
Abstract
The new WHO classification of adrenal cortical proliferations reflects translational advances in the fields of endocrine pathology, oncology and molecular biology. By adopting a question–answer framework, this review highlights advances in knowledge of histological features, ancillary studies, and associated genetic findings that increase the understanding of the adrenal cortex pathologies that are now reflected in the 2022 WHO classification. The pathological correlates of adrenal cortical proliferations include diffuse adrenal cortical hyperplasia, adrenal cortical nodular disease, adrenal cortical adenomas and adrenal cortical carcinomas. Understanding germline susceptibility and the clonal-neoplastic nature of individual adrenal cortical nodules in primary bilateral macronodular adrenal cortical disease, and recognition of the clonal-neoplastic nature of incidentally discovered non-functional subcentimeter benign adrenal cortical nodules has led to redefining the spectrum of adrenal cortical nodular disease. As a consequence, the most significant nomenclature change in the field of adrenal cortical pathology involves the refined classification of adrenal cortical nodular disease which now includes (a) sporadic nodular adrenocortical disease, (b) bilateral micronodular adrenal cortical disease, and (c) bilateral macronodular adrenal cortical disease (formerly known primary bilateral macronodular adrenal cortical hyperplasia). This group of clinicopathological entities are reflected in functional adrenal cortical pathologies. Aldosterone producing cortical lesions can be unifocal or multifocal, and may be bilateral with no imaging-detected nodule(s). Furthermore, not all grossly or radiologically identified adrenal cortical lesions may be the source of aldosterone excess. For this reason, the new WHO classification endorses the nomenclature of the HISTALDO classification which uses CYP11B2 immunohistochemistry to identify functional sites of aldosterone production to help predict the risk of bilateral disease in primary aldosteronism. Adrenal cortical carcinomas are subtyped based on their morphological features to include conventional, oncocytic, myxoid, and sarcomatoid subtypes. Although the classic histopathologic criteria for diagnosing adrenal cortical carcinomas have not changed, the 2022 WHO classification underscores the diagnostic and prognostic impact of angioinvasion (vascular invasion) in these tumors. Microscopic angioinvasion is defined as tumor cells invading through a vessel wall and forming a thrombus/fibrin-tumor complex or intravascular tumor cells admixed with platelet thrombus/fibrin. In addition to well-established Weiss and modified Weiss scoring systems, the new WHO classification also expands on the use of other multiparameter diagnostic algorithms (reticulin algorithm, Lin–Weiss–Bisceglia system, and Helsinki scoring system) to assist the workup of adrenal cortical neoplasms in adults. Accordingly, conventional carcinomas can be assessed using all multiparameter diagnostic schemes, whereas oncocytic neoplasms can be assessed using the Lin–Weiss–Bisceglia system, reticulin algorithm and Helsinki scoring system. Pediatric adrenal cortical neoplasms are assessed using the Wieneke system. Most adult adrenal cortical carcinomas show > 5 mitoses per 10 mm2 and > 5% Ki67. The 2022 WHO classification places an emphasis on an accurate assessment of tumor proliferation rate using both the mitotic count (mitoses per 10 mm2) and Ki67 labeling index which play an essential role in the dynamic risk stratification of affected patients. Low grade carcinomas have mitotic rate of ≤ 20 mitoses per 10 mm2, whereas high-grade carcinomas show > 20 mitoses per 10 mm2. Ki67-based tumor grading has not been endorsed in the new WHO classification, since the proliferation indices are continuous variables rather than being static thresholds in tumor biology. This new WHO classification emphasizes the role of diagnostic and predictive biomarkers in the workup of adrenal cortical neoplasms. Confirmation of the adrenal cortical origin of a tumor remains a critical requirement when dealing with non-functional lesions in the adrenal gland which may be mistaken for a primary adrenal cortical neoplasm. While SF1 is the most reliable biomarker in the confirmation of adrenal cortical origin, paranuclear IGF2 expression is a useful biomarker in the distinction of malignancy in adrenal cortical neoplasms. In addition to adrenal myelolipoma, the new classification of adrenal cortical tumors has introduced new sections including adrenal ectopia, based on the potential role of such ectopic tissue as a possible source of neoplastic proliferations as well as a potential mimicker of metastatic disease. Adrenal cysts are also discussed in the new classification as they may simulate primary cystic adrenal neoplasms or even adrenal cortical carcinomas in the setting of an adrenal pseudocyst.
Similar content being viewed by others

Introduction
The new WHO classification of adrenal cortical proliferations reflects translational advances in the fields of endocrine pathology, endocrine oncology, and molecular biology and recognizes the importance of structural and functional correlations. A group of clinicopathological entities are reflected in functional and non-functional adrenal cortical pathologies. By adopting a practical question–answer framework, this review highlights advances in knowledge of histological features, ancillary studies, and associated genetic findings that increase the understanding of adrenal cortex pathologies that are now reflected in the 2022 WHO classification.
Question 1: Are There Any Nomenclature Changes or Any New Diagnostic Categories in the New WHO Classification of Adrenal Cortical Proliferations?
The pathological correlates of adrenal cortical proliferations include diffuse adrenal cortical hyperplasia, adrenal cortical nodular disease, adrenal cortical adenomas and adrenal cortical carcinomas [1].
In the new 5th edition of the WHO classification of adrenal cortical disease, the most significant nomenclature change in adrenal cortical pathology is the refined classification of adrenal cortical nodular disease (Fig. 1). The 2022 WHO classification no longer endorses the use of “nodular adrenal cortical hyperplasia” for incidentally discovered sporadic non-functional adrenal cortical nodules. The latter stems from recognition of the clonal/neoplastic nature of incidentally discovered non-functional subcentimeter benign adrenal cortical nodules [1,2,3,4,5]. The new classification endorses the term sporadic nodular adrenocortical disease for such manifestations (Fig. 2). Historical terms of primary bilateral micronodular or macronodular adrenocortical hyperplasia are no longer used as the nodules are independent clonal proliferations and referred to as bilateral micro- or macro-nodular adrenal cortical disease and classified among benign adrenal cortical tumors [1,2,3,4,5]. Virtually all bilateral micronodular and a significant fraction of bilateral macronodular adrenal cortical diseases are caused by genetic susceptibility [4,5,6,7,8,9,10,11,12,13,14]. These findings have resulted in redefining the clinicopathologic spectrum of adrenal cortical nodular disease. Consistently, the new WHO classification subtypes adrenal cortical nodular disease as: (a) sporadic nodular adrenocortical disease, (b) bilateral micronodular adrenal cortical disease, and (c) bilateral macronodular adrenal cortical disease (Fig. 1). The distinction between these diagnostic categories reflects diverse clinical manifestations [4, 5, 15].

The new WHO classification of adrenocortical nodular disease (Created with BioRender.com)

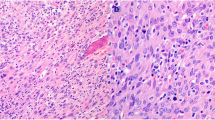
Sporadic nodular adrenocortical disease. This photomicrograph illustrates a sporadic nodular adrenocortical disease The green circle outlines the lesion
In the 2022 WHO classification, apart from diffuse compact cell adrenal cortical hyperplasia identified in adrenal glands and adrenal rests from patients with congenital adrenal hyperplasia, the diagnostic category of adrenal cortical hyperplasia is now restricted to bilateral diffuse adrenal cortical hyperplasia, which can be driven by a pituitary corticotroph tumor (corticotroph pituitary neuroendocrine tumor) or hypothalamic neoplasms or ectopically with ACTH- or CRH-dependent pathogenesis (e.g., ACTH- or CRH-producing neuroendocrine neoplasm, paraganglioma) [1, 15]. Pituitary ACTH-dependent diffuse adrenal cortical hyperplasia results in diffuse expansion of the adrenal cortex in both adrenal glands with preserved adrenal cortical zonation [15], while ectopic ACTH-driven adrenal cortical hyperplasia lacks distinct adrenal cortical zonation due to diffuse compact cell hyperplasia resulting in a pink appearance to the adrenal cortices due to lipid depletion [15].
Another significant change in the new WHO classification is the pathologic evaluation of adrenal glands removed for primary aldosteronism. As primary aldosteronism may be due to a single lesion or multiple lesions involving one or both adrenal glands, the new WHO classification endorses the nomenclature of the HISTALDO classification (Fig. 3), which uses CYP11B2 immunohistochemistry to identify functional sites of aldosterone production which help predict the risk of bilateral disease in primary aldosteronism [16,17,18,19,20,21].

HISTALDO classification scheme: graphic depiction of the HISTALDO classification model. Aldosterone producing adrenal cortical carcinoma (APACC) and adenoma (APA) are solitary lesions clearly visible by both routine hematoxylin–eosin (H&E) and immunohistochemical (IHC) staining for CYP11B2 (aldosterone synthase). Smaller solitary lesions (sub-centimeter) visible by H&E and IHC are denoted aldosterone producing nodules (APNs), while the counterpart that may be hard to distinguish using H&E but always visualized on IHC are entitled aldosterone producing micronodules (APMs) (formerly known as aldosterone producing cell clusters). When multifocal, these entities are termed “multiple APN” (MAPN) and “multiple APM” (MAPM), respectively—corresponding to the older term “micronodular hyperplasia.” Finally, aldosterone producing diffuse hyperplasia is characterized by a continuous CYP11B2 staining along the zona glomerulosa. Image created with BioRender.com. Reproduced from Juhlin CC, Bertherat J, Giordano TJ, Hammer GD, Sasano H, Mete O. What Did We Learn from the Molecular Biology of Adrenal Cortical Neoplasia? From Histopathology to Translational Genomics. Endocr Pathol. 2021 Mar;32(1):102–133
The new WHO bluebook also covers three new separate chapters on adrenal cysts, adrenal ectopia, and myelolipoma. The details of most of the new sections and other updates are discussed in questions of this review.
Question 2: Why Do Pathologists Need to Know Ectopic or Heterotopic Locations for Adrenal Cortical Tissue?
The 2022 WHO classification of adrenal cortical tumors has a new chapter on adrenal ectopia, based on the potential role of ectopic tissue as the source of neoplastic proliferations as well as a potential mimicker of metastatic disease. For practicing pathologists, it is helpful to know the existence, location and main histological features of these conditions. Adrenal ectopia has been variably addressed as adrenal heterotopia, rests, remnants or choristoma.
Adrenal ectopia is defined as isolated benign adrenal cortical tissue outside orthotopic adrenal glands. Most adrenal rests have been identified in anatomic locations that are aligned with their embryologic developmental tract. Adrenal cortical choristoma is a related term to adrenal ectopia and is defined as mature adrenal cortical elements whose location cannot be explained with the normal embryogenesis of adrenal cortex. Adrenal rests may become clinically relevant when they are associated with disorders such as congenital adrenal hyperplasia. Adrenal rest tumor is a term generally restricted to nodular proliferations in the gonads, and it is not a term for ectopic adrenal cortical tissue.
In surgical pathology, most adrenal rests are discovered as incidental microscopic findings in various surgical specimens including kidneys or gonads (particularly, spermatic cord [22], rete testis [23,24,25], epididymis [26], ovary/paraovarian [22, 27,28,29,30,31,32,33], salpinx and parasalpinx [22, 34, 35] ). A recent surgical series from adults identified ectopic adrenal rests in both males and females with a more frequent presentation in para-ovarian tissues [22]. Rare locations of adrenal rests include the placenta [36], broad ligament [22, 37, 38], retroperitoneum [39, 40], urinary bladder and prostate [41], groin [42, 43], choroid plexus [44], spine [45,46,47], and lung [48, 49]. Among various locations, adrenal cortical choristomas have also been described within TPIT-lineage pituitary neuroendocrine tumors (corticotroph PitNETs) [50, 51].
Adrenal cortical rests are characterized by small subcentimeter, isolated, yellow nodules in the fibrofatty tissue surrounding normal components at the individual location [22,23,24, 52, 53]. Histologically, aggregates of large eosinophilic or clear cells, with no or bland atypia resembling orthotopic adrenal cortical cell populations are observed (Fig. 4). Conversely, the medullary component of the adrenal is rarely observed, and generally restricted to cases found in the ectopias surrounding normal adrenal medulla, with even rarer exceptions (e.g., a mediastinal location of functioning cortical and medullary cells) [54].

