygote mutations of p.R1441H. Of these, we conducted brain autopsies of three patients: two homozygotes and one heterozygote. Our findings provide a new perspective of brain pathologies in patients with LRRK2 mutations.
Materials and methods
Subjects
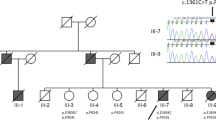
This study was approved by the ethics committee of the Juntendo University School of Medicine, in accordance with The Code of Ethics of the World Medical Association (Declaration of Helsinki). All participants for genetic and clinical analyses gave full written informed consent before participation. The inheritance mode was defined as autosomal dominant when the family members of at least two consecutive generations were affected, and as autosomal recessive when one same-generation sibling was affected. The diagnosis of PD was established using clinical criteria [18]. A good responder to levodopa was defined as a patient whose symptoms improved with levodopa treatment. The two families, A and B, were from Makurazaki city in the Kagoshima prefecture, located in the southwest of Japan, with a population of approximately 20,000 in 2018 (Fig. 1a). The parents of each family were first cousins and born in Makurazaki. Our definition of inheritance confirmed family A as autosomal recessive inheritance, and family B as autosomal dominant inheritance (Fig. 1b).

Illustrated location of Makurazaki city and the family trees of family A and B. a Geographical illustration of Japan. Makurazaki is a small city in the Kagoshima prefecture, located in the southernmost tip of Kyushu island. b Family trees of the two families harboring p.R1441H in LRRK2 mutation and living in Makurazaki city. Parkinson’s disease is shown as black and schizophrenia is shown as half black and half white. W/W = wildtype, W/M = heterozygous of c.4322G > A, M/M = homozygous of c.4322G > A. Diagonal line denotes deceased individuals. Asterisk represents autopsied cases
Genetic analyses
We collected genomic DNA using QIAamp DNA Blood Midi Kit (QIAGEN, Hilden, Germany) from eight individuals in family A, which included four patients with PD, one patient with schizophrenia without parkinsonism, and three healthy siblings. From family B we collected genomic DNA from four individuals, which included three patients with PD and one healthy sibling. We selected four patients with PD (A-II-2, A-II-6, B-III-2, and B-III-6) for whole genome sequencing (WGS). WGS was performed using TruSeq DNA PCR-Free Library Prep Kit (Illumina, San Diego, CA, USA) and paired-end sequencing (150 bp × 2) on a HiSeq X Ten (Illumina). Sequence reads from WGS were trimmed by Trimmomatic (version 0.36) [2] and aligned to the GRCh37 human reference genome using BWA (version 0.7.17) [27]. Duplicated reads were removed by Picard (https://broadinstitute.github.io/picard/). Variants calling was performed using GATK (version 4.0.1.1) [31], and variants were annotated using ANNOVAR (version 2017Jul17) [49]. LRRK2 exon 31 was sequenced using the Sanger method, previously reported by Zimprich et al. [54]. Haplotypes were constructed using genetic markers including four SNPs and eight microsatellites mapped onto the flanking region of LRRK2. These genetic markers were genotyped by Sanger method or fragment analysis using fluorescence-labeled primers, as reported previously [4].
Neuropathological analyses
We obtained brain autopsies from A-II-3, A-II-6, and B-III-2 and carried out neuropathological examinations (Fig. 1b). Brains were fixed with 15% neutral buffered formalin and the selected tissues were embedded in paraffin. The paraffin embedded blocks were sliced 6-μm thick. Brain sections were stained with hematoxylin and eosin (H&E), Klüber-Barrera (KB), methenamine-silver stain, Gallyas-Braak stain, and immunohistochemical stains using several antibodies for proteins related to neurodegenerative diseases. For immunohistochemistry, brain sections underwent antigen retrieval either by heat activation in a microwave oven or by reaction in formic acid, before being incubated overnight at 4 °C in primary antibody. The primary antibodies used were against phosphorylated alpha-synuclein (pSyn#64, monoclonal, diluted 1:10,000, Wako, Osaka, Japan), phosphorylated tau (AT8, monoclonal, diluted 1:100, Thermo Fisher Scientific, Waltham, MA, USA), amyloid beta (1–42, polyclonal, diluted 1:100, IBL, Gunma, Japan), Ubiquitin (Ubi-1, monoclonal, diluted 1:200, Abcam, Cambridge, UK), phosphorylated TAR DNA binding protein 43 (TDP43, Ser 409/410, monoclonal, diluted 1:1000, Cosmo Bio, Tokyo, Japan), LRRK2 (NB300–268, polyclonal, diluted 1:1000, Novus Biologicals, Littleton, CO, USA), tyrosine hydroxylase (TH, monoclonal, diluted 1:1000, Sigma-Aldrich, St. Louis, MO, USA), glial fibrillary acidic protein (GFAP, G-25-8-3, monoclonal, diluted 1:200, IBL), and ionized calcium-binding adapter molecule 1 (Iba1, polyclonal, diluted 1:1000, Wako) were used. Bound antibodies were visualized using the peroxidase-polymer-based method using a Histofine Simple Stain MAX-PO kit (Nichirei, Tokyo, Japan) with diaminobenzidine as the chromogen.
