
Abstract
Endolymphatic sac tumor (ELST) is a rare neoplasm arising in the temporal petrous region thought to originate from endolymphatic sac epithelium. It may arise sporadically or in association with Von-Hippel-Lindau syndrome (VHL). The ELST prevalence in VHL ranges from 3 to 16% and may be the initial presentation of the disease. Onset is usually in the 3rd to 5th decade with hearing loss and an indolent course. ELSTs present as locally destructive lesions with characteristic computed tomography imaging features. Histologically, they show papillary, cystic or glandular architectures. Immunohistochemically, they express keratin, EMA, and variably S100 and GFAP. Currently it is recommended that, given its rarity, ELST needs to be differentiated from other entities with similar morphologic patterns, particularly other VHL-associated neoplasms such as metastatic clear cell renal cell carcinoma (ccRCC). Nineteen ELST cases were studied. Immunohistochemistry (18/19) and single nucleotide polymorphism microarray testing was performed (12/19). Comparison with the immunophenotype and copy number profile in RCC is discussed. Patients presented with characteristic bone destructive lesions in the petrous temporal bones. Pathology of tumors showed characteristic ELST morphology with immunoexpression of CK7, GFAP, S100, PAX-8, PAX-2, CA-9 in the tumor cells. Immunostaines for RCC, CD10, CK20, chromogranin A, synaptophysin, TTF-1, thyroglobulin, and transthyretin were negative in the tumor cells. Molecular testing showed loss of 3p and 9q in 66% (8/12) and 58% (7/12) cases, respectively. Immunoreactivity for renal markers in ELST is an important diagnostic caveat and has not been previously reported. In fact, renal markers are currently recommended in order to rule out metastatic RCC although PAX gene complex and CA-9 have been implicated in the development of the inner ear. Importantly copy number assessment of ELST has not been previously reported. Loss of 3p (including the VHL locus) in ELST suggests similar mechanistic origins as ccRCC.
Similar content being viewed by others
Introduction
Von-Hippel Lindau (VHL) is an autosomal dominant hereditary cancer predisposition syndrome characterized by abberations in the VHL tumor supressor gene at chromosome location 3p25.3. Patients with VHL are at increased risk of developing a variety of neoplasasms such as central nervous system (CNS) hemangioblastomas, clear cell renal cell carcinomas (ccRCC), pheochromocytomas and extra-adrenal paragangliomas, pancreatic neuroendocrine tumors and adenomas, and endolymphatic sac tumors (ELST) of the inner ear [7, 32].
ELST are very rare tumors of neuroectodermal origin, thought to arise from the rugose, intraosseous portion of the endolymphatic sac. The endolymphatic sac represents an extension of the membranous labyrinth that follows the endolymphatic duct. It has both intraosseous and extraosseous components and ends in a blind pouch in the dura mater lining the posterior surface of the temporal bone. Histologically, the endolymphatic sac is composed of a single layer of flat to cuboidal to low and tall columnar cells resting on a basement membrane with folds and papillae formation in the inferomedial aspect of the intraosseous portion [1, 3, 18]. ELSTs more commonly arise sporadically, but also may arise in association with VHL, with a prevalence of up to approximately 16%. ELST typically occur around the 3rd to 5th decade and present with sensorineural hearing loss, tinnitus, and cranial nerve palsies on the affected side [2, 7]. Radiographically on computed tomography (CT), the tumors are characterized by heterogenous bone destruction centered on the posterior portion of the temporal bone. On magnetic resonance imaging (MRI), ELST demostrate T1 post-contrast enhancement and bright T2 signal [3, 7]. Grossly, tumors are described as blue to red in color and hypervascular [7, 37]. The histologic appearance of ELST ranges from a follicular growth pattern with colloid-filled cystic spaces to a papillary arrangement with solid and hypercellular areas, and occasionally an epithelioid clear cell pattern. The neoplastic cells are arranged in a single cuboidal layer with uniform nuclei and minimal pleomorphism, mitotic activity, or necrosis. Tumors are cytokeratin positive and immunoexpress epithelial membrane antigen (EMA), vimentin, neuron specific enolase (NSE), glialfibrillary acidic protein (GFAP), and variable S100, vascular endothelial growth factor (VEGF), and synaptophysin [14, 39].