Adrenal ectopia. This composite photomicrograph illustrates two examples of ectopic adrenal cortical tissue involving the fibroadipose tissue surrounding the spermatic cord (A) and the para-salpinx (B). The inset on photomicrograph B represents Melan A expression in the adrenal cortical cells
Adrenal cortical choristomas are composed of adrenal cortical cells with bland cytomorphology, and they occur either isolated or in aggregates within normal or neoplastic cell populations (e.g., corticotroph PitNET) of the involved organ [50, 51]. The mechanism underlying their development is incompletely understood. It may be the result of misplacement of adrenal tissue far from its anatomic location during embryogenesis [50, 51]. Alternatively, at least in pituitary locations, a role of SF1 (a transcription factor implicated in both pituitary and adrenal cortex functions), might be envisaged, possibly acting on uncommitted stem cells within the sella [51].
Rarely, adrenal cortical neoplasms arise from ectopic adrenal tissues. Adenomas of adrenal cortical origin have been reported in the liver [55] and the gastric wall [56]. In addition, prolonged hormonal hyperstimulation can induce adrenal cortical cell hyperplasia-to-neoplasia sequence and may result in “adrenal rest tumors.” Adrenal ectopic tissue must be distinguished from Leydig cell and other steroid producing gonadal tumors and non-steroidogenic neoplasms (e.g., renal cell carcinoma and hepatocellular carcinoma) [23, 24, 53, 57, 58].
Adrenal cortical rests and choristomas share immunohistochemical features of adrenal cortical cells as they express SF1 and other non-specific biomarkers of adrenal cortical differentiation [22, 51, 59, 60]. The distinction of adrenal cortical rest-related proliferations from other steroidogenic neoplasms poses a diagnostic challenge and may require the use of adrenal cortex-related steroidogenic enzyme immunohistochemistry as discussed in Question 10.
Question 3: What are the Pathological Correlates of Adrenal Cysts?
The new WHO classification has introduced a new chapter on adrenal cysts, based on their potential of simulating primary cystic adrenal neoplasms or even adrenal cortical carcinomas in the setting of an adrenal pseudocyst. For practicing pathologists, it is therefore relevant to be aware of their different types, main histological features and relationships with cystic neoplasms. Adrenal cysts are defined as benign, circumscribed, fluid-containing masses or nodules. They are rare with a reported incidence of 0.06% [61]. They are usually detected as incidental findings at the time of imaging studies, and account for up to 4% of adrenal masses [62,63,64,65]. A slight female predilection and a mean cyst size of 7.9 cm have been reported in a review of the literature [62].
There are four distinct types of adrenal cysts: (a) pseudocyst, (b) endothelial (vascular cyst), (c) epithelial cyst, and (d) parasitic cyst [62, 63, 65,66,67] These are associated with different etiologies and pathogenetic mechanisms. The most common adrenal cysts are pseudocysts accounting for up to 80% of cases in some series and probably resulting from hemorrhagic or traumatic events of various etiologies in the adrenal area, including COVID-19-related hemorrhage [62, 63, 66]. While parasitic cysts are related to Echinococcus infections, endothelial cysts develop from dilated and thrombosed blood or lymphatic vessels, and epithelial cysts are associated to mesothelial remnants [63]. Irrespective of the subtype and origin they may reach relevant size and mimic a tumor [62, 68, 69].
Histologically, adrenal cysts can be relatively easily classified, based on the cyst wall structure. First, the presence of a cell lining is to be assessed. In its absence, an adrenal pseudocyst is considered, generally containing a fibrino-hemorrhagic fluid and delimited by a more or less thickened fibrous tissue [62, 69] (Fig. 5). When a lining is detected, it is generally a monolayer of flat endothelial (Fig. 6) or mesothelial cells (Fig. 7). The former is related to lymphatic rather than blood endothelium and occasional intraluminal protrusions can be observed [61, 62]. The latter are flat or cuboidal mesothelial cells, devoid of atypia. Parasitic cysts can reach remarkable size [68] and contain clear fluid in single or multiple cystic spaces and a fibro-calcific wall with PAS positive parasitic membranes [68, 70, 71] (Fig. 8). The described histological features generally do not pose diagnostic problems with neoplastic conditions, in the absence of solid areas and/or cell atypia. However, extensive sampling of the cyst wall is recommended to exclude an associated neoplasm. This is particularly important as majority of grossly cystic adrenal glands are adrenal cortical neoplasms or pheochromocytoma with cystic hemorrhagic degeneration [65].

Adrenal pseudocyst: the adrenal tissue (superior part of the photomicrograph) is compressed by a mass demarcated by a thick fibrous wall and containing fibrino-hemorrhagic material

Vascular (endothelial) cyst: a multiloculated cyst contains red blood cells and is lined by bland endothelial cells (A) that are positive for CD31 (B)

Adrenal epithelial (mesothelial) cyst: A uniloculated cyst has a thin fibrous wall and is lined by a single layer of flat cuboidal cells (A) that are positive for calretinin (B)

Parasitic cyst: a large cyst is observed in the periadrenal adipose tissue, compressing the gland and containing amorphous and lamellar material (A). Manifestations restricted to the adrenal parenchyma also occur. Parasitic membranes are better highlighted by PAS staining at higher power (B)
In specific instances, particularly for epithelial and endothelial cysts, immunohistochemical markers may help classify the cyst. Conventional mesothelial (calretinin, WT1) and endothelial (D2-40, CD31, ERG) markers can readily define the epithelial/mesothelial or endothelial origin, respectively [72] (Figs. 6 and 7). Pseudocysts and parasitic cysts generally do not require specific immunoprofiling.
Several primary and metastatic tumors in the adrenal gland may develop cystic changes, thus entering in the differential diagnosis with adrenal cysts and pseudocysts. These include cystic regressive and hemorrhagic degenerative changes in adrenal cortical adenoma, adrenal cortical carcinoma, pheochromocytoma, hemangioma, lymphangioma, angiosarcoma, vascular malformations, renal cell carcinoma, mature teratomas, ectopic thyroid nodules, and even in metastases [62, 63, 65, 67, 73,74,75]. The morphological distinction is generally straightforward in the presence of a conventional histological component of the individual neoplasms. However, a source of confusion is represented by the rare co-occurrence of a true cystic lesion with an adrenal neoplasm, mostly adrenal cortical adenoma [76,77,78], or, even more rarely, with aldosterone-producing micronodules [79].
Question 4: What are the Pathological Correlates of Adrenocortical Nodular Disease in the 2022 WHO Classification?
The new WHO classification classifies adrenocortical nodular disease into 3 categories: (a) sporadic nodular adrenocortical disease, (b) bilateral micronodular adrenocortical disease, and (c) bilateral macronodular adrenocortical disease (Fig. 1). While sporadic nodular adrenocortical disease may be non-hormone producing and occur as unilateral or bilateral adrenal manifestations, the bilateral micro- and macronodular forms usually contribute to hypercortisolism and involve both adrenal glands. The latter two entities are often associated with germline variants in specific susceptibility genes, making them important to identify for appropriate patient follow-up and further genetic counseling [4].
Sporadic nodular adrenocortical disease may be discovered incidentally via imaging in patients of all age groups and is composed of non-hormone producing adrenal cortical nodules measuring < 1 cm [1] (Fig. 2). The underlying etiology is largely unknown but they likely represent non-functional adrenal cortical microadenomas. Sporadic nodular adrenal cortical disease can be solitary or multifocal with a sparse distribution. The histology is usually similar to that of non-functional adrenal cortical adenoma, and if several nodules are discovered in a patient with a known functional adenoma, the distinction of functional adrenal cortical adenoma(s) (irrespective of their size) from sporadic nodular adrenocortical disease requires functional immunohistochemistry targeting enzymes of hormone production, such as CYP11B1, CYP11B2, and CYP17 [18, 80].
Bilateral micronodular or macronodular adrenocortical disease (Figs. 9, 10, 11, and 12) is much more infrequently observed than sporadic nodular adrenocortical disease, and it is estimated that the two former entities only account for approximately 2% of all patients with endogenous hypercortisolism [81, 82]. Bilateral micronodular adrenocortical nodular disease mainly occur in children and young adults, with the majority of cases being female [81,82,83,84]. Radiology may underestimate the presence of bilateral adrenal gland involvement in these patients, which is probably due to the discrete nodular formations (sizes < 1 cm) [1, 83] (Fig. 9). In this aspect, functional imaging using cholesterol-based scintigraphy techniques may be of value [85].