Results
Genetic analyses
We identified 13 consensus variants by WGS analysis (Table 1 and Additional file 1). Among them, 11 of the 13 variants were insertion/deletion, which might be misaligned false positive variant calls. The remaining two variants were MUC5B (c.7843G > A:p.G2615S) and LRRK2 (c.4322G > A:p.R1441H). Mucin 5B, oligomeric mucus/gelforming (MUC5B) has been reported as a susceptibility gene for pulmonary fibrosis [4]. Therefore, the results of WGS indicated that LRRK2 (c.4322G > A: p.R1441H) is a causative mutation for families A and B. Sanger sequencing validation identified eight symptomatic patients (A-II-2, A-II-3, A-II-5, A-II-6, A-II-7, B-III-2, B-III-6, and B-III-8) with p.R1441H, and four asymptomatic individuals (A-II-1, A-II-9, A-III-1, B-III-3) without p.R1441H, which signals complete segregation of p.R1441H in families A and B (Fig. 1b). In addition, Sanger sequencing revealed a homozygous mutation in five patients (A-II-3, A-II-5, A-II-6, B-III-6, and B-III-8) and a heterozygous mutation in three patients (A-II-2, A-II-7, and B-III-2; Additional file 1: Figure S1). There were no pathogenic mutations, as well as risk variants and haplotypes, including SNCA, PAKR16, BST1, and MAPT, related to familial PD except LRRK2 p.R1441H in our WGS reads. Haplotype analysis indicated that patients from families A and B shared a common haplotype in the region of between D12S2080 and D12S2522, which signals a founder effect (Additional file 1: Table S2).
Case presentations
The parents of family A (A-I-1 and A-I-2) and family B (B-II-1 and B-II-2) married with consanguinity. All patients indicated as black symbols in the family trees were clinically diagnosed with typical PD (Fig. 1b). A-II-7 was diagnosed as schizophrenia without parkinsonism. Age at onset of PD was 61.25 ± 9.19 (± SD) in all patients, 61.60 ± 7.23 in patients with homozygous mutations, and 68.50 ± 6.54 in patients with heterozygous mutations. There were no differences in age at onset between homozygous and heterozygous mutations (p > 0.05). The clinical history of the three autopsied cases is described in the following sections.
A-II-3
This patient had a homozygous mutation of p.R1441H in LRRK2. At 68 years of age, he noticed left-side dominant tremor in his upper and lower limbs. The attending doctor observed his rigidity, tremor, and masked face and diagnosed him with PD. Following diagnosis, his motor symptoms showed a good response to levodopa carbidopa 200 mg and cabergoline 2 mg per day. In the following few years, he remained in good condition and with good activity of daily living, without support from anyone. At 73 years of age, the wearing off phenomenon emerged and at 74 years of age, he died suddenly at home.
A-II-6
This patient had a homozygous mutation of p.R1441H in LRRK2. At the age of 60 years, he noticed resting tremor in his right upper and lower limbs. The next year, he went to the local hospital and was diagnosed with PD, and had a good response to levodopa/benserazide, trihexyphenidyl, and pramipexole. His parkinsonism slowly progressed, and by 72 years of age, visual and auditory hallucinations, diurnal fluctuations, dyskinesia, and camptocormia became prominent. At 74 years of age, he was bed-ridden. At his final admission at Juntendo Hospital, his intelligence was well preserved, with a Mini-Mental State Examination (MMSE) score of 26/30. He exhibited marked fluctuations in consciousness, bradyphrenia, visual hallucinations, hypersexuality, and delusions of persecution. We observed disuse-induced atrophy in the upper and lower limbs, and contracture and pes equinus in the lower limbs, due to the long duration of his bed-ridden state. Deep tendon reflexes were decreased in the upper and lower limbs, and dysautonomia such as orthostatic hypotension, urinary incontinence, and constipation was not seen. Rigidity and tremor were mild even at the end stage. Brain magnetic resonance imaging (MRI) indicated no abnormalities. 123I-metaiodobenzilguanidine (MIBG) myocardial scintigraphy revealed no decreasing rate with 3.00 at early and 3.40 at delay of heart to mediastinum (H/M) ratio (normal value: over 2.2). Dopamine transporter (DAT) imaging showed a severe decrease in DAT densities. A nerve conduction study indicated axonal polyneuropathy. At the age of 74 years, the patient died from aspiration pneumonia.
B-iii-2
This patient had a heterozygous mutation of p.R1441H in LRRK2. At the age of 72, he became aware of a resting tremor in his right extremities. He was diagnosed with PD, and presented a good response to levodopa/benserazide, entacapone, and selegiline over 10 years. At the age of 85, wearing off was prominent, but he could walk alone. At 87 years of age, he found walking difficult after the onset of a lumbar fracture. Following this injury, he repeatedly experienced aspiration pneumonia. In the same year, his consciousness was disturbed due to hypoxia and aspiration pneumonia, and he was admitted to the hospital. After comprehensive treatment, he died from the progression of pneumonia, multiple organ failure, and hypoxia. At his final hospital admission, delusion was present, but cognitive function was preserved (MMSE: 25/30). His brain MRI indicated no abnormalities, while his 123I-MIBG myocardial scintigraphy indicated normal uptake, with 2.51 in early and 2.79 in delay of H/M ratio. A DAT scan demonstrated a severe decrease in uptake with left-side-dominant laterality. A nerve conduction study indicated axonal polyneuropathy.