The radiographic and histologic appearance of ELST raises a broad differential diagnosis and one must be aware of other more common conditions that may mimic such in order to avoid diagnostic pitfalls [18]. In fact, ELSTs are often not diagnosed until after the initial interpretation is questioned clinically [3]. Entities to consider in the work-up of suspected ELST include choroid plexus tumors, paragaganglioma, and metastatic papillary thyroid carcinoma, tumors which would be expected to immunoexpress transthyretin, chromogranin/synapthophysin, and TTF-1/thyroglobulin, respectively [7, 39]. Of particular importance to rule out are other VHL-associated neoplasms such as metastatic ccRCC. Immunohistochemistry for paired box (PAX) transcription factors PAX-8 and PAX-2, carbonic anhydrase 9 (CA-9), RCC, and CD10 has been suggested for this purpose and are reportedly negative in ELST [7, 25, 32, 39].
In this paper we characterize a cohort of ELST and demostrate immunoreactivity for renal cell markers as well as molecular evidence of predominant 3p and 9q loss which has not been previously described. Loss of 3p (including the VHL locus) in ELST suggests similar mechanistic origins as ccRCC. These findings are important, both to correct the previous assumption that renal cell immunohistochemical markers should not be expressed by ELST, which is important for diagnosis, and also to further characterize this rare neoplasm in order to better understand its pathogenesis.
Materials and Methods
Cases of ELST were identified via search of the laboratory information system and details regarding patient demographics, presentation, and imaging were collected through electronic medical record review with the approval of the institutional review board from all institutions. Hematoxylin and eosin (H&E) stained slides were reviewed and the diagnosis confirmed. The best tumor block was selected for DNA extraction. All immunohistochemical and molecular testing was performed on formalin-fixed, paraffin embedded (FFPE) tissues except in one case where only fresh frozen tumor tissue was available.
Immunohistochemistry
Antibodies were validated according to protocol with appropriate tissue controls. Four μm sections were prepared for immunohistochemical evaluation with the following antibodies (clone, dilution, antigen retrieval, supplier): CK7 (OV-TL 12/30, 1:500, citrate, Cell Marque, Rocklin, California, USA), CK20 (Ks20.8, 1:500, EDTA, Cell Marque, Rocklin, CA, USA), PAX-8 (MRQ-50, 1:3000, EDTA, Cell Marque, Rocklin, CA, USA), RCC (PN-15, 1:500, protease, Cell Marque, Rocklin, CA, USA), CD10 (56C6, 1:1000, EDTA, Cell Marque, Rocklin, CA, USA), CA-9 (MRQ-54, 1:2000, EDTA, Cell Marque, Rocklin, CA, USA), GFAP (EP672Y, 1:200, EDTA, Cell Marque, Rocklin, CA, USA), thyroglobulin (2H11+6E1, 1:5000, EDTA, Cell Marque, Rocklin, CA, USA), S100 (4C4.9, 1:4000, EDTA, Cell Marque, Rocklin, CA, USA), chromogranin A (LK2H10, 1:6000, citrate, Cell Marque, Rocklin, CA, USA), synaptophysin (MRQ-40, 1:5000, citrate, Cell Marque, Rocklin, CA, USA), PAX-2 (EP235, 1:1000, citrate, Cell Marque, Rocklin, CA, USA), transthyretin (rabbit polyclonal, 1:15000, citrate, Boster Biological Technology, Pleasanton, CA, USA), TTF-1 (EP229, 1:500, EDTA, Cell Marque, Rocklin, CA, USA), and Ki-67 (SP6, 1:500, EDTA, Cell Marque, Rocklin, CA, USA). Visualization was performed using the HiDef Detection™ HRP Polymer System (Cell Marque, Rocklin, CA, USA) with diaminobenzidine substrate (Cell Marque, Rocklin, CA, USA) and with hematoxylin counterstain in order to visualize the antibody-antigen complex and background tissue, respectively.
Single nucleotide polymorphism (SNP)-microarray
Genomic DNA extraction for SNP-microarray analysis was performed using the Maxwell® CSC DNA FFPE Kit (Promega, Madison, WI, USA) as detailed by the manufacturer. Microarray-based chromosome analysis of copy number and genotype data was performed according to the manufacturer’s protocol using the IScan System with the Infinium CytoSNP-850 K v1.1 BeadChip (Illumina, Inc., San Diego, CA, USA) and analyzed using GenomeStudio (Illumina, Inc), and Nexus, version 9.0 (BioDiscovery, Inc., El Segundo, CA, USA) software. The signal intensity was determined using the log2 ratio and was used along with the specific allele (B-allele) frequency to evaluate copy number patterns of aberrations (clonal changes in less than 100% of cells including deletion, duplication, loss of heterozygosity, ploidy) and genotype, and assessed visually using the KaryoStudio and Nexus files, by comparing with standard curve data charts generated by computer modeling of mosaicism (SiDCoN [simulated DNA copy number]) [26]. Non-mosaic aberrations (those found in 100% of cells) were not included in data analysis as they were considered to represent constitutional changes.