Gross findings in a primary pigmented micronodular adrenocortical disease (PPNAD). PPNAD is composed of multiple bead-like pigmented adrenal cortical micronodules (< 10 mm, often 2–5 mm) throughout the adrenal cortex

Primary pigmented adrenocortical nodular disease (PPNAD). PPNAD is characterized by multiple, subcentimeter micronodules that are composed of eosinophilic adrenal cortical cells with variable pigment deposition (A, inset). Inter-nodular cortical atrophy is usually observed (A). The micronodules are positive for CYP11B1, confirming cortisol production (B)

Gross findings in a bilateral macronodular adrenocortical disease. Bilateral macronodular adrenocortical disease is often associated with multiple lipid-rich adrenal cortical macronodules (> 10 mm) that result in marked adrenal gland enlargement

Bilateral macronodular adrenocortical disease. This photomicrograph illustrates a bilateral macronodular adrenocortical disease in a patient with a pathogenic ARMC5 germline variant leading to Cushing syndrome. Depicted here is the resected left adrenal, displaying multiple clear cell-rich nodules intermingling with eosinophilic cells, each nodule > 1.0 cm. The inset shows clear cell adrenal cortical cells
The new WHO classification describes two types of bilateral micronodular disease: (a) primary pigmented nodular adrenocortical disease (PPNAD) (Figs. 9 and 10) and (b) isolated micronodular adrenocortical disease (i-MAD) [1, 4, 5, 15]. Patients with these conditions have multiple small adrenocortical nodules on histology, each measuring < 1 cm [1, 5]. These micronodules are often cortisol-producing and composed of lipid-poor cortical cells normally located to the zona fasciculata or to the zona fasciculata-reticularis junction [5]. Primary pigmented adrenocortical disease (PPNAD) is reported in most individuals with Carney complex [83, 86], and hence, this syndrome should be suspected and ruled in/out in patients with PPNAD. However, it should be stressed that except for Carney complex-associated PPNAD (entitled “c-PPNAD”), PPNAD can also occur in patients without this syndrome (denoted as “isolated PPNAD; i-PPNAD) [4, 15, 87]. The entity i-MAD is very rarely encountered in clinical practice, and the distinction between i-MAD and PPNAD is based on histological findings, in which the latter entity displays multiple adrenocortical nodules with cytoplasmic pigmentation and inter-nodular cortical atrophy (Fig. 10), while the pigmentation and inter-nodular adrenal cortical atrophy are generally absent in i-MAD [4, 5, 15, 81, 88]. Most patients with bilateral micronodular adrenocortical disease carry pathogenic germline variants in genes normally associated with the regulation of the protein kinase A (PKA) pathway involved in the physiological response to ACTH stimulation [4, 5]. For c-PPNAD and i-PPNAD, the most frequently mutated gene is PRKAR1A, whereas a subset of patients with i-PPNAD and i-MAD may exhibit constitutional PRKACA copy number gain. Moreover, individuals with i-MAD may display variants in PDE8B and PDE11A, which will also cause an increase in PKA signaling [5, 81].
Bilateral macronodular adrenocortical disease (formerly known as “primary bilateral macronodular adrenal cortical hyperplasia”) is most often diagnosed in adults, although rare pediatric cases have been reported [4, 5, 15, 81]. More than 90% of individuals with bilateral macronodular adrenocortical disease have varying degrees of endogenous hypercortisolism, but a few are non-producing [9]. Adrenal imaging usually detects massive bilateral adrenal enlargement [1, 89] (Fig. 11). Histologically, bilateral macronodular adrenocortical hyperplasia is composed of numerous nodules measuring > 1 cm each (Fig. 12). The nodules are by lipid-rich (clear) cells with occasional eosinophilic cells [4, 5, 15, 90]. Via next-generation sequencing studies, we now know that this entity is caused by germline variants in one out of several susceptibility genes, often with a somatic-type “second hit” on the trans allele, thereby strongly arguing for a neoplastic condition rather than a “hyperplastic” disease, the latter which is a misnomer in this aspect. Of note, constitutional ARMC5 gene variants are the most commonly reported aberration in this context, occurring in 25–55% of cases [9, 10]. Moreover, germline variants in MEN1 (causing the multiple endocrine neoplasia type 1 syndrome) [7, 13, 91], FH (causing the hereditary leiomyomatosis and renal cell cancer syndrome) [12, 92], and APC (causing familial adenomatosis polyposis) [11, 12] have also been reported in individuals with bilateral macronodular adrenocortical disease. Moreover, additional somatic and/or germline gene variants have also been identified in a subset of individuals with this condition [5, 7, 8, 12, 14, 93]. Intricate molecular mechanisms, for example aberrant G-protein-coupled receptor expression or dysregulation of the ACTH receptor may explain subsets of cases [94,95,96,97,98].
For individuals with endogenous hypercortisolism, the new WHO classification restricts the terminology of “adrenal cortical hyperplasia” to ACTH-dependent diffuse adrenal cortical hyperplasia in which the cortical zonation is intact, but usually shows an expansion of the zona reticularis layer [1]. Since bilateral micronodular or macronodular adrenal cortical disease is a collective term describing the development of several adrenocortical lesions that often are driven by germline variants usually associated with the protein kinase A pathway, the term “hyperplasia” should be reserved for a physiological response to increased ACTH levels rather than arbitrarily used for multifocal nodules driven by clonal expansions.
Question 5: What is New in the Classification of Primary Aldosteronism in the 2022 WHO Classification of Adrenal Cortical Tumors?
Primary aldosteronism (PA) is a leading cause of secondary hypertension and is characterized by aldosterone overproduction with supressed renin-angiotensin system. Hypokalemia may not occur in some patients [99]. The laboratory diagnosis of primary aldosteronism is typically based on elevated aldosterone-to-renin ratio [99].
The histopathological correlates of primary aldosteronism include (a) aldosterone-producing bilateral diffuse hyperplasia, (b) aldosterone-producing adrenal cortical adenomas (including microscopic nodular lesions, which can be bilateral and/or multifocal), and (c) aldosterone-producing adrenal cortical carcinoma [4, 100]. Aldosterone producing diffuse hyperplasia and (micro)nodules are the most common clinical manifestations that cause bilateral primary aldosteronism [4, 5]. Since aldosterone-producing adrenal cortical carcinomas are exceptionally rare [101, 102], aldosterone-producing adrenal cortical adenomas are the most frequent pathologic correlates of unilateral primary aldosteronism [4, 99].
Aldosterone is produced in the zona glomerulosa by aldosterone synthase (CYP11B2, cytochrome P450 family 11, subfamily B, member 2). The production of specific monoclonal antibodies against CYP11B2 [103] and its immunolocalization in resected adrenals has refined our understanding of the morphologic spectrum of this disorder [16]. This approach also helps us appreciate the link between CYP11B2-positive adrenal cortical lesions and somatic ion channel mutations leading to increased intracytoplasmic calcium levels that lead to autonomous CYP11B2 transcription [4, 5, 104, 105] (see Question 11).
The progress in the field of primary aldosteronism has also brought opportunities to address reporting of the histological entities that fall into the spectrum of aldosterone-producing adrenal cortical proliferations [16]. The recently proposed HISTALDO classification introduced a simplified approach by combining CYP11B2 immunohistochemistry and morphological findings to define clinically relevant diagnostic categories [16] (Fig. 3). This approach has been shown to predict better the risk of biochemical recurrence despite preoperative diagnostic workup [16]. The 2022 WHO classification also endorses the use of this approach to ensure accurate distinction of aldosterone-producing cortical lesions. The diagnostic categories in the HISTALDO classification include (a) aldosterone-producing adrenal cortical carcinoma (APACC), (b) aldosterone-producing adrenal cortical adenoma (APA), (c) aldosterone-producing nodule (APN), (d) aldosterone-producing micronodule (APM), (e) multifocal APN and/or APM, (f) aldosterone-producing diffuse hyperplasia (APDH).
Aldosterone-producing adrenal cortical carcinoma (APACC)
This rare diagnostic category is applied to an adrenal cortical carcinoma that is positive for CYP11B2 [16].
Aldosterone-producing adrenal cortical adenoma (APA)
APA is a CYP11B2-positive benign adrenal cortical neoplasm that measures ≥ 1 cm. It is composed of clear (zona fasciculata-like cells) and compact (zona reticularis-like) adrenal cortical cells [16] (Fig. 13).

Aldosterone producing adrenal cortical adenoma. This photomicrograph an aldosterone-producing adrenal cortical adenoma composed of lipid-rich adrenal cortical cells (A). The tumor cells are diffusely positive for CYP11B2 (B)
Aldosterone-producing nodule (APN)
APN is a morphologically distinguished and CYP11B2-positive benign adrenal cortical lesion that measures < 1 cm (Fig. 14). This lesion represents a form of aldosterone-producing microadenoma at a molecular level; however, it is distinguished from APA by the gradient CYP11B2 reactivity from the external to internal fronts of the proliferation [16, 106, 107].

Aldosterone-producing nodule. This composite photomicrograph illustrates a morphologically distinct well-delineated adrenal cortical nodule (N) enriched in lipid-rich adrenal cortical cells (A). This nodule measures 1.3 mm. CYP11B2 shows a gradient reactivity with a more intense reactivity at the outer part of the nodule (B)
Aldosterone-producing micronodule (APM) (formerly known aldosterone producing cell clusters; APCC)
APM is a CYP11B2-positive benign adrenal cortical lesion that measures < 1 cm (often few millimetres) (Fig. 15). APMs are composed exclusively of zona glomerulosa-like adrenal cortical cells underneath the adrenal capsule. APMs may be difficult to distinguish on hematoxylin and eosin-stained sections; thus, they are typically distinguished using CYP11B2 immunohistochemistry. At a molecular level, APMs also represent a form of aldosterone-producing micro-adenomas given the high-frequency of ion channel mutations [4, 5]. Similar to APN, a gradient of CYP11B2 reactivity from the external to internal fronts of the cellular proliferation occurs in APM [16].