Pathological analysis
The neuropathological findings of three patients (individual A-II-3, A-II-6, and B-III-2) are presented. The brain weights of A-II-3, A-II-6, and B-III-2 were 1350 g, 1350 g, and 1126 g, respectively.
Macroscopically, the substantia nigra (SN) was markedly depigmented in our cases, compared with age-matched controls (Fig. 2 a-c), but the LC was preserved. Atrophy and morphological changes were not observed in other brain regions. In H&E, KB, GFAP, and IBA1 staining, neuronal cell loss and astrogliosis were apparent in the SN (Fig. 2 d–i). Almost all microglia exhibited a ramified shape (resting form) in the SN of all three cases; small numbers of ameboid-shaped cells (reactive form) appeared only in B-III-2 (Fig. 2 j–l). These findings were considered common pathologies of chronic nigral degeneration. In TH stains, loss of dopaminergic neurons was apparent in the SNpc, compared with age matched-controls (Fig. 3 a–d). In contrast, there was no neuronal cell loss or gliosis in the LC in any case. The remains of neurons showed no Lewy bodies in H&E sections. In anti-phosphorylated alpha-synuclein (p-alpha-synuclein) and ubiquitin immunostains, there were no Lewy pathologies (bodies, neurites, or dots) in the SN, compared with sporadic PD (Fig. 3 e–h). Moreover, p-alpha-synuclein-positive pathologies were not detected at all in the brainstem, limbic area, subcortical nuclei, white matter, or neocortex in any individual. Phosphorylated-tau (p-tau)-positive neurofibrillary tangles (NFT) and threads were limited to within the parahippocampus and hippocampus (Braak NFT stage II) [3]. Amyloid-beta-positive senile plaques were detected within the range of normal aging in the neocortex. These tau pathologies were considered primary age-related tauopathy (PART) [6]. There were no TDP43-positive pathologies or LRRK2-positive pathologies. These pathological findings were common to all three patients. In addition to these common findings, a small number of p-tau-positive structures were observed in the brainstem and cerebellar dentate nucleus of individual B-III-2, while the peripheral nerve (sural nerve) was also evaluated in A-II-6 and demonstrated preserved axons and myelin. In summary, the major pathological feature of all three individuals was isolated nigral degeneration without Lewy or other pathological inclusions.

Neuronal loss and astrogliosis in the substantia nigra of cases with the LRRK2 p.R1441H mutation. A macroscopic picture of the midbrains (a: age-matched control, b: A-II-6, c: B-III-2). The substantia nigra of A-II-6 and B-III-2 were markedly depigmented, compared with the age-matched control (a macroscopic photograph of A-II-3 was not available). Black arrowheads indicate the pigmented nigral areas. The substantia nigra of all cases showed apparent neuronal loss and astrogliosis in hematoxylin and eosin (H&E) stain (d: A-II-3, e: A-II-6, f: B-III-2) and glial fibrillary acidic protein (GFAP) stain (g: A-II-3, h: A-II-6, i: B-III-2), but proliferations of reactive microglia were absent or mild (j: A-II-3, k: A-II-6, l: B-III-2). The scale bars represent d–f: 500 μm, g–i: 200 μm, and j–l: 200 μm, respectively

Dopaminergic cell loss and absence of alpha-synuclein pathologies in the substantia nigra of cases with the LRRK2 p.R1441H mutation. Severe losses of dopaminergic neurons were present in the substantia nigra of cases with the mutation, compared with age-matched controls, using a tyrosine hydroxylase (TH) immunostain (a: age-matched control, b: A-II-3, c: A-II-6, d: B-III-2). However, sporadic PD with Lewy pathology corresponding to Braak stage 5 showed typical alpha-synuclein-positive neuronal inclusions in the substantia nigra; there was no Lewy pathology in the substantia nigra or other brain regions of cases with the mutation, as assessed with immunohistochemical analysis for alpha-synuclein (e: age-matched control, f: A-II-3, g: A-II-6, h: B-III-2). The scale bars represent a–d: 1 mm, and e–h: 200 μm, respectively
Discussion
In the present study, we investigated two families that harbored homozygous and heterozygous mutations of p.R1441H in LRRK2. All affected members had a founder effect, suggesting a common haplotype in two different families. The two families came from a narrow district in Makurazaki city, with consanguinity. The environment incidentally generated both heterozygous and homozygous mutations of p.R1441H in two families. All PD patients presented late-onset parkinsonism with a good response to levodopa and a mild disease course, without apparent cognitive decline or dysautonomia. The clinical manifestations were similar to those in patients with sporadic PD or PD with other LRRK2 mutations. Most PD patients with LRRK2 mutations are heterozygous missense mutations with an autosomal dominant inheritance mode, which we infer as a gain-of-toxic-function pathological mechanism of LRRK2 mutations. In the families in our study, there were no differences in clinical symptoms, age at onset, or brain pathologies between patients with heterozygous and homozygous mutations. Previous reports also described no apparent differences in symptoms between homozygous and heterozygous mutations in LRRK2 p.G2019S [1, 24, 25]. The patients with LRRK2 p.G2019S homozygous mutations had middle-aged onset, tremor-dominant and mixed type of tremor, and rigid akinesia, similar to heterozygous patients [24]. It has also been reported that homozygous p.R1441C knock-in mice present a normal phenotype, with no dopaminergic neurodegeneration [48]. It remains unclear why homozygote and heterozygote patients with LRRK2 mutations have similar clinical manifestations.