Statistical methods and bioinformatics
Descriptive statistics were performed in Microsoft® Excel® 2013 (15.0.4971.1000), 64-bit. Gene mapping and visualization was performed using the hg19 assembly, UCSC Genome Browser tool suite [17, 19]. Pathway analysis and protein interactions was performed using Ingenuity Pathway Analysis (IPA) v. 01–07 (Qiagen Inc., https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis/) [20].
Results
Clinical data
Nineteen ESLT from 10 males and 9 females were analyzed. The median age at diagnosis was 45.1 years (mean: 42.8, range: 14.7–63.3). One patient had known VHL disease confirmed by sequencing (VHL exon 2 heterozygous deletion identified) with multiple small nodular cerebellar lesions on imaging suggestive of hemangioblastomas (biopsy not performed). None of the patients had clinical history of RCC. Abdominal imaging when available (9/19) showed no evidence of a renal mass. Patients presented with symptoms including hearing loss, balance difficulties, and unilateral facial paralysis.
Imaging
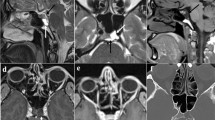
On CT, all patients showed unilateral expansile bone-destructive lesions involving the posterior petrous segment of the temporal bones. Tumors reached up to 6.4 cm in largest dimension (Case 10). Larger lesions were complex in nature with combined solid and cystic patterns (Fig. 1, Case 10 and 17) and mass effect in the posterior cranial fossa (Case 17). On MRI the lesions showed heterogeneous T1 and T2 signal and post-contrast enhancement on T1-weighted sequences (Fig. 1, Cases 1, 4, 10, 17). Some tumors demonstrated regions of intrinsic T1 hyperintensity, which has been described and is thought to correlate with internal hemorrhage or proteinaceous material (Fig. 1Ab, Db, Hb) [6, 23].

Imaging Findings: Computed tomography shows bone destructive lesions involving the posterior aspect of the petrous temporal bones (Aa, B, C, Da, E-G, Ha, I, J, L, M, Na, O). Magnetic resonance imaging shows expansile complex partially solid and cystic masses centered on the petrous segment of the temporal bones demonstrating heterogeneous intrinsic T1 (Ab, Db, Hb) and T2 signal (Hd, Nb) and post-contrast enhancement (Ac, Dc, Hc). (# represents case number). Note: All images are pre-operative except K which is an axial T1-weighted post-operative (at recurrence) image
Gross features
Tumors were tan-pink to red in color and many were described intraoperatively as “vascular” appearing. Some were pulsating intraoperatively, and were initially thought to represent glomus jugulare tumors.
Microscopy
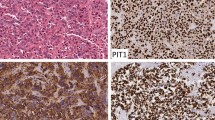
The histopathological appearance of ELST displayed characteristic papillary architecture with a single-cell epithelial lining and central fibrovascular cores. The surface epithelial cells were cuboidal to cylindrical, bland in appearance, with vacuolated to clear cytoplasm and round to elongated nuclei. Several cases showed cystic growth with glandular spaces filled with an eosinophilic colloid-like material (Fig. 2). Immunohistochemically, the neoplastic cells showed diffuse expression of CK7 (18/18, 100%), CA-9 (19/19, 100%), focal to regional GFAP (15/18, 83.33%), PAX-8 (18/18, 100%), PAX-2 (15/18, 83.33%), and S100 (15/18, 83.33%). The tumor cells were negative for CK20, synaptophysin, chromogranin A, transthyretin, TTF-1, RCC, thyroglobulin, and CD10. Ki-67 (MIB-1) showed a < 10% proliferative index for all cases (1000 tumor nuclei counted) (median: 1.1, mean: 1.97, range: 0–9.4) (Figs. 2 and 3).