CYP11B2 immunohistochemistry in the distinction of aldosterone-producing diffuse hyperplasia from multiple aldosterone-producing micronodules. Diffuse hyperplasia (DH) of the zona glomerulosa (ZG) is defined as diffuse continuous hyperplasia of CYP11B2-positive ZG (A). This pattern may also contain areas of micronodules (A). Multiple aldosterone-producing micronodules are characterized by CYP11B2-expressing micronodules in the absence of continuous (linear) ZG layer (B). Reprinted from Mete O, Asa SL, Giordano TJ, Papotti M, Sasano H, Volante M. Immunohistochemical Biomarkers of Adrenal Cortical Neoplasms. Endocr Pathol. 2018 Jun;29(2):137–149
Multifocal APN and/or APM
This category refers to the occurrence of synchronous multifocal APN and/or APM in an adrenal gland (Fig. 15).
Aldosterone-producing diffuse hyperplasia (APDH)
The HISTALDO classification defines APDH as a relatively broad and uninterrupted linear strip of zona glomerulosa cells with more than 50% of these cells showing CYP11B2 reactivity [16] (Fig. 15). This is the most common cause of bilateral idiopathic primary aldosteronism that often requires lifelong anti-mineralocorticoid therapy [4, 5, 99]. The so-called “paradoxical zona glomerulosa layer hyperplasia,” which is typically seen in the non-lesional adrenal cortex adjacent to APAs and APNs, should not be mistaken for diffuse hyperplasia. Unlike the diffuse hyperplasia, the paradoxical hyperplasia is negative for CYP11B2 [4, 5]. The latter finding supports the non-functional status of the paradoxical zona glomerulosa layer hyperplasia.
Based on the HISTALDO classification, the identification of a solitary APA or APN refers to a classic histology whereas adrenals with APDH, multifocal APN and/or APM are considered to have non-classic histology [16]. This distinction is of clinical significance since biochemical disease recurrence (due to bilateral disease) occurred in around 42% of patients with non-classic histology findings, compared to less than 5% in patients with classic histology [16].
Question 6: What are the Pathological Correlates of Adrenal Cortical Adenomas in the 2022 WHO Classification?
Adrenal cortical adenoma is a neoplasm of adrenocortical cell derivation that lacks morphologic features of malignancy. Adrenal cortical adenomas are characterized by heterogeneity of pathological and clinical presentations; thus, their characteristics are influenced by the different pathogenetic and functional scenarios, as well as histopathological variants. Adrenal cortical adenomas may be hormonally inactive or synthesize/secrete steroid hormones with subclinical or overt clinical manifestations. Cortisol and aldosterone secreting-adrenal cortical adenomas are the most frequent functional correlates of adrenal cortical neoplasms, although some patients may have concurrent primary aldosteronism and Cushing syndrome [4, 5, 108].
Grossly, most adrenal cortical adenomas are homogenous and well-delineated cortical neoplasms that are enriched in lipid-rich adrenal cortical cells resulting in a yellow color (Fig. 13). Oncocytic adrenal cortical adenomas have a characteristic Mahogany brown appearance, while “black adenomas” are composed of tumor cells with lipofuscin pigment deposition [1]. Aldosterone-producing adrenal cortical adenomas (especially those harboring KCNJ5 mutations) typically have a canary (golden) yellow appearance [1]. Most non-functional adrenal cortical adenomas have no distinct gross appearance. Non-tumorous cortical atrophy is a characteristic gross and microscopic feature of cortisol-secreting adrenal cortical adenomas (Fig. 16). Adrenal cortical adenomas are often solitary nodules; however, multifocal and/or bilateral manifestations can occur. Sporadic nodular adrenal cortical disease (sub-centimeter non-functional benign adrenal cortical nodular proliferation) [1] is indistinguishable from functional adrenal cortical adenomas that measure < 1.0 cm. The large tumor size and weight (> 5 cm and > 100 g) as well as irregular border and heterogenous cut surface (e.g., necrosis, hemorrhage, fibrosis or gelatinous appearance) should alert the pathologist to the possibility of malignancy; therefore, extensive sampling or complete submission of the tumor is encouraged [1].

Non-tumorous adrenal cortical atrophy. In the absence of exogenous cortisol intake, the presence of adrenal cortical atrophy in the non-tumorous adrenal cortex is a hallmark of adrenal Cushing syndrome due to an autonomous cortisol-secreting adrenal cortical neoplasms. Reduced adrenal cortical thickness (< 2 mm) in association with significantly reduced to absent zona reticularis layer is a diagnostic feature of adrenal cortical atrophy
Histologically, adrenal cortical adenomas are composed of lipid-rich clear cells in a variable mixture with eosinophilic/compact cells. Clear-cut malignancy-related features such as vascular invasion, local invasion into adjacent structures, tumor necrosis unrelated to a former manipulation, atypical mitotic figures (even a single one), increased mitotic activity (> 5 mitoses per 10 mm2) and a marked loss of reticulin framework (unrelated to an underlying degeneration or hemorrhage) are not seen in adrenal cortical adenomas. However, some other features included in multiparameter diagnostic scoring schemes may be present, thus posing problems in the differential diagnosis with adrenal cortical carcinoma. Random nuclear atypia (corresponding to renal cell carcinoma Fuhrman/ISUP grade 3 or 4) may be focally identified. However, endocrine atypia should not be mistaken for nuclear atypia. Infarct-type necrosis may be detected as a result of involutional changes and should by no means be considered suggestive for malignancy, alone. If carefully searched, myelolipomatous areas are often detected in these cases [109] (Fig. 17). The term “oncocytoma” is no longer a recommended terminology for oncocytic adrenal cortical neoplasms. Irrespective of their biological behavior, oncocytic adrenal cortical neoplasms are usually associated with diffuse growth pattern and presence of macronucleoli. Since these features are not indicative of malignancy in oncocytic cortical neoplasms, a diagnosis of malignancy cannot be rendered based on the Weiss scoring system, which incorporates these parameters. However, oncocytic adrenal cortical neoplasms are assessed using other diagnostic algorithms which are detailed in the next question.

Infarcted adrenal cortical adenoma with central hemorrhage. This composite photomicrograph illustrates a well-delineated adrenal cortical adenoma with infarct-type necrosis (A, B) as well as foci of fibro-calcifications (B, C) and myelolipomatous change (C, inset)
Adrenal cortical origin should always be confirmed in all non-functional adrenal lesions and in adrenal neoplasms with predominant oncocytic features (even in an otherwise morphologically benign lesion) to prevent clinically relevant diagnostic pitfalls, the erroneous misinterpretation with pheochromocytoma being the most common [110] (Fig. 18). When an adrenal cortical neoplasm shows a predominant myxoid change, the risk of underestimating malignancy has been well-documented [111]. However, myxoid change does not indicate malignancy, but the behavior of myxoid adrenal cortical neoplasms cannot be predicted with standard Weiss scoring systems as myxoid adrenal cortical tumors with a Weiss score of 1 have been associated with fatal disease [1].

Oncocytic adrenal cortical adenoma. This photomicrograph illustrates an oncocytic adrenal cortical adenoma (A) that was mislabelled as an adrenal paraganglioma (pheochromocytoma) based on intense synaptophysin (B). The tumor is negative for chromogranin-A (C) and is positive for SF1 (D). These findings confirm the adrenal cortical origin of this tumor. It is also of note that alpha-inhibin can be expressed in paragangliomas. Therefore, synaptophysin and alpha-inhibin expression are not reliable biomarkers in the distinction of adrenal cortical origin from medullary origin
Non-functioning adrenal cortical adenomas do not show characteristics histologic features, except for their relatively larger size as compared to functional adrenal cortical adenomas. Cortisol-secreting adrenal cortical adenomas are the most frequent cause of ACTH-independent Cushing syndrome [15]. Clinical features of cortisol excess are extremely heterogeneous and include central obesity, rounded facies, hirsutism, poor wound healing, skin striae, weight gain, proximal muscle weakness, hypertension, hyperglycemia, osteoporosis, and susceptibility to infections [15]. However, the clinical features may be subtle and pre-operative dynamic endocrine work-up may be indeterminate or even not being performed. As a consequence, a subset of adrenal cortical adenomas with mild autonomous cortisol secretion (also known as subclinical Cushing syndrome) may simulate a non-functional adrenal cortical adenoma [5, 15]. Subsets of pigmented “black” adrenal cortical adenomas can show mild autonomous cortisol secretion [1]. In the absence of exogenous cortisol intake, the presence of adrenal cortical atrophy in the non-tumorous adrenal cortex is a hallmark of adrenal Cushing syndrome due to an autonomous cortisol-secreting adrenal cortical neoplasm [1, 5, 15, 112]. Reduced adrenal cortical thickness in association with significantly reduced to absent zona reticularis layer is a diagnostic feature of adrenal cortical atrophy [1, 5, 15] (Fig. 16). In patients with mild autonomous cortisol secretion, an intermittent absence of zona reticularis can be the first sign of cortical atrophy. Therefore, pathologists should carefully assess the non-tumorous adrenal cortex [112].
The former question focused on the morphological spectrum of CYP11B2-positive clonal and hyperplastic adrenal cortical proliferations leading to primary aldosteronism. Aldosterone producing cortical lesions can be unifocal or multifocal, and may be bilateral [1, 4, 5]. More importantly, not all grossly or radiologically identified adrenal cortical lesions can be the source of aldosterone excess [5, 113]. It is also not uncommon to have microscopic clonal nodular proliferations that cannot be identified on imaging studies [16, 107]. For this reason, the new WHO classification endorses the use CYP11B2 immunohistochemistry in the workup of all adrenalectomy specimens from patients with primary aldosteronism to identify functional sites of aldosterone secretion for the appropriate distinction of bilateral from unilateral aldosterone secreting lesions and predicting possible biochemical recurrence [114]. It is also important to recognize that APAs and APNs have distinct cytomorphological features that are reflected in their genotype–phenotype correlations [1, 4, 5]. For instance, KCNJ5-mutant APAs and APNs are enriched in zona fasciculata-like clear cells whereas KCNJ5 wild-type tumors have been enriched in lipid-poor cells similar to zona reticularis [4, 5] (Fig. 19). In cases associated with primary aldosteronism treated with spironolactone, the drug may form eosinophilic concentrically laminated electron dense inclusions, both in the tumor (Fig. 20) and the adjacent adrenal cortex (particularly in the zona glomerulosa), the so-called spironolactone bodies [1, 99]. Spironolactone bodies can be highlighted using Luxol-Fast blue histochemistry [99] (Fig. 20). Interestingly, similar inclusions are not detected in patients treated with other aldosterone antagonists, such as eplerenone [115].

Aldosterone-producing adrenal cortical nodule (microadenoma). KCNJ5 wild-type tumors are enriched in lipid poor adrenal cortical cells. This adrenalectomy specimen shows a solitary aldosterone-producing adrenal cortical nodule, which is an adrenal cortical microadenoma. CYP11B2 is not illustrated in this example. The adjacent non-tumorous cortex shows paradoxical zona glomerulosa layer hyperplasia

Spironolactone bodies in an aldosterone-producing adrenal cortical adenoma. Tumor cells show intracytoplasmic concentric lamellated eosinophilic inclusions, consistent with spironolactone bodies. The inset shows Luxol Fast blue reactivity in spironolactone bodies
Sex steroid-producing adrenal cortical adenomas are exceptional, especially in the adult population, and the presence of virilization or feminization should always increase the suspicion of malignancy [4, 116].
Question 7: Have the Pathologic Criteria for Adult Adrenal Cortical Carcinomas Changed in the 2022 WHO Classification?
Adrenal cortical carcinomas (ACCs) in adults may be clinically apparent due to the functional status with hormone secretion in approximately half or an abdominal mass, although approximately 10–15% may be incidentally detected [1, 117, 118]. Functional ACCs are usually glucocorticoid producing or produce both glucocorticoids and sex steroids [1]. An adrenal cortical neoplasm associated with virilization or feminization is highly worrisome for malignancy [1, 119], while aldosterone producing adrenal cortical carcinomas are rare [101, 102].
The classic pathologic criteria for the diagnosis of adult ACCs have not changed (Figs. 21, 22, 23, 24, and 25); however, the 2022 WHO classification underscores the diagnostic and prognostic impact of angioinvasion (vascular invasion) in these tumors [120]. Vascular invasion is assessed at the intersection of the tumor and adrenal capsule or beyond the adrenal capsule [1, 120, 121] (Fig. 21). While gross or clinically detected large vessel invasion is a finding of advanced ACCs [121] (Fig. 22), microscopic angioinvasion is defined when tumor cells invade through a vessel wall and form a thrombus/fibrin-tumor complex or intravascular tumor cells admixed with platelet thrombus/fibrin [120, 121] (Fig. 21). While the new classification emphasizes the diagnostic and predictive role of ancillary biomarkers in ACCs (see Questions 9 and 10), the role of CD61 immunohistochemistry in the detection of platelets at sites of angioinvasion is also noted in the new WHO classification.