Arginine (R) is located at position 1441within the Roc domain of the LRRK2 protein. The missense mutation in R1441 induces different amino acid changes of glycine (G), cysteine (C), or histidine (H) [30, 33, 54]. LRRK2 mutations of p.R1441C/G/H induce late-onset parkinsonism with good response to levodopa, and closely resemble sporadic PD or PD with LRRK2 p.G2019S mutations [20, 30, 34]. The p.R1441H mutations have been identified in four families from Asia, Europe, and North America; it is present in diverse ethnicities [12, 30, 42, 53]. One rare case with LRRK2 p.R1441H has been related to progressive supranuclear palsy [42]. However, our three autopsy cases showed homogeneity in their symptoms; they had preserved cognitive function until the end of their lives, with the appearance of persistent psychosis in the advanced stage, and without dysautonomia. These findings may arise from their consanguinity, a founder effect, and their descent from a small city population.
A summary of 55 autopsied cases with LRRK2 mutations (p.G2019S, p.I2020T, p.R1441C, and others) has been previously reported [40]. Most pathologies involved SNpc degeneration associated with Lewy pathologies. However, other patients presented broad types of pathology, including pure nigral degeneration without any inclusions, MSA-like-synucleinopathy, PSP-like tauopathy, frontotemporal lobar degeneration with ubiquitin-positive-inclusions, and coexistence with TDP-43-positive inclusions. The pathological heterogeneity suggests that LRRK2 mutations may cause a cascade of effects to induce neuronal deterioration.
To our knowledge, this study is the first report of neuropathology in LRRK2 p.R1441H mutations. In addition, there have been no pathological reports of any homozygous mutations in LRRK2. Our pathological presentation of both homozygous and heterozygous mutations in LRRK2 is crucial for considering LRRK2-associated pathogenesis. Isolated nigral (SNpc) degeneration without alpha-synuclein, tau, amyloid-beta, ubiquitin, and TDP43 pathology was a prominent feature in all three of our autopsy cases. Previous studies have reported pure nigral degeneration, without inclusions of disease-specific proteins, in p.G2019S, p.R1441G, and p.I2020T mutations [15, 17, 19, 29]. In addition, all patients showed typical sporadic PD-like parkinsonism, similar to our cases. Considering the clinicopathological relationship, their parkinsonism might be caused by neurodegeneration of the SNpc, irrespective of expression levels of alpha-synuclein pathologies. Nigral degeneration not associated with Lewy pathologies was also observed in the brains of most parkin RBR E3 ubiquitin protein ligase (PRKN) mutations and one PTEN induced putative kinase 1 (PINK1) mutation previously [10, 45]. Parkin and PINK1 collaborate in mitophagy to remove damaged mitochondria and maintain mitochondrial quality [44]. There is convincing evidence linking LRRK2 with autophagy/mitophagy [5, 23, 32, 37, 39, 47], with regulating roles for LRRK2 involving mTOR signaling [11, 22, 28, 39, 52] as well as Parkin/PINK1 mitophagy. In contrast, LRRK2, a large multi-domain protein, plays an important role in phosphorylation of itself and of target substrates in cellular signaling. Some mutations in the kinase domain, especially p.G2019S, are considered to augment kinase activity, and excess kinase activity is known to induce neurotoxicity [41, 50]. Alternatively, mutations in the Roc-COR domain can cause a decrease in GTPase activity and are thought to play an important role in neurodegeneration [51]. In addition, in relation to alpha-synuclein and tau pathologies, several studies have shown that LRRK2 affects these protein aggregations, but alpha-synuclein- or tau-aggregation-related pathogenesis in brains with LRRK2 mutations remains poorly understood [26]. Our three cases, and twelve previously reported cases (p.R1441H and p.R1441G in the Roc domain, p.G2019S and p.I2020T in the kinase domain), all showed SNpc degeneration without Lewy or other pathological inclusions, although these enzymes act at a steady rate and the biological aberrations induced by these mutations are quite different. Recently, a linkage between Ras analogue in brain (Rab) GTPases and alpha-synuclein, LRRK2, and vacuolar sorting protein (VPS) 35 associations with PD pathogenesis was highlighted [16]. One study reported that p.R1441C in the GTPase domain enhances GTP binding and stimulates LRRK2 activity through interaction with Rab29 and the Golgi apparatus [35]. These data suggest that p.R1441H in the GTPase domain can also cause LRRK2-associated neurotoxicity induced by Rab29-mediated Golgi recruitment.