Histological and immunophenotypical findings: All tumors showed the characteristic papillary architecture (a, b, 100X) with bone invasion (b) and some showed follicular morphology (c, 200X). All tumors immunoexpressed GFAP (d, 400X), CK7 (e, 200X), PAX-8 (f, 200X), PAX-2 (g, 200X), and CA-9 (h, 200X). S100 was focally immunoexpressed (i, 400X) in all but one case

Overview of the clinical, immunohistochemical, and molecular results. Abbreviations: mo.-months; N-no; Y-yes; yr.-years
Copy number alterations
Of 12 cases tested by SNP-microarray, 8 demonstrated loss of 3p of which 7 also showed loss of 9q. A detailed summary of the chromosomal regions involved in presented in Figs. 3, 4 and Table 1. Four cases had no abnormalities detected on SNP-microarray.

Overview of the SNP-microarray findings: Loss of 3p in 8/12 cases and loss of 9q in 7/12 cases, copy number changes present in clear cell renal cell carcinoma. Legend: chromosomal gains are depicted in blue, losses are depicted in red
Pathway and protein interaction analyses
Using 584 and 686 genes mapped on 3p and 9q chromosomal regions, respectively, we investigated which of the encoded proteins potentially interact with PAX-2, PAX-8, CA-9, HIF-1, and GFAP. Relevant genes on chromosome 3p were: CTNNB1, CAND2, VHL, MIFT, WNT7A, PDCD6IP, TGFBR2, PRKCD, and MST1. Relevant genes on chromosome 9q were: LMX1B, GOLGA2, HSPA5, LCN2, RAD23B, TLR4, KLF4, and NOTCH1. The protein products of these genes are involved in cancer activation pathways like WNT, mTOR, HIF-1alpha, renal cell carcinoma, and p53 signaling, regulation of epithelial to mesenchymal transition, and neuroinflammation signaling (Additional file 1). Based on interactions with PAX-2, PAX-8, and CA-9 the main candidates for ELST tumorigenesis were VHL (on 3p), KLF4 (on 9q), and CTNNB1 (beta-catenin) (on 3p).
Treatment and follow-up
Treatment involved surgical resection of the mass in all except 2 cases (3 and 12) that underwent biopsy only (Fig. 3). None of the patients with complete medical records (15/19) received chemotherapy. Two patients received radiation therapy. One patient (case 9) received gamma knife radiotherapy to what was considered to be an intraoperative macroscopic glomus jugulare tumor (tissue was not sent to pathology), 8 and 5 years before a histologic diagnosis of ELST was rendered. Another patient (case 11) received radiation at initial diagnosis and at recurrence. Seven patients experienced recurrence of the ELST (Fig. 3), 4 of which required additional surgery (cases 6, 7, 13, and 19), and three of which underwent radiation therapy (cases 6, 11, and 18). One of the patients who experienced tumor recurrence passed away (case 7); however, it is not known with certainty whether this was related to disease or not. One patient was lost to follow-up after his initial resection (case 15) (Fig. 3).
Discussion
In this paper we present a cohort of patients with ELSTs and show that the majority of tumors immunoexpress renal cell markers. This novel finding is an important diagnostic caveat for general surgical pathology practice and we emphasize the importance of correlating the clinical pathological and radiological findings before rendering a final diagnosis for tumors involving the posterior temporal bone. In addition, we demonstrate combined loss of 3p (including the VHL locus) and 9q in the majority (58%, 7/12) of the tested cases by SNP-microarray analysis. These novel molecular findings suggest similar mechanistic origins between sporadic ELST and ccRCC with VHL likely playing a central role.
Immunoexpression of CA-9, PAX-8, and PAX-2 prove an important diagnostic caveat when attempting to rule out metastatic RCC in the work-up of suspected ELST. While these markers are more commonly associated with RCC, there are a number of syndromes that link the kidney to the inner ear suggesting certain embryological and functional similarities between the two organ systems [12, 36]. Torban et al. investigated the parallel functions between these two organ systems and categorized diseases affecting both the kidney and ear into groups that A) arose from mutations in shared developmental genes [e.g. Branchio-Oto-renal (BOT) syndrome, Hypoparathyroidism, Deafness, and Renal Dysplasia (HDR) syndrome, Townes-Brocks syndrome (TBS), Kallmann syndrome], B) involved defective ciliary function (e.g. Bardet-Biedl syndrome–associated hearing loss, Alstrom syndrome, nephronophthisis-associated inner ear dysfunction) and C) were due to disruption of specialized transport or structural proteins [e.g. Distal Renal Tubular Acidosis with Deafness (dRTA), Alport syndrome, Bartter syndrome with deafness] [36].