Microscopic vascular invasion in adrenal cortical carcinoma. The identification of bona fide angioinvasion (vascular invasion) is a reliable diagnostic biomarker of malignancy in adrenal cortical carcinomas. This photomicrograph illustrates venous angioinvasion characterized by intravascular tumor cells admixed with fibrin/thrombus. Microscopic angioinvasion is also a prognostic factor in adrenal cortical carcinomas

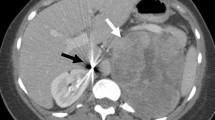
Gross vascular invasion in adrenal cortical carcinoma. This photograph illustrates a large adrenal cortical carcinoma invading a large vessel. The asterisk illustrates an intravascular tumor component. This finding qualifies with a pT4 disease in the 8th edition of the AJCC TNM classification

Tumor necrosis in adrenal cortical carcinomas. Tumor necrosis is typically identified in high grade adrenal cortical carcinomas. This composite photograph illustrates a large heterogenous adrenal mass with irregular border (A). The tumor shows both gross (A) and histologically confirmed (B) tumor necrosis

Increased mitotic activity in adrenal cortical carcinomas. In adrenal cortical pathology, increased mitotic activity is defined when an adrenal cortical tumor shows a mitotic count that exceeds 5 mitoses per 10 mm2 (50 high-power fields). This cutoff has been widely applied in all traditional multiparametric diagnostic schemes in adults. A mitotic count that exceeds 15 mitoses per 4 mm2 (20 high-power fields) has been used in pediatric manifestations. This composite photomicrograph illustrates increased mitotic activity in a conventional adrenal cortical carcinoma (A). Mitotic figures are circled. PhosphoHistone-H3 immunohistochemistry (B) may facilitate mitotic count in low-grade adrenal cortical carcinomas. This biomarker also facilitates the distinction of mitotic figures from apoptotic figures and may also help identify atypical mitotic figures

Atypical mitotic figures in adrenal cortical carcinomas. Atypical mitotic figure is a component of several multiparametric diagnostic schemes in adrenal cortical neoplasia. In the Lin–Weiss–Bisceglia system, it is one of the major criteria that would warrant a diagnosis of oncocytic adrenal cortical carcinoma. This photomicrograph illustrates atypical mitotic figures in an oncocytic adrenal cortical carcinoma. This tumor had also an increased mitotic activity (> 5 per 10 mm2) and angioinvasion, and Ki67 labeling index > 15% (not shown)

Subtypes of adrenal cortical carcinomas. Adrenal cortical carcinomas are subtyped based on their morphological features to include conventional (A), oncocytic (B), myxoid (C), and sarcomatoid subtypes
ACCs are subtyped based on their characteristic cytomorphological features to include conventional [1], oncocytic [122], myxoid [111, 123, 124], and sarcomatoid ACCs [1, 125] (Fig. 26). Oncocytic ACCs are composed of oncocytic tumor cells that account for more than 90% of the tumor [121, 122]. Myxoid ACCs are characterized by prominent extracellular mucin deposition [111, 123, 124]. Some Weiss parameters (e.g., lack of diffuse growth, nuclear atypia or lymphatic invasion) may be difficult to assess in myxoid adrenal cortical neoplasms. Sarcomatoid carcinomas resemble sarcomatoid carcinomas of various organs and often show adrenal cortical differentiation [126, 127]. Sarcomatoid ACCs that are unassociated with other subtypes of ACCs need to be distinguished from sarcomas.
The classic criteria of Weiss and colleagues described in 1984 and modified (also known as modified Weiss system) in 1989 continue to be used for the classification of conventional adrenal cortical neoplasms in adults [128, 129] (Tables 1 and 2). For conventional ACCs in adults, 3 of the 9 histologic parameters in the Weiss criteria need to be present for a diagnosis of malignancy (Table 1). The new WHO classification also expands the use of other multiparameter diagnostic algorithms to assist the workup of adrenal cortical neoplasms in adults. These include (a) reticulin algorithm which can be used for conventional, oncocytic and myxoid adrenal cortical neoplasms [120, 121, 130, 131], (b) Lin–Weiss–Bisceglia system for oncocytic adrenal cortical neoplasms [122], and (c) Helsinki scoring system which can be used for conventional, oncocytic and myxoid adrenal cortical neoplasms [121, 132, 133].
The reticulin algorithm (Table 3) has gained popularity in the workup of adrenal cortical neoplasms given its reproducibility [1, 120, 121, 130]. A diagnosis of ACC is rendered when an altered reticulin network (see Question 10) (Fig. 27), as demonstrated by the Gordon Sweet silver histochemical stain, is seen in association with one of the following parameters: (i) the presence of mitotic rate > 5 mitoses per 10 mm2 (50 high-power fields), (ii) tumor necrosis, or (iii) vascular invasion (angioinvasion) [120, 130, 131, 134].

Reticulin histochemistry in adrenal cortical carcinomas. A practical tool in the diagnostic workup of adrenal cortical carcinomas is the demonstration of reticulin alterations using the Gordon–Sweet Silver histochemistry. An altered reticulin framework forms the basis of the Reticulin algorithm Most adrenal cortical carcinomas tend to show a quantitative alteration (loss of reticulin framework) (A, C) but may have associated areas displaying qualitative alterations (mesh-like pericellular pattern) (A, B). The qualitative alterations alone do not stand out as a concerning feature of carcinoma since focal/variable qualitative reticulin alterations may occur in adrenal cortical adenomas which are associated with a preserved reticulin framework
The Lin–Weiss–Bisceglia system (Table 4) has been developed to evaluate oncocytic adrenal cortical neoplasms [122, 128, 135]. Of note, it is important that oncocytic adrenal cortical neoplasms are extensively sampled to be certain that they do fit into the category of pure oncocytic tumors to use this classification system. Greater than 90% of the tumor must be oncocytic for it to be regarded as a pure oncocytic adrenal cortical neoplasm. If it is not a pure oncocytic adrenal cortical neoplasm, then the criteria applied to conventional adrenal cortical carcinomas should be used. The “Lin–Weiss–Bisceglia” system is comprised of major (high mitotic rate, atypical mitosis, or vascular invasion) and minor criteria (large size and huge weight, necrosis, capsular invasion, or sinusoidal invasion) [122]. The diagnosis of malignancy requires the presence of at least one major criteria, whereas the presence of at least one minor criterion would indicate a tumor of uncertain malignant potential, and no major or minor criteria would be an oncocytic adrenal cortical adenoma.
The Helsinki score (Table 5) integrated the numeric value of Ki67 labeling index (using an automated image analysis nuclear algorithm) by adding scores assigned to increased mitotic rate (score 3 for mitotic rate greater than 5 mitoses per 10 mm2) and tumor necrosis (score 5). A Helsinki score > 8.5 is diagnostic of ACC, and a score of > 17 has been suggested to be helpful in predicting metastasis in adrenal cortical tumors [132, 133].
A variety of immunohistochemical and molecular biomarkers have been evaluated diagnostically and prognostically in ACCs [18, 120, 127, 136,137,138]. While IGF2 immunohistochemistry (Fig. 28) is a useful translational diagnostic immunohistochemical biomarker of ACC [120], the role of Ki67 (Fig. 29) as a diagnostic and prognostic biomarker [120, 139,140,141,142] has been expanded in this new classification. The details on the mitotic tumor grade, Ki67 labeling index, and other immunohistochemical and molecular biomarkers are discussed in subsequent sections of this review.

Paranuclear IGF2 expression in adrenal cortical carcinomas. Irrespective of the tumor grade or cytomorphological features (e.g., oncocytic versus conventional), a juxtanuclear granular IGF2 expression (optimized at 1/3000–1/6000 dilutions) has been shown to be the best diagnostic ancillary tool in adrenal cortical carcinomas. This photomicrograph illustrates a characteristic IGF2 reactivity in a conventional adrenal cortical carcinoma