Conclusions
It was noteworthy that our three autopsied series showed homogeneous pathology. Regardless of the homozygous or heterozygous mutations of p.R1441H, the pathological findings and the clinical features were similar, suggesting that any possibility of a gene dosage effect could be excluded. Two consanguineous families with LRRK2 p.R1441H had a founder effect; the similarity might be caused by their homogeneous genetic background. Our pathological findings indicated that isolated nigral degeneration is essential pathology for LRRK2 p.R1441H mutations. When considering previous reports of neuropathologies from LRRK2 mutations, SNpc degeneration was a constant finding in all cases. Other findings of protein pathologies, such as alpha-synuclein, tau, ubiquitin, and TDP43, were inconsistent. Thus, we consider that neuronal degeneration in the SNpc is primary degeneration, caused by neurotoxicity of pathogenic LRRK2 mutants. We speculate that abnormal protein aggregation, such as of alpha-synuclein, tau, ubiquitin, or TDP43, would be downstream in the pathological cascade of LRRK2. Our three autopsy cases strongly support this theory, showing isolated nigral degeneration with LRRK2 mutation. In addition, it is important to investigate the possibility of proteopathic aggregations that have not been identified previously in LRRK2 mutated pathologies, in future pathological and biochemical studies.
Abbreviations
- AD:
-
Alzheimer’s disease
- COR:
-
C-terminal of Roc
- DAT:
-
Dopamine transporter
- GFAP:
-
Glial fibrillary acidic protein
- H&E:
-
Hematoxylin and eosin
- H/M:
-
Heart to mediastinum
- Iba1:
-
Ionized calcium-binding adapter molecule 1
- KB:
-
Klüber-Barrera
- LC:
-
Locus coeruleus
- LRR:
-
leucine-rich repeat
- LRRK2:
-
Leucine-rich repeat kinase 2
- MIBG:
-
Metaiodobenzilguanidine
- MMSE:
-
Mini-Mental State Examination
- MRI:
-
Magnetic resonance imaging
- MSA:
-
Multiple system atrophy
- MUC5B:
-
Mucin 5B, oligomeric mucus/gelforming
- NFT:
-
neurofibrillary tangles
- P-alpha-synuclein:
-
Phosphorylated alpha-synuclein
- PART:
-
primary age-related tauopathy
- PD:
-
Parkinson’s disease
- PINK1:
-
PTEN induced putative kinase 1
- PRKN:
-
parkin RBR E3 ubiquitin protein ligase
- P-tau:
-
Phosphorylated-tau
- Rab:
-
Ras analogue in brain
- Roc:
-
Ras in complex proteins
- SN:
-
Substantia nigra
- SNCA:
-
synuclein alpha
- SNpc:
-
Substantia nigra pars compacta
- TDP43:
-
TAR DNA binding protein 43
- TH:
-
Tyrosine hydroxylase
- VPS:
-
Vacuolar sorting protein
- WGS:
-
Whole genome sequencing
References
Alcalay RN, Mirelman A, Saunders-Pullman R, Tang MX, Mejia Santana H, Raymond D, Roos E, Orbe-Reilly M, Gurevich T, Bar Shira A et al (2013, 1966) Parkinson disease phenotype in Ashkenazi Jews with and without LRRK2 G2019S mutations. Mov Disord 28:–1971. https://doi.org/10.1002/mds.25647
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. https://doi.org/10.1093/bioinformatics/btu170
Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82:239–259
Chandra S, Chen X, Rizo J, Jahn R, Sudhof TC (2003) A broken alpha -helix in folded alpha -Synuclein. J Biol Chem 278:15313–15318. https://doi.org/10.1074/jbc.M213128200
Cherra SJ 3rd, Steer E, Gusdon AM, Kiselyov K, Chu CT (2013) Mutant LRRK2 elicits calcium imbalance and depletion of dendritic mitochondria in neurons. Am J Pathol 182:474–484. https://doi.org/10.1016/j.ajpath.2012.10.027
Crary JF, Trojanowski JQ, Schneider JA, Abisambra JF, Abner EL, Alafuzoff I, Arnold SE, Attems J, Beach TG, Bigio EH et al (2014) Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol 128:755–766. https://doi.org/10.1007/s00401-014-1349-0
Deng H, Wang P, Jankovic J (2018) The genetics of Parkinson disease. Ageing Res Rev 42:72–85. https://doi.org/10.1016/j.arr.2017.12.007
Di Fonzo A, Wu-Chou YH, Lu CS, van Doeselaar M, Simons EJ, Rohe CF, Chang HC, Chen RS, Weng YH, Vanacore N et al (2006) A common missense variant in the LRRK2 gene, Gly2385Arg, associated with Parkinson's disease risk in Taiwan. Neurogenetics 7:133–138. https://doi.org/10.1007/s10048-006-0041-5
Dickson DW (2018) Neuropathology of Parkinson disease. Parkinsonism Relat Disord 46(Suppl 1):S30–S33. https://doi.org/10.1016/j.parkreldis.2017.07.033