Paired box genes encode a family of transcription factors with roles in organogenesis [12]. PAX-8 activation is tightly linked to PAX-2 (Additional file 1) and these proteins are essential for proper ear [4] and kidney development [27]. In particular, PAX-2 has been shown to play a role in the induction of inner ear development and commitment of progenitor cells to the formation of the otic placode, the earliest structure identified in the morphogenesis of the inner ear, calling into question its anecdotic specificity for the kidney in the surgical pathology community [9]. PAX-2 is highly expressed in proliferating areas of the early developing inner ear (otic placode and otic vesicle) and is downregulated in areas of apoptosis at later stages of development and in maturing, differentiated hair cells [21, 22]. Its important role in cochlear development has been demonstrated using knockout mice [4, 5]. PAX-8 is one of the earliest markers for the ectodermally-derived otic placode and intermediate mesoderm, having a central role in auditory and urinary system development [12, 27]. It seems that in the neoplastic cells of ELSTs PAX-2 and PAX-8 become upregulated, reproducing the proliferative stages of early development. We speculate the role of an upstream activation mechanism which possibly involves VHL-KLF4-CTNNB1 protein interactions (Additional file 1). VHL is a tumor suppressor with known roles in tumorigenesis [7, 32]. VHL physically interacts with Krüppel-like factor 4 (KLF4), a transcription factor with roles in tumorigenesis [8]. Human KLF4 induces increased mouse PAX-2 and CTNNB1 mRNA expression [31] and, importantly, KLF4 binds to CTNNB1 inhibiting WNT signaling [13, 34]. In addition, it has been shown that gain-of-function CTNNB1 mutant protein promotes increased expression of PAX-8 mRNA in mice [28].
The enzyme carbonic anhydrase is distributed in a wide variety of organ systems, including the renal tubules and the inner ear. Within the ear, carbonic anhydrase has activity in the cochlear hair cells, supporting cells surrounding the sensory hair cells in the vestibule, in the stria vascularis, and in the epithelial cells of the rugose portion of the endolymphatic sac. It is thought to play a role in regulating the pH and ionic balance of the endolymph [15]. CA-9 is upregulated in hypoxic conditions and has been shown to play a role in tumorigenesis by altering pH to promote tumor growth and survival in a number of tumors, notably renal cell carcinoma [24, 29]. CA-9 expression is regulated by VHL. Briefly, under normal conditions VHL binds HIF-1alpha and degrades it, preventing its binding to HIF-1beta. Mutant VHL or normal VHL under hypoxic conditions facilitates HIF-1alpha binding to HIF1-beta and HIF-1 protein complex formation which causes downstream transcription of hypoxia-inducible genes such as CA-9. CA-9 is a marker of hypoxia [16, 29, 30, 32, 33]. CA-9 immunoexpression in ELST along with 3p loss provides supporting evidence of VHL deficiency in the mechanism of ELST development.
Furthermore, both syndromic and sporadic ELSTs have been shown to harbor mutations in the VHL gene [35, 38]; however, to the best of our knowledge molecular profiling of ELST has not yet been attempted [3, 18]. Our finding of 3p loss in ELST links it to ccRCC, which is characterized by variably sized deletions in the short arm of chromosome 3, including the VHL tumor suppressor gene [10, 11, 29]. Loss of 9q is also frequently seen in ccRCC, where it, along with 14q deletions, denote a poorer prognosis [10]. In our cohort, only one ELST patient had combined 3p/9q/14q losses but no tumor recurrence after 1.6 years of follow-up (Case 16). While 3p deletions in ccRCC may encompass the entire chromosome arm or only a small portion around the VHL locus, the 8 ELST cases in our cohort harboring 3p alterations showed whole p arm losses. Moreover, the 7 cases with concurrent 9q deletions showed large partial chromosomal losses of similar size among all tested tumors. From this, we can speculate the possibility of a derivative (3;9) resulting from an unbalanced translocation as the common mechanism of chromosomal alteration in ELST.
The molecular similarities between ccRCC and ELST may make the diagnosis of ELST even more challenging and we emphasize the importance of integrating the clinical presentation with the radiological features of this tumor. Certainly, the presence of 3p loss in both tumors suggests a similar mechanistic origin of tumorigenesis between the two entities that warrants further investigation. Furthermore, ELST may benefit from therapies targeting the same molecular pathways that lead to RCC development including HIF-1 and its targets such as VEGF, CA-9, and platelet derived growth factor (PDGF) [24, 29].