Ki67 immunohistochemistry in adrenal cortical carcinoma. Most adrenal cortical carcinomas show a Ki67 labeling index that typically exceeds 5%. Clinical series from adult and pediatric adrenal cortical carcinomas underscored the prognostic role of Ki67 in both adult and pediatric adrenal tumors. Therefore, it is a required element in the diagnostic workup of all adrenal cortical carcinomas. This photomicrograph illustrates an elevated Ki67 labeling index in an adrenal cortical carcinoma. Please note that any intensity and nucleolar reactivity should also be counted as a positive reactivity when assessing this biomarker
Question 8: What are the Clinicopathological Correlates of Pediatric Adrenal Cortical Carcinomas in the 2022 WHO Classification?
Pediatric ACCs are uncommon adrenal cortical neoplasms, with an annual incidence of 0.2 to 0.3 cases per million population or 25 patients per year in the USA [143]. Compared to adult forms, pediatric ACCs show distinct clinical, molecular, and pathologic characteristics [139].
In terms of epidemiologic correlates, pediatric ACC have two age peaks, one before the age of 5 years and a second in adolescents. While very rare, they have a 15-fold increased frequency in the southern part of Brazil, due to the occurrence of a TP53 p.R337H founder mutation [144, 145]. In this age range, ACCs more frequently present with excess hormone secretion than in adults, with excess androgen secretion being the most frequent presentation in up to 80% of patients, leading to signs of virilization in both boys and girls, with pubic hair growth, enlargement of penis or clitoris and hirsutism [146]. Less commonly, excess cortisol secretion leads to Cushing syndrome [147].
In contrast to adults, up to 80% of ACCs in children are linked to pathogenic germline TP53 variants as the most common genetic abnormality, not only in geographic areas with increased incidence, but also elsewhere [148]. Indeed, ACC is a well-recognized entity in the context of Li-Fraumeni syndrome [127, 148,149,150,151]. Genomic instability caused by loss of p53 leads to loss of heterozygosity of 11p15 with resultant IGF2 overexpression, which may act as a driver for ACC development. Interestingly, the hereditary variant of 11p15 abnormalities, Beckwith-Wiedemann syndrome, may also present with pediatric ACCs, underlining the importance of this pathogenetic pathway [151]. Additional somatic mutations in ATRX and CTNNB1 have been described, which may result in poor outcome [152].
The macroscopic features of pediatric ACC largely resemble those of adult ACCs, with large tumors showing signs of hemorrhage, necrosis, and gross tumor extension beyond the adrenal gland. This is in contrast to benign adrenal cortical tumors which lack such signs. However, most adrenal cortical tumors are limited to the adrenal gland and thus gross appearance only infrequently contributes to tumor classification.
The histological characteristics of pediatric ACC are not different than their adult counterparts; however, the interpretation of histological findings and consequent classification are different (Table 6). Cytologically, they are composed of adrenal cortical cells with eosinophilic or clear cytoplasm, sometimes with oncocytic differentiation. Nuclear pleomorphism may be pronounced, with prominent nucleoli and/or giant nuclei. Mitoses may be frequent. The presence of atypical mitoses is a very helpful criterion to establish a diagnosis of ACC, although these are frequently not present. Architecturally, tumors are characterized by nodular, trabecular or diffuse growth, with areas of necrosis and sometimes signs of vascular invasion (angioinvasion), lymphatic invasion, or adrenal capsule invasion. Unfortunately, the classification of pediatric adrenal cortical tumors is fraught with difficulty, and classification on the basis of the Weiss criteria, used for adult counterparts, leads to overdiagnosis of tumors with benign clinical behavior as ACC in children [153]. Thus, the currently preferred criteria are those described by Wieneke et al. in 2003 [154] (Table 6). However, additional studies are required to validate the role of various other multiparameter systems. Recently, Ki67 (MIB1) immunohistochemistry (see Question 9) has been proposed as an ancillary biomarker in the distinction of pediatric adrenal cortical adenomas from ACC as well as in the prediction of tumor behavior, with a labeling index less than 10% associated with benign disease and a labeling index of greater than 15% related to higher risk of malignancy or poor outcome [139, 155].
Although the AJCC TNM staging system is widely used in ACCs, a clinical staging scheme has been proposed by the Children’s Oncology Group (COG), according to tumor weight, completeness of resection and presence of metastases [156,157,158] (Table 7). When present, the brain, liver, lungs, and bone are the organs most frequently affected by metastases [159]. In addition to the role of Ki67 immunohistochemistry, meta-analyses have also confirmed the prognostic significance of the following parameters: age, stage, tumor weight > 200 g, extra-adrenal extension, incomplete resection, metastatic disease, and cortisol secretion [152, 155, 157].
Question 9: What is the Role of Mitotic Tumor Grade and the Ki67 Index in Adult and Pediatric Adrenal Cortical Carcinomas?
The 2022 WHO classification emphasizes the importance of an accurate assessment of tumor proliferation rate using both the mitotic count (as per 10 mm2) (Fig. 24) and Ki67 labeling index (Fig. 29) in the dynamic risk stratification of tumors [1, 121]. In adrenal cortical pathology, increased mitotic activity is defined when an adrenal cortical tumor shows a mitotic count that exceeds 5 mitoses per 10 mm2 (50 high-power fields) [129, 132, 160,161,162]. This cutoff has been widely applied in all traditional multiparametric diagnostic schemes in adults [129, 132, 160,161,162]; however, a mitotic count that exceeds 15 mitoses per 4 mm2 (20 high-power fields) has been used in pediatric manifestations given their distinct features [154].
The 2022 WHO classification of adrenal cortical carcinomas adopts the mitotic tumor grade that was originally introduced by Weiss et al. [128]. This grading system uses the cut-off of 20 mitoses per 10 mm2 to distinguish low- and high-grade carcinomas. Low-grade adrenal cortical carcinomas have a mitotic activity ≤ 20 mitoses per 10 mm2, whereas high-grade adrenal cortical carcinomas show > 20 mitoses per 10 mm2 [120, 128, 163]. The prognostic value of this approach has been validated in cohorts from adult tumors [120, 121, 163]; however, it remains to be validated in pediatric adrenal cortical carcinomas [164]. Two independent series from Duregon et al. [142] and Mete et al. [120] also showed that a cut-off of 10 mitoses per 10 mm2 shows a better performance; however, further validation of the optimum cut-off points is encouraged.
Counting mitoses especially in the setting of low-grade adrenal cortical carcinomas may be facilitated using phosphoHistone-H3 immunohistochemistry [1, 18, 142] (Fig. 24). The original study on adrenal cortical carcinoma employed a methodology that is based on mitotic count from 10 high-power fields on 5 different sections (50 high-power fields in toto) with high-mitotic density; however, it is also acceptable to count mitoses in 10 mm2 (50 high-power fields) from hot spots in multiple individual slides with high mitotic density.
In terms of the Ki67 proliferation index, most adrenal cortical carcinomas show a labeling index that typically exceeds 5% [4, 18, 120, 139, 155, 165,166,167,168] (Fig. 29). Clinical series from adult and pediatric adrenal cortical carcinomas underscored the prognostic role of Ki67 in both adult and pediatric adrenal tumors [120, 139, 141, 142, 155]. While some large clinical series introduced a three-tiered Ki67 cutoffs (< 10%, 10–19%, ≥ 20%) for prognostication of adrenal cortical carcinomas [141], others have applied different prognostic cutoffs (< 20%, 20–50%, > 50%) [142]. In addition, two recently published independent cohorts of pediatric adrenal cortical carcinomas underscored the cutoff of 15% as of prognostic significance [139, 155].
The recently validated S-GRAS (Stage, Grade based on the Ki67 labeling index, Resection status and Symptoms) scoring scheme has shown a better prognostic significance compared to the tumor stage and Ki67 proliferation index in patients with adrenal cortical carcinomas [169]. The Ki67 proliferation index is a component of the Helsinki scoring system that uses the proliferation index as a continuous numeric value [132]. Ki67-based tumor grading has not been endorsed in the 2022 WHO classification of adrenal cortical carcinomas, since the proliferation indices are continuous variables rather than being static thresholds in tumor biology. However, the WHO classification of adrenal cortical carcinoma classification recommends diagnosticians specify the numeric value for mitotic count (as per 10mm2) and Ki67 labeling index in all adrenal cortical carcinomas.
Another point to remember is adrenal cortical carcinomas frequently show proliferative heterogeneity [1, 121]; thus, the identification of hot spots is an important step to ensure accurate assessment of the mitotic tumor grade and Ki67 proliferation index in these tumors. Given the impact of the Ki67 labeling index in the dynamic risk stratification that also rationalizes the of adjuvant mitotane therapy [1, 118], the new WHO classification does not suggest eye-balling for reporting of the Ki67 proliferation index. Similar to all neuroendocrine neoplasms, the assessment of Ki67 proliferation index is done with either manual count or automated image analysis nuclear algorithms [1, 121, 140, 170] (Fig. 30). Pathologists should document the methodology used in their assessment of Ki67 and mitotic count [171].

Assessment of Ki67 labeling index in adrenal cortical carcinomas. The new WHO classification does not suggest eye-balling for reporting of the Ki67 proliferation index. Similar to all neuroendocrine neoplasms, the assessment of Ki67 proliferation index is done with either manual count or automated image analysis nuclear algorithm. Pathologists should document the methodology used in their assessment of Ki67. This photomicrograph illustrates an automated image analysis nuclear algorithm in the assessment of Ki67 proliferation index
Question 10: Which Ancillary Tools should Pathologists Consider Utilizing in the Workup of Benign and Malignant Adrenal Cortical Proliferations?
The 2022 WHO classification emphasizes the role of ancillary tools in the workup of adrenal cortical proliferations to ensure accurate tumor classification. Several ancillary tools may be used by pathologists in the diagnostic assessment of adrenal cortical lesions. Molecular studies may be utilized as diagnostic and/or prognostic ancillary tools in select clinical settings [1, 4, 127, 172,173,174,175]; however, molecular studies do not form the basis of the standard of clinical practice. Therefore, the modern endocrine pathology practice relies on the application of immunohistochemical biomarkers and/or histochemical stains (e.g., Gordon Sweet Silver in the workup of malignancy, Luxol Fast Blue in the distinction of spironolactone bodies) that are interpreted in association with morphological and clinical findings [1, 18, 120, 130]. Therefore, diagnosticians should be aware of the various situations that may require the application of specific biomarker studies to meet clinical practice.
In adrenal cortical proliferations, ancillary tools are typically used to address specific needs such as the (i) distinction of the adrenal cortical origin, (ii) confirmation of functional adrenal cortical lesions especially in the context of primary aldosteronism, (iii) distinction of malignancy, (iv) risk stratification in ACC, (v) providing theranostic information in ACC, and (vi) rationalization of the need for genetic screening for underlying germline pathogenic variants [1, 18, 120].
The confirmation of the adrenal cortical origin is a critical task especially when dealing with non-functional adrenal lesions which may be mistaken for a primary adrenal cortical neoplasm. Therefore, the new WHO classification emphasizes the confirmation of the adrenal cortical origin as an important step in the diagnostic workup of these tumors. From that perspective, the use of SF1 immunohistochemistry (Fig. 31) is encouraged since it has been shown to be the most reliable and specific biomarker of adrenal cortical origin [18]. Other non-specific biomarkers such as Melan-A, synaptophysin, alpha-inhibin and calretinin may only be used in a panel approach; however, it is important to recognize their limitations [18]. Similar to synaptophysin, alpha-inhibin cannot be used in the distinction of an adrenal cortical lesion from a pheochromocytoma (adrenal paraganglioma) since paraganglioma/pheochromocytoma with a pseudohypoxia-driven pathogenesis often show reactivity for alpha-inhibin [176].

Confirmation of the adrenal cortical differentiation. The new WHO classification emphasizes the confirmation of the adrenal cortical origin as an important step in the diagnostic workup of these tumors. From that perspective, the use of SF1 immunohistochemistry is encouraged since it has been shown to be the most reliable and specific biomarker of adrenal cortical origin. This photomicrograph illustrates diffuse strong nuclear reactivity for SF1 in an oncocytic adrenal cortical carcinoma

p53 in adrenal cortical carcinoma. An abnormal p53 expression (often overexpression and rarely global loss) may be identified in a subset of adrenal cortical carcinomas which are typically enriched in high-grade carcinomas that are reflected in poor-prognostic molecular clusters. This photomicrograph illustrates p53 overexpression in a high-grade adrenal cortical carcinoma

Beta-catenin in adrenal cortical carcinomas. Similar to p53, nuclear beta-catenin expression may be identified in a subset of adrenal cortical carcinomas which are typically enriched in high-grade carcinomas that are reflected in poor-prognostic molecular clusters. This composite photomicrograph illustrates beta-catenin expression in two adrenal cortical carcinomas. The tumor on the left side (A) shows membranous beta-catenin expression whereas the tumor on the right side shows nuclear and cytoplasmic beta-catenin expression (B)