Doherty KM, Hardy J (2013) Parkin disease and the Lewy body conundrum. Mov Disord 28:702–704. https://doi.org/10.1002/mds.25486
Ferree A, Shirihai O (2012) Mitochondrial dynamics: the intersection of form and function. Adv Exp Med Biol 748:13–40. https://doi.org/10.1007/978-1-4614-3573-0_2
Ferreira JJ, Guedes LC, Rosa MM, Coelho M, van Doeselaar M, Schweiger D, Di Fonzo A, Oostra BA, Sampaio C, Bonifati V (2007) High prevalence of LRRK2 mutations in familial and sporadic Parkinson’s disease in Portugal. Mov Disord 22:1194–1201. https://doi.org/10.1002/mds.21525
Funayama M, Hasegawa K, Kowa H, Saito M, Tsuji S, Obata F (2002) A new locus for Parkinson's disease (PARK8) maps to chromosome 12p11.2-q13.1. Ann Neurol 51:296–301
Funayama M, Li Y, Tomiyama H, Yoshino H, Imamichi Y, Yamamoto M, Murata M, Toda T, Mizuno Y, Hattori N (2007) Leucine-rich repeat kinase 2 G2385R variant is a risk factor for Parkinson disease in Asian population. Neuroreport 18:273–275. https://doi.org/10.1097/WNR.0b013e32801254b6
Gaig C, Marti MJ, Ezquerra M, Rey MJ, Cardozo A, Tolosa E (2007) G2019S LRRK2 mutation causing Parkinson's disease without Lewy bodies. J Neurol Neurosurg Psychiatry 78:626–628. https://doi.org/10.1136/jnnp.2006.107904
Gao Y, Wilson GR, Stephenson SEM, Bozaoglu K, Farrer MJ, Lockhart PJ (2018) The emerging role of Rab GTPases in the pathogenesis of Parkinson's disease. Mov Disord 33:196–207. https://doi.org/10.1002/mds.27270
Giasson BI, Covy JP, Bonini NM, Hurtig HI, Farrer MJ, Trojanowski JQ, Van Deerlin VM (2006) Biochemical and pathological characterization of Lrrk2. Ann Neurol 59:315–322. https://doi.org/10.1002/ana.20791
Gibb WR, Lees AJ (1988) The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson's disease. J Neurol Neurosurg Psychiatry 51:745–752
Hasegawa K, Stoessl AJ, Yokoyama T, Kowa H, Wszolek ZK, Yagishita S (2009) Familial parkinsonism: study of original Sagamihara PARK8 (I2020T) kindred with variable clinicopathologic outcomes. Parkinsonism Relat Disord 15:300–306. https://doi.org/10.1016/j.parkreldis.2008.07.010
Haugarvoll K, Rademakers R, Kachergus JM, Nuytemans K, Ross OA, Gibson JM, Tan EK, Gaig C, Tolosa E, Goldwurm S et al (2008) Lrrk2 R1441C parkinsonism is clinically similar to sporadic Parkinson disease. Neurology 70:1456–1460. https://doi.org/10.1212/01.wnl.0000304044.22253.03
Healy DG, Falchi M, O'Sullivan SS, Bonifati V, Durr A, Bressman S, Brice A, Aasly J, Zabetian CP, Goldwurm S et al (2008) Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case-control study. Lancet Neurol 7:583–590. https://doi.org/10.1016/s1474-4422(08)70117-0
Herzig MC, Kolly C, Persohn E, Theil D, Schweizer T, Hafner T, Stemmelen C, Troxler TJ, Schmid P, Danner S et al (2011) LRRK2 protein levels are determined by kinase function and are crucial for kidney and lung homeostasis in mice. Hum Mol Genet 20:4209–4223. https://doi.org/10.1093/hmg/ddr348
Hindle SJ, Elliott CJ (2013) Spread of neuronal degeneration in a dopaminergic, Lrrk-G2019S model of Parkinson disease. Autophagy 9:936–938. https://doi.org/10.4161/auto.24397
Hulihan MM, Ishihara-Paul L, Kachergus J, Warren L, Amouri R, Elango R, Prinjha RK, Upmanyu R, Kefi M, Zouari M et al (2008) LRRK2 Gly2019Ser penetrance in Arab-Berber patients from Tunisia: a case-control genetic study. Lancet Neurol 7:591–594. https://doi.org/10.1016/s1474-4422(08)70116-9
Ishihara L, Warren L, Gibson R, Amouri R, Lesage S, Durr A, Tazir M, Wszolek ZK, Uitti RJ, Nichols WC et al (2006) Clinical features of Parkinson disease patients with homozygous leucine-rich repeat kinase 2 G2019S mutations. Arch Neurol 63:1250–1254. https://doi.org/10.1001/archneur.63.9.1250
Islam MS, Moore DJ (2017) Mechanisms of LRRK2-dependent neurodegeneration: role of enzymatic activity and protein aggregation. Biochem Soc Trans 45:163–172. https://doi.org/10.1042/BST20160264
Li H, Durbin R (2009) Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 25:1754–1760. https://doi.org/10.1093/bioinformatics/btp324
Manzoni C, Mamais A, Dihanich S, Abeti R, Soutar MPM, Plun-Favreau H, Giunti P, Tooze SA, Bandopadhyay R, Lewis PA (2013) Inhibition of LRRK2 kinase activity stimulates macroautophagy. Biochim Biophys Acta 1833:2900–2910. https://doi.org/10.1016/j.bbamcr.2013.07.020