While this study is limited by small sample size due to the rarity of ELST, we believe the novelty of our findings and the implications for avoiding diagnostic pitfalls lend its strength. Additionally, the opportunity to better understand the pathogenesis of such a rare neoplasm as ELST make this a unique and meaningful investigation. Other potential limitations of the study include the small amount of tissue available for testing in some cases, the age of some samples impairing the quality of DNA, and the unavailability of tissue for more extensive molecular testing in several cases.
Although ELST are uncommon tumors, generally associated with VHL disease, the majority of cases in our cohort had no history of VHL disease or other associated neoplasms. While ELST may be the initial presentation of the disease, it is more likely that these are sporadic cases of ELST. As such, our findings may be generalizable to both sporadic and syndromic ELST cases. Furthermore, loss of 3p has been seen in a number of other human cancers and our findings of 3p loss in ELST further support the presence of one or more tumor suppressor genes including VHL in this region of the chromosome.
Conclusion
In conclusion, our findings dispel the previously reported misconception that ESLT are negative for expression of PAX-2, PAX-8, and CA-9. Likewise, copy number profiling will not help differentiate ELST from metastatic ccRCC. Based on our panel of immunohistochemical stains, CD10 and RCC may prove more useful in discriminating between the two entities. These noteworthy findings have important implications for the diagnosis, study, and possibly treatment of ELST and further investigation into the molecular pathways involved in tumorigenesis is warranted.
Abbreviations
- CA-9:
-
Carbonic anhydrase 9
- ccRCC:
-
clear cell renal cell carcinoma
- CNS:
-
Central nervous system
- CT:
-
Computed tomography
- ELST:
-
Endolymphatic sac tumor
- EMA:
-
Epithelial membrane antigen
- FFPE:
-
Formalin-fixed paraffin embedded
- GFAP:
-
Glial fibrillary acidic protein
- H&E:
-
Hematoxylin and eosin
- MRI:
-
Magnetic resonance imaging
- NSE:
-
Neuron specific enolase
- PAX:
-
Paired box
- PDGF:
-
Platelet derived growth factor
- RCC:
-
Renal cell carcinoma
- SNP:
-
Single nucleotide polymorphism
- VEGF:
-
Vascular endothelial growth factor
- VHL:
-
Von-Hippel-Lindau
References
Bagger-Sjöbäck D, Friberg U, Rask-Anderson H (1986) The human endolymphatic sac. An ultrastructural study. Arch Otolaryngol Head Neck Surg 112:398–409. https://doi.org/10.1001/archotol.1986.03780040038008
Bausch B, Wellner U, Peyre M, Boedeker CC, Hes FJ, Anglani M, de Campos JM, Kanno H, Maher ER, Krauss T et al (2016) Characterization of endolymphatic sac tumors and von Hippel-Lindau disease in the international endolymphatic sac tumor registry. Head Neck 38(Suppl 1):E673–E679. https://doi.org/10.1002/hed.24067
Bell D, Gidley P, Levine N, Fuller GN (2011) Endolymphatic sac tumor (aggressive papillary tumor of middle ear and temporal bone): sine qua non radiology-pathology and the University of Texas MD Anderson Cancer Center experience. Ann Diagn Pathol 15:117–123. https://doi.org/10.1016/j.anndiagpath.2010.08.009
Bouchard M, de Caprona D, Busslinger M, Xu P, Fritzsch B (2010) Pax2 and Pax8 cooperate in mouse inner ear morphogenesis and innervation. BMC Dev Biol 10:89. https://doi.org/10.1186/1471-213x-10-89
Burton Q, Cole LK, Mulheisen M, Chang W, Wu DK (2004) The role of Pax2 in mouse inner ear development. Dev Biol 272:161–175. https://doi.org/10.1016/j.ydbio.2004.04.024
Choyke PL, Glenn GM, Walther MM, Patronas NJ, Linehan WM, Zbar B (1995) von Hippel-Lindau disease: genetic, clinical, and imaging features. Radiology 194:629–642. https://doi.org/10.1148/radiology.194.3.7862955