IGF2 expression in a low-grade oncocytic adrenal cortical carcinoma
In the past, ultrastructure examination has helped in the distinction of aldosterone-producing adrenal cortical proliferations from other steroidogenic adrenal cortical lesions by assessing characteristics of the mitochondrial cristae. This distinction was based on aldosterone-producing cells typically showing plate-like cristae, whereas tubulovesicular cristae featured cortisol-producing adrenal cortical cells [112]. However, the modern endocrine pathology practice applies functional immunohistochemistry in the distinction of certain functional states of adrenal cortical proliferations [18]. Several antibodies against steroidogenic enzymes have shown value in the workup of adrenal cortical proliferations. These include (but not limited to) HSD3B1 (3β-hydroxysteroid dehydrogenase type 1), HSD3B2 (3β-hydroxysteroid dehydrogenase type 2), CYP11B1 (cytochrome P450 family 11 subfamily B member 1), CYP11B2 (cytochrome P450 family 11 subfamily B member 2, also known as aldosterone synthetase), and CYP21A2 (cytochrome P450 family 21 subfamily A member 2) [1, 18]. Among these, the most popular ones have been CYP11B1 and CYP11B2. While CYP11B1 is considered as a functional biomarker of the zona fasciculata-like cells, CYP11B2 (aldosterone synthase) has gained popularity due to its important role in the distinction of aldosterone-producing cortical lesions which may be difficult on H&E-stained sections [16, 18, 120]. As a consequence, the new WHO classification endorses the use of CYP11B2 immunohistochemistry, which forms the basis of HISTALDO classification, that provides clinicopathologically relevant diagnostic categories in the workup of primary aldosteronism [16].
Although SF1 is the most reliable biomarker in the confirmation of adrenal cortical origin, this biomarker is also expressed in gonadotroph tumors (pituitary neuroendocrine tumors with gonadotroph cell differentiation) as well as in a subset of steroidogenic gonadal neoplasms (e.g., steroid cell tumor, Leydig cell tumor) [177]. These pitfalls should be kept in mind when assessing metastatic tumors with SF1 expression or when assessing adrenal rest-related manifestations including adrenal cortical choristomas in anatomic sites beyond the developmental pathway (e.g., pituitary corticotroph tumor with adrenal cortical choristomas) [50, 51]. When the distinction of an adrenal cortical origin from a steroidogenic gonadal tumor is required, adrenal cortex-specific steroidogenic enzymes may also provide additional value [1]. For instance, the application of CYP11B1 and CYP21A2 has shown promise in the distinction of TARTs (testicular adrenal rest tumors) from Leydig cell tumors [58]. Although most laboratories currently do not have access to the full spectrum of functional immunohistochemistry tools, it is important to know that there are additional biomarkers (e.g., abundance of DLK1 and lack of INSL3 expression) that may be used in the distinction of TARTs from Leydig cell tumors [58].
Several biomarkers can be used to support the diagnosis of malignancy in the background of appropriate morphological features. As discussed in the former question, ACCs typically exceed a Ki67 labeling index of 5% [4, 18, 120, 139, 155, 165,166,167,168], and the value of Ki67 also matters in terms of tumor prognostication. An abnormal p53 expression (overexpression or global loss) (Fig. 32) and/or nuclear beta-catenin expression (Fig. 33) may be identified in a subset of ACCs, which are typically enriched in high-grade carcinomas that are reflected in poor-prognostic molecular clusters [127]. However, such tumors are easily diagnosed based on conventional morphological features, whereas the distinction of a low-grade ACC with no angioinvasion or extra-adrenal tumor spread may remain a diagnostic challenge [120]. Therefore, the confirmation of increased proliferation (Ki67 labeling index and phosphoHistone-H3 assisted mitotic count) may assist the diagnosis of malignancy in association with other ancillary tools. Evidence suggests that irrespective of the tumor grade or cytomorphological features (e.g., oncocytic versus conventional) (Figs. 28 and 34), a juxtanuclear granular IGF2 expression (optimized at 1/3000–1/6000 dilutions) has been shown to be the best diagnostic ancillary tool in ACCs [120]. This finding occurs in around 80% of ACCs in adult series [28877067] and is thought to reflect IGF2 overexpression-related impaired translation and processing of the IGF2 molecule in the Golgi apparatus [178].
While access of IGF2 immunohistochemistry is not widely available, an additional practical tool is the demonstration of reticulin alterations using the Gordon-Sweet Silver histochemistry (Fig. 27). As discussed before, an altered reticulin framework forms the basis of the reticulin algorithm which combines altered reticulin framework with one of the followings: (i) necrosis, (ii) vascular invasion (angioinvasion), and (iii) increased mitotic activity (> 5 per 10 mm2) [120, 121, 130, 131]. However, diagnosticians should recognize potential pitfalls with respect to the assessment of reticulin histochemistry. Most ACCs tend to show a quantitative alteration (loss of reticulin framework) but may have associated qualitative alterations (mesh-like pericellular pattern) [120]. The qualitative alterations alone do not stand out as a concerning feature of carcinoma since focal/variable qualitative reticulin alterations may occur in adrenal cortical adenomas which are associated with a preserved reticulin framework [120]. In addition, areas of degeneration including sites of hemorrhage or biopsy sites in benign cortical proliferations can show disrupted reticulin [1].Therefore, it is important to match reticulin alterations with morphological findings.
Global loss of DAXX and/or ATRX expression (Fig. 35) may also be identified in ACCs, and this has shown to have prognostic value [18, 120, 136]. The application of MMR proteins (MLH1, MSH2, MSH6 and PMS2) has gained popularity not only for the purpose of screening for an extra-colonic manifestation of Lynch syndrome [18, 179, 180] (Fig. 36), but also to provide additional guidance on the use of immunotherapy trials [4, 181]. Similarly, the PDL1/PD1 expression status of ACCs has been an area of interest. There are also other theranostic biomarkers including CYP2B6 polymorphism that have shown variable promise with respect to the prediction of mitotane response in patients with ACCs [18, 182, 183]; however, these biomarkers are not widely used as the standard of practice in most diagnostic laboratories.

Loss of ATRX expression in adrenal cortical carcinomas. A subset of adrenal cortical carcinomas may show global loss of ATRX. This has shown to have prognostic value

MMR immunohistochemistry in an adrenal cortical carcinoma. This composite photomicrograph illustrates global loss of PMS2 and MLH1 expression in an adrenal cortical carcinoma from an adult patient that was found to have Lynch syndrome
Similar to the role of MMR immunohistochemistry in Lynch syndrome (Fig. 36), a number of biomarkers may be used to facilitate the screening of genetic susceptibility in the setting of appropriate clinical and morphological context [1]. These include (a) nuclear beta-catenin and loss of APC expression in FAP-related adrenal cortical neoplasia), (b) loss of SDHB expression in adrenal cortical neoplasia that occur in the setting of SDH-deficient paraganglioma syndrome, (c) abnormal p53 expression (Li-Fraumeni syndrome), (d) loss of menin expression (MEN1-related adrenal cortical neoplasia), and (e) loss of p27 expression (MEN4-related adrenal cortical neoplasia) [1, 4, 184].
Question 11: What are the Molecular Correlates of Adrenal Cortical Proliferations in the 2022 WHO Classification? Are There Any Diagnostic or Prognostic Molecular Studies in the Workup of Adrenal Cortical Carcinoma?
The molecular background of adrenal cortical neoplasia is multifaceted and heterogenous. In aldosterone- and cortisol-producing adrenal cortical adenomas, the underlying genetic aberrancies are often missense mutations that often hijack the physiological pathways leading to cell proliferation and hormone secretion—which is governed by an amplified cell membrane depolarization in aldosterone-secreting adenomas and increased protein kinase A (PKA) pathway signaling in cortisol-producing adenomas [4, 5]. Adrenal cortical carcinoma often displays multiple gross chromosomal alterations as well as mutations and deletions of genes regulating classical cancer-associated signaling networks, such as the Wnt and P53 pathways [4]. As it is expected that molecular analyses will become an integrated part of the work-up of these lesions in the future, an understanding of the background genetics propelling these tumors will be necessary in order to provide an optimal diagnostic and prognostic assessment, as well as to identify possible therapeutic options for non-responding cases. In this question, we focus on common genetic aberrancies underlying the development of primary hyperaldosteronism, cortisol-producing adrenal cortical adenomas as well as ACCs. As cortisol-producing adrenal cortical nodular disease patients often carry germline variants in susceptibility genes, the genetics of these lesions are separately discussed in the next question.
Molecular Correlates of Primary Aldosteronism
The advent of next-generation sequencing has led to groundbreaking molecular findings in aldosterone-producing adrenal cortical lesions and has helped establish this tumor entity as a disease of defective ion channels. In the absence of extracellular angiotensin II, the normal zona glomerulosa cell is in a resting state facilitated by G-protein activated inward rectifier potassium channel 4 (GIRK4) encoded by the KCNJ5 gene, a membrane-bound K+ channel that allows K+ to exit the cell and ensure a membrane hyperpolarization. The build-up of this membrane potential will mediate entry into the G0 phase of the cell cycle, and no CYP11B2 (aldosterone synthase) enzyme will be produced. Upon physiological response to angiotensin II binding its receptor, the GIKR4/KCNJ5 channels close, and depolarization is mediated through the accumulation of intracellular K+. This will initiate an intracellular signaling cascade, leading to cell cycle activation and aldosterone production [4]. In aldosterone-producing adrenal cortical adenomas (irrespective of the size of the cortical proliferation), this process is mimicked through somatic mutations in genes encoding several different ion channel proteins. These mutations will cause constitutive membrane depolarization, even in the absence of extracellular angiotensin II, leading to augmented cell proliferation (Fig. 37). Mutations in KCNJ5 [185, 186], CACNA1D [187,188,189], and CACNA1H [187, 190, 191] will cause depolarization through the influx of Na + (KCNJ5) or Ca2 + (CACNA genes), while mutations in ATP1A1 [185, 188, 192] and ATP2B3 [185, 192] will impede the normal function of these ion channels, i.e., to leak Na+ and Ca2+ respectively to the extracellular compartment—thus leading to membrane depolarization (Fig. 37). Intriguingly, subsets of patients with autosomal dominant forms of familial hyperaldosteronism have also been linked to pathogenic germline variants in KCNJ5, CACNA1D, CACNA1H, as well as the CLCN2 chloride channel gene [4].