Marti-Masso JF, Ruiz-Martinez J, Bolano MJ, Ruiz I, Gorostidi A, Moreno F, Ferrer I, Lopez de Munain A (2009) Neuropathology of Parkinson’s disease with the R1441G mutation in LRRK2. Mov Disord 24:1998–2001. https://doi.org/10.1002/mds.22677
Mata IF, Kachergus JM, Taylor JP, Lincoln S, Aasly J, Lynch T, Hulihan MM, Cobb SA, Wu RM, Lu CS et al (2005) Lrrk2 pathogenic substitutions in Parkinson’s disease. Neurogenetics 6:171–177. https://doi.org/10.1007/s10048-005-0005-1
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M et al (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20:1297–1303. https://doi.org/10.1101/gr.107524.110
Orenstein SJ, Kuo SH, Tasset I, Arias E, Koga H, Fernandez-Carasa I, Cortes E, Honig LS, Dauer W, Consiglio A et al (2013) Interplay of LRRK2 with chaperone-mediated autophagy. Nat Neurosci 16:394–406. https://doi.org/10.1038/nn.3350
Paisan-Ruiz C, Jain S, Evans EW, Gilks WP, Simon J, van der Brug M, Lopez de Munain A, Aparicio S, Gil AM, Khan N et al (2004) Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 44:595–600. https://doi.org/10.1016/j.neuron.2004.10.023
Paisan-Ruiz C, Lewis PA, Singleton AB (2013) LRRK2: cause, risk, and mechanism. J Parkinsons Dis 3:85–103. https://doi.org/10.3233/JPD-130192
Purlyte E, Dhekne HS, Sarhan AR, Gomez R, Lis P, Wightman M, Martinez TN, Tonelli F, Pfeffer SR, Alessi DR (2018) Rab29 activation of the Parkinson’s disease-associated LRRK2 kinase. EMBO J 37:1–18. https://doi.org/10.15252/embj.201798099
Rajput A, Dickson DW, Robinson CA, Ross OA, Dachsel JC, Lincoln SJ, Cobb SA, Rajput ML, Farrer MJ (2006) Parkinsonism, Lrrk2 G2019S, and tau neuropathology. Neurology 67:1506–1508. https://doi.org/10.1212/01.wnl.0000240220.33950.0c
Ramonet D, Daher JP, Lin BM, Stafa K, Kim J, Banerjee R, Westerlund M, Pletnikova O, Glauser L, Yang L et al (2011) Dopaminergic neuronal loss, reduced neurite complexity and autophagic abnormalities in transgenic mice expressing G2019S mutant LRRK2. PLoS One 6:e18568. https://doi.org/10.1371/journal.pone.0018568
Ross OA, Toft M, Whittle AJ, Johnson JL, Papapetropoulos S, Mash DC, Litvan I, Gordon MF, Wszolek ZK, Farrer MJ et al (2006) Lrrk2 and Lewy body disease. Ann Neurol 59:388–393. https://doi.org/10.1002/ana.20731
Schapansky J, Nardozzi JD, Felizia F, LaVoie MJ (2014) Membrane recruitment of endogenous LRRK2 precedes its potent regulation of autophagy. Hum Mol Genet 23:4201–4214. https://doi.org/10.1093/hmg/ddu138
Schneider SA, Alcalay RN (2017) Neuropathology of genetic synucleinopathies with parkinsonism: review of the literature. Mov Disord 32:1504–1523. https://doi.org/10.1002/mds.27193
Smith WW, Pei Z, Jiang H, Dawson VL, Dawson TM, Ross CA (2006) Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nat Neurosci 9:1231–1233. https://doi.org/10.1038/nn1776
Spanaki C, Latsoudis H, Plaitakis A (2006) LRRK2 mutations on Crete: R1441H associated with PD evolving to PSP. Neurology 67:1518–1519. https://doi.org/10.1212/01.wnl.0000239829.33936.73
Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M (1997) Alpha-synuclein in Lewy bodies. Nature 388:839–840. https://doi.org/10.1038/42166
Stolz A, Dikic I (2014) PINK1-PARKIN interplay: down to ubiquitin phosphorylation. Mol Cell 56:341–342. https://doi.org/10.1016/j.molcel.2014.10.022
Takanashi M, Li Y, Hattori N (2016) Absence of Lewy pathology associated with PINK1 homozygous mutation. Neurology 86:2212–2213. https://doi.org/10.1212/wnl.0000000000002744
Tan EK, Zhao Y, Skipper L, Tan MG, Di Fonzo A, Sun L, Fook-Chong S, Tang S, Chua E, Yuen Y et al (2007) The LRRK2 Gly2385Arg variant is associated with Parkinson's disease: genetic and functional evidence. Hum Genet 120:857–863. https://doi.org/10.1007/s00439-006-0268-0
Tong Y, Giaime E, Yamaguchi H, Ichimura T, Liu Y, Si H, Cai H, Bonventre JV, Shen J (2012) Loss of leucine-rich repeat kinase 2 causes age-dependent bi-phasic alterations of the autophagy pathway. Mol Neurodegener 7:2. https://doi.org/10.1186/1750-1326-7-2
Tong Y, Pisani A, Martella G, Karouani M, Yamaguchi H, Pothos EN, Shen J (2009) R1441C mutation in LRRK2 impairs dopaminergic neurotransmission in mice. Proc Natl Acad Sci U S A 106:14622–14627. https://doi.org/10.1073/pnas.0906334106
Wang K, Li M, Hakonarson H (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38:e164. https://doi.org/10.1093/nar/gkq603