Devaney KO, Ferlito A, Rinaldo A (2003) Endolymphatic sac tumor (low-grade papillary adenocarcinoma) of the temporal bone. Acta Otolaryngol 123:1022–1026
Gamper AM, Qiao X, Kim J, Zhang L, DeSimone MC, Rathmell WK, Wan Y (2012) Regulation of KLF4 turnover reveals an unexpected tissue-specific role of pVHL in tumorigenesis. Mol Cell 45:233–243. https://doi.org/10.1016/j.molcel.2011.11.031
Groves AK, Bronner-Fraser M (2000) Competence, specification and commitment in otic placode induction. Development 127:3489–3499
Hagenkord JM, Gatalica Z, Jonasch E, Monzon FA (2011) Clinical genomics of renal epithelial tumors. Cancer Genet 204:285–297. https://doi.org/10.1016/j.cancergen.2011.06.001
Hamilton HH, McDermott A, Smith MT, Savage SJ, Wolff DJ (2015) Clinical utility of concurrent single-nucleotide polymorphism microarray on fresh tissue as a supplementary test in the diagnosis of renal epithelial neoplasms. Am J Clin Pathol 144:731–737. https://doi.org/10.1309/ajcpjt7f5vnrxxpf
Heller N, Brandli AW (1999) Xenopus Pax-2/5/8 orthologues: novel insights into Pax gene evolution and identification of Pax-8 as the earliest marker for otic and pronephric cell lineages. Dev Genet 24:208–219. https://doi.org/10.1002/(sici)1520-6408(1999)24:3/4<208::Aid-dvg4>3.0.Co;2-j
Hoffmeyer K, Raggioli A, Rudloff S, Anton R, Hierholzer A, Del Valle I, Hein K, Vogt R, Kemler R (2012) Wnt/beta-catenin signaling regulates telomerase in stem cells and cancer cells. Science 336:1549–1554. https://doi.org/10.1126/science.1218370
Horiguchi H, Sano T, Toi H, Kageji T, Hirokawa M, Nagahiro S (2001) Endolymphatic sac tumor associated with a von Hippel-Lindau disease patient: an immunohistochemical study. Mod Pathol 14:727–732. https://doi.org/10.1038/modpathol.3880380
Hsu CJ, Nomura Y (1985) Carbonic anhydrase activity in the inner ear. Acta Otolaryngol Suppl 418:1–42
Ivanov SV, Kuzmin I, Wei MH, Pack S, Geil L, Johnson BE, Stanbridge EJ, Lerman MI (1998) Down-regulation of transmembrane carbonic anhydrases in renal cell carcinoma cell lines by wild-type von Hippel-Lindau transgenes. Proc Natl Acad Sci U S A 95:12596–12601
Karolchik D, Hinrichs AS, Furey TS, Roskin KM, Sugnet CW, Haussler D, Kent WJ (2004) The UCSC table browser data retrieval tool. Nucleic Acids Res 32:D493–D496. https://doi.org/10.1093/nar/gkh103
Kempermann G, Neumann HP, Volk B (1998) Endolymphatic sac tumours. Histopathology 33:2–10
Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D (2002) The human genome browser at UCSC. Genome Res 12:996–1006. https://doi.org/10.1101/gr.229102
Kramer A, Green J, Pollard J Jr, Tugendreich S (2014) Causal analysis approaches in ingenuity pathway analysis. Bioinformatics 30:523–530. https://doi.org/10.1093/bioinformatics/btt703
Kwan KY, Shen J, Corey DP (2015) C-MYC transcriptionally amplifies SOX2 target genes to regulate self-renewal in multipotent otic progenitor cells. Stem Cell Reports 4:47–60. https://doi.org/10.1016/j.stemcr.2014.11.001
Li H, Liu H, Corrales CE, Mutai H, Heller S (2004) Correlation of Pax-2 expression with cell proliferation in the developing chicken inner ear. J Neurobiol 60:61–70. https://doi.org/10.1002/neu.20013
Lo WW, Applegate LJ, Carberry JN, Solti-Bohman LG, House JW, Brackmann DE, Waluch V, Li JC (1993) Endolymphatic sac tumors: radiologic appearance. Radiology 189:199–204. https://doi.org/10.1148/radiology.189.1.8372194
McDonald PC, Winum JY, Supuran CT, Dedhar S (2012) Recent developments in targeting carbonic anhydrase IX for cancer therapeutics. Oncotarget 3:84–97. https://doi.org/10.18632/oncotarget.422
Megerian CA, Pilch BZ, Bhan AK, McKenna MJ (1997) Differential expression of transthyretin in papillary tumors of the endolymphatic sac and choroid plexus. Laryngoscope 107:216–221
Nancarrow DJ, Handoko HY, Stark MS, Whiteman DC, Hayward NK (2007) SiDCoN: a tool to aid scoring of DNA copy number changes in SNP chip data. PLoS One 2:e1093. https://doi.org/10.1371/journal.pone.0001093