Main molecular aberrations in aldosterone- and cortisol-producing adrenal cortical adenoma. Left: Mutations in various genes encoding membrane-bound ion channels underlie the development of aldosterone-producing adenoma. Mutations confer an increased passage of cations into the cytosol (KCNJ5, CACNA1D/1H) alternatively inhibit the leakage of cations out of the cell (ATP1A1, ATP2B3). In either way, depolarization follows, which in turn stimulate cellular proliferation as well as aldosterone synthase expression. Right: Various genetic aberrancies in the protein kinase A (PKA) pathway (gain-of-function mutations in GNAS or PRKACA or deleterious PRKAR1A and phosphodiesterase (PDE) family mutations) will activate the pathway and stimulate proliferation and cortisol synthesis. + , activating mutations; -, deleterious mutations. Created with BioRender.com
Somatic ion channel mutations were initially reported in approximately half of sporadic aldosterone-producing adrenal cortical adenomas, of which somatic KCNJ5 mutations alone account for 40% of cases—notably with an overrepresentation in younger female patients with marked hormonal activity, with possible correlations also to the adrenal vein sampling lateralization index as well as the overall histology—notably zona fasciculata-like clear cells [193,194,195]. In contrast, KCNJ5 wild-type tumors are more frequent in males with a later age at presentation and recurrently display smaller tumor sizes and not seldom a compact cell morphology [5]. Despite the early work suggesting ion channel mutations in approximately 50% of patients, the true proportion of patients with is probably much higher, probably between 90 and 96% as indicated by more recent work relying on positive CYP11B2 immunoreactivity as an inclusion criterion [187, 196, 197]. Moreover, mutations in ion channels are not only restricted to conventional aldosterone-producing adrenal cortical adenomas but are also found in other benign neoplastic proliferations referred to as aldosterone-producing nodules (multiple adenomas) and micronodules (multiple microadenomas) [198, 199]. The very rare aldosterone-producing ACCs do not exhibit mutations in well-characterized ion channel genes, and additional molecular aberrancies is likely to drive the overexpression of CYP11B2 in these cases [101].
While ion channel mutations occur in the vast majority of benign, aldosterone-producing lesions, approximately 5% of sporadic aldosterone-producing adrenal cortical adenomas display mutually exclusive CTNNB1 mutations [200, 201]. This gene encodes beta-catenin, a Wnt signaling pathway effector, and activating CTNNB1 mutations allows this protein to escape proteasome-mediated degradation and translocate to the nucleus, where it will associate to various transcription factors and initiate gene transcription. In adrenal cortical disease, CTNNB1 mutations are best-known to be recurrently observed in adrenal cortical carcinoma, as well as in non-functional adrenal cortical adenomas and adrenal cortical adenomas with mild autonomous cortisol production (also known as subclinical Cushing syndrome) [127, 202,203,204]. Interestingly, while CTNNB1 mutations are rare, a general Wnt pathway activation seems to be a regular theme in aldosterone-producing adrenal cortical adenoma [205]. While the exact mechanisms coupling the Wnt pathway to aldosterone production is not known, there have been studies indicating a link between aberrant beta-catenin expression and up-regulation of angiotensin II receptors as well as the CYP11B2 enzyme [205].
Molecular Correlates of Cortisol-Producing Adrenal Cortical Adenomas
Just like aldosterone-producing adrenal cortical lesions hijack the physiological pathways of angiotensin II stimulation via mutations, cortisol-producing adenomas display genetic aberrancies in the ACTH response mediated protein kinase A (PKA) pathway. In the normal zona fasciculata, the cells respond to ACTH stimulation via the melanocortin 2 receptor (MC2R), thus activating a cascade in which cAMP is produced from ATP via the enzyme adenylyl cyclase. The cAMP molecules in turn will inhibit regulatory subunits of PKA, allowing the catalytic subunits to stimulate proliferation and cortisol production (Fig. 37).
In cortisol-producing adenomas, mutations in PRKACA, PRKAR1A, GNAS, and PRKACB are observed—and these aberrations will in different ways lead to over-activation of the PKA pathway [206,207,208,209,210,211,212].
Recent pan-genomic work regarding benign correlates of adrenal Cushing syndrome suggest that these entities segregate into three distinct clusters, including (a) PPNADs and cortisol-producing adrenal cortical adenomas coupled to an activated PKA pathway, (b) adrenal cortical adenomas with mild autonomous cortisol secretion associated to the activation of the Wnt signaling pathway, and (c) ARMC5-driven primary bilateral macronodular adrenal cortical disease with a coupling to a specific ovarian gene expression signature [172].
Adrenal Cortical Carcinoma
Most ACCs arising in the adult population do so through an accumulation of somatic genetic aberrances. The pinpointing of genes associated to the development of this disease is an effort that spans over several decades and include gene-specific studies as well as pan-genomic analyses. In comparison to adrenal cortical adenomas, ACCs commonly display mutually exclusive driver gene alterations associated to the Wnt and P53 pathways, as well as aberrant IGF2 signaling and complex gross chromosomal aberrations—all features those adrenal cortical adenomas regularly lack [4] (Fig. 38). Moreover, ACCs are characterized by a global hypomethylation pattern, as opposed to adenomas that display global hypermethylation.

Schematic overview of genetic differences between adrenal cortical adenoma and carcinoma. While adenomas mainly develop through mutations in ion channel genes or in genes encoding members of the PKA pathway and display a placid chromosomal landscape, carcinomas regularly display Wnt and P53 pathway mutations, aberrant IGF2 signaling and advanced chromosomal aberrations. Moreover, adenomas recurrently exhibit global DNA hypermethylation compared to carcinomas. Created with BioRender.com
Recurrent genetic events in ACCs include activating CTNNB1 mutations and deleterious ZNRF3 mutations (alternatively homozygous ZNRF3 copy number loss), which will lead to an activation of the oncogenic Wnt signaling pathway. Moreover, cell cycle related gene mutations in TP53, RB1, MDM2, and CDKN2A are also commonly seen. TP53 mutations alone are found in 20% of ACCs, and the deleterious nature of the mutations abrogates the normal function of the P53 protein, including cell cycle control, DNA repair, as well as regulation of cellular aging and apoptosis (Fig. 39) [213]. Moreover, patients with germline TP53 mutations predispose for the Li-Fraumeni syndrome, in which patients have an increased risk of developing ACC—thus further solidifying the association between this gene and malignant cortical tumorigenesis [214].

P53 normal functions and effects of TP53 mutations. In the wild-type state, P53 is activated upon DNA damage and inhibited via oncogene activation. P53 has many important cellular roles, including the regulation of cell cycle progression, DNA repair as well as regulation of cellular senescence and apoptosis. When inactivated by mutations or deletions, DNA damage accumulate, and cell cycle progression and inhibition of apoptosis is seen. Created with BioRender.com
Subsets of cases also display mutations in chromatin remodeling genes such as DAXX, ATRX, and MEN1, and some may also exhibit PKA pathway associated mutations (PRKAR1A) [127, 202, 215, 216].
In terms of chromosomal events, loss of the maternal 11p15 allele adjoined by paternal allele duplication is commonly noted in ACC [217, 218]. This region encompasses the H19 and IGF2 genes, which are imprinted and thus expressed selectively from the maternal and paternal allele respectively [219]. This selective loss leads to the down-regulation of H19, a gene encoding a long non-coding RNA with tumor suppressor abilities, and cause up-regulation of oncogenic IGF2. Of note, Beckwith-Wiedemann syndrome patients display uniparenteral disomy (with two inherited paternal alleles) of this locus and display an increased risk of developing ACC. Apart from this chromosomal aberrancy, ACCs are either seen with whole chromosomal losses, or with numerous copy number alterations and whole-genome doubling (Fig. 40) [127].

Molecular risk triaging of adrenal cortical carcinoma. By recent pan-genomic analyses, carcinomas in general adhere to one out of three main clusters (“Cluster of Clusters; CoC”) I-III. While CoC I tumors are clinically more indolent and display low levels of Wnt/P53 pathway mutations and a CpG island methylator phenotype (CIMP) low phenotype, CoC III tumors are considered high risk lesions, and associate to a different expressional cluster and carry frequent Wnt/P53 signaling pathway mutations, a hypermethylated genome and frequent chromosomal aberrancies, including whole-genome doubling. Created with BioRender.com
Next-generation sequencing techniques have prompted a considerable leap in our understanding of ACCs, in which several molecular subclassification schemes with prognostic relevance have been presented [127, 174, 175, 202, 215, 220,221,222]. Although the studies differ in terms of case numbers and techniques employed, adrenal cortical carcinomas adhere to either low-, intermediate-, or high-risk clusters when profiling their mutational, expressional, DNA methylation, and gross chromosomal landscapes (Fig. 40) [127, 215, 223]. While low progression rate tumors rarely exhibit Wnt/P53 pathway mutations, display low levels of DNA methylation (“CpG island methylator phenotype (CIMP) low”) and associate to mRNA expressional clusters normally associated to a more indolent clinical course, high-risk ACCs carry frequent Wnt/P53 pathway mutations, an expression pattern often associated to worse outcome, a CIMP-high profile as well as extensive chromosomal alterations with whole-genome doubling (Fig. 40) [127].
While the pan-genomic classification of ACC is promising for the future integration in clinical work-up of these lesions, there is still a gap between what we are able to do in the research laboratory compared to our everyday clinical routine. The new WHO classification encourages diagnosticians to provide common immunohistochemical correlates of poor prognostic molecular clusters (e.g., P53 and beta-catenin); however, routine use of targeted genetic analyses and/or prognostic molecular analyses (e.g., transcriptome or methylome studies) is anticipated to aid the management in the near future. Indeed, the TCGA study on ACCs identified a smaller methylation assay consisting of 68 probes may be helpful in the identification of high-risk ACCs [127], and subsequent studies provided additional value to the assessment of the G0/G1 Switch 2 (G0S2) gene hypermethylation as a surrogate marker for CIMP-high cases endowed with poor prognosis [127, 173].
Question 12: What are Correlates of Genetic Predisposition in Adrenal Cortical Proliferations?
Although most adrenal cortical proliferations are clonal expansions due to the acquisition of somatic mutations, subsets of cases arise in patients with an underlying genetic predisposition. Basic knowledge regarding syndromic variants of adrenal cortical lesions is imperative in order to detect these cases in the clinical setting, allowing genetic counseling and adequate patient follow-up. In this section, a brief overview of germline alterations coupled to the development of adrenal cortical disease is summarized.
Congenital Adrenal Hyperplasia (CAH)
CAH is an autosomal recessive genetic disorder in which patients display germline mutations in genes encoding various enzymes in the adrenal cortical steroidogenesis pathway. The dominant gene in this aspect is CYP21A2, which is found mutated in 95–99% of CAH patients [224, 225]. The CYP21A2 gene encodes the 21-hydroxylase enzyme involved in both the production of mineralocorticoids and glucocorticoids, and individuals with bi-allelic mutations in CYP21A2 will impede the ability to produce aldosterone and cortisol, while instead driving excess production of androgens [226]. This may cause significant virilization. Also, the reduced production of cortisol will lift the negative feedback on the hypothalamus-pituitary axis, thereby leading to elevated ACTH levels, which in turn will cause the adrenals to become hyperplastic (Fig. 41). Mutations in other genes may also cause CAH, including CYP11B1 (encoding 11-beta-hydroxylase), HSD3B2 (encoding 3-beta-hydroxysteroid dehydrogenase), and CYP17A1 (encoding 17-alpha-hydroxylase) [224]. Adrenal cortical tumor formation is noted in approximately 24% of patients with genetically confirmed CAH, and almost 40% of patients may exhibit adrenal myelolipoma (Fig. 41) [227, 228]. The reason for the overrepresentation of myelolipomas in CAH is not known.

Morphological correlates