West AB, Moore DJ, Biskup S, Bugayenko A, Smith WW, Ross CA, Dawson VL, Dawson TM (2005) Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci U S A 102:16842–16847. https://doi.org/10.1073/pnas.0507360102
Xiong Y, Coombes CE, Kilaru A, Li X, Gitler AD, Bowers WJ, Dawson VL, Dawson TM, Moore DJ (2010) GTPase activity plays a key role in the pathobiology of LRRK2. PLoS Genet 6:e1000902. https://doi.org/10.1371/journal.pgen.1000902
Yakhine-Diop SM, Bravo-San Pedro JM, Gomez-Sanchez R, Pizarro-Estrella E, Rodriguez-Arribas M, Climent V, Aiastui A, Lopez de Munain A, Fuentes JM, Gonzalez-Polo RA (2014) G2019S LRRK2 mutant fibroblasts from Parkinson’s disease patients show increased sensitivity to neurotoxin 1-methyl-4-phenylpyridinium dependent of autophagy. Toxicology 324:1–9. https://doi.org/10.1016/j.tox.2014.07.001
Zabetian CP, Samii A, Mosley AD, Roberts JW, Leis BC, Yearout D, Raskind WH, Griffith A (2005) A clinic-based study of the LRRK2 gene in Parkinson disease yields new mutations. Neurology 65:741–744. https://doi.org/10.1212/01.wnl.0000172630.22804.73
Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB et al (2004) Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44:601–607. https://doi.org/10.1016/j.neuron.2004.11.005
Acknowledgements
We thank Bronwen Gardner, PhD, and Ryan Chastain-Gross, PhD, from Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript, as well as Mrs. Akiko Sumii for technical help regarding neuropathology.
Funding
This work was supported by JSPS KAKENHI Grant Numbers 20790625 (to MF), 22790829 (to MF), 16 K09676 (to MF), 18 K07536 (to HY), 16 K09700 (to YL), 16 K09678 (to KN), and 18H04043 (to NH). We are very grateful for the following grants: AMED-CREST (Japanese Agency of Medical Research and Development) (to NH), the Platform Program for Promotion of Genome Medicine (P3GM), Advanced Genome Research and Bioinformatics Study to Facilitate Medical Innovation (GRIFIN) from AMED; 17km0405206h0002 (to NH), and the Program for the Strategic Research Foundation at Private Universities from the Japanese Ministry of Education, Culture, Sports, Science and Technology (MEXT). This work was partially supported by the program for Brain Mapping by Integrated Neurotechnologies for Disease Studies (Brain/MINDS) from AMED.
Availability of data and materials
The data are not available for public access because of patient privacy concerns, but are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
MT, MF, KN, and NH designed the concepts of this study and wrote the manuscript, MT analyzed neuropathology, MF, HY, and YL performed genetic analysis, KN, EM, and HT provided and summarized the clinical data, TS performed autopsy. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was approved by the ethics committee of the Juntendo University School of Medicine, in accordance with The Code of Ethics of the World Medical Association (Declaration of Helsinki).
Consent for publication
All participants for genetic and clinical analyses gave full written informed consent before participation.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Table S1. Consensus nonsynonymous variants detected by whole genome sequencing. A portion of the output from ANNOVAR is shown. Most of the variants are indel and might be misaligned variant calls. NA: not applicable. Figure S1. Sequence electropherogram of LRRK2 c.4322G > A:p.R1441H. Three representative sequences are shown. Healthy sibling A-II-1 has wild type (W/W), patient B-III-2 has heterozygous (W/M), and patient A-II-3 has homozygous (M/M) c.4322G > A variant (arrowhead) in LRRK2 exon 31. Table S2 Comparison of haplotypes of the LRRK2 region. All patients of families A and B harboring LRRK2 c.4322G > A:p.R1441H shared a common haplotype from D12S2080 to D12S2522 (in bold). (DOCX 365 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Takanashi, M., Funayama, M., Matsuura, E. et al. Isolated nigral degeneration without pathological protein aggregation in autopsied brains with LRRK2 p.R1441H homozygous and heterozygous mutations. acta neuropathol commun 6, 105 (2018). https://doi.org/10.1186/s40478-018-0617-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-018-0617-y