Narlis M, Grote D, Gaitan Y, Boualia SK, Bouchard M (2007) Pax2 and pax8 regulate branching morphogenesis and nephron differentiation in the developing kidney. J Am Soc Nephrol 18:1121–1129. https://doi.org/10.1681/asn.2006070739
Park JS, Valerius MT, McMahon AP (2007) Wnt/beta-catenin signaling regulates nephron induction during mouse kidney development. Development 134:2533–2539. https://doi.org/10.1242/dev.006155
Rathmell WK, Chen S (2008) VHL inactivation in renal cell carcinoma: implications for diagnosis, prognosis and treatment. Expert Rev Anticancer Ther 8:63–73. https://doi.org/10.1586/14737140.8.1.63
Shen T, Shi Q, Velosa C, Bai S, Thompson L, Simpson R, Wei S, Brandwein-Gensler M (2015) Sinonasal renal cell-like adenocarcinomas: robust carbonic anhydrase expression. Hum Pathol 46:1598–1606. https://doi.org/10.1016/j.humpath.2015.06.017
Shu J, Wu C, Wu Y, Li Z, Shao S, Zhao W, Tang X, Yang H, Shen L, Zuo X et al (2013) Induction of pluripotency in mouse somatic cells with lineage specifiers. Cell 153:963–975. https://doi.org/10.1016/j.cell.2013.05.001
Sommaruga SAOV (2014) Neuropathology of von Hippel Lindau disease. J Transl Med Epidemiol 2:1011
Stillebroer AB, Mulders PF, Boerman OC, Oyen WJ, Oosterwijk E (2010) Carbonic anhydrase IX in renal cell carcinoma: implications for prognosis, diagnosis, and therapy. Eur Urol 58:75–83. https://doi.org/10.1016/j.eururo.2010.03.015
Thylur RP, Senthivinayagam S, Campbell EM, Rangasamy V, Thorenoor N, Sondarva G, Mehrotra S, Mishra P, Zook E, Le PT et al (2011) Mixed lineage kinase 3 modulates beta-catenin signaling in cancer cells. J Biol Chem 286:37470–37482. https://doi.org/10.1074/jbc.M111.298943
Tibbs RE Jr, Bowles AP Jr, Raila FA, Fratkin JD, Hutchins JB (1997) Should endolymphatic sac tumors be considered part of the von Hippel-Lindau complex? Pathology case report. Neurosurgery 40:848–855 discussion 855
Torban E, Goodyer P (2009) The kidney and ear: emerging parallel functions. Annu Rev Med 60:339–353. https://doi.org/10.1146/annurev.med.60.052307.120752
Virk JS, Randhawa PS, Saeed SR (2013) Endolymphatic sac tumour: case report and literature review. J Laryngol Otol 127:408–410. https://doi.org/10.1017/s0022215113000327
Vortmeyer AO, Huang SC, Koch CA, Governale L, Dickerman RD, McKeever PE, Oldfield EH, Zhuang Z (2000) Somatic von Hippel-Lindau gene mutations detected in sporadic endolymphatic sac tumors. Cancer Res 60:5963–5965
Wenig BM (2016) Atlas of head and neck pathology. Philadelphia: Elsevier Inc.
Funding
This research was funded by an intramural grant from the MUSC Department of Pathology and Laboratory Medicine to support research by residents and clinical fellows.
Author information
Authors and Affiliations
Contributions
Study conception and design: AO. Data collection/organization: RJ, IZ, MG, SP, TT, MA, CMH, MR, DJW, RL, WM, FJR, JM, AO. Bench work and experiments: RJ, IZ, MG, JNR, RK. Data analysis: RJ, IZ, AO. Writing of manuscript: RJ, AO. Critical manuscript review: IZ, MG, JNR, RK, SP, TT, MA, CMH, MR, DJW, RL, WM, FJR, JM. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This research was approved by the institutional review board with a waiver of informed consent.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Pathway and protein interactions in endolymphatic sac tumor. (PDF 52855 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Jester, R., Znoyko, I., Garnovskaya, M. et al. Expression of renal cell markers and detection of 3p loss links endolymphatic sac tumor to renal cell carcinoma and warrants careful evaluation to avoid diagnostic pitfalls. acta neuropathol commun 6, 107 (2018). https://doi.org/10.1186/s40478-018-0607-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-018-0607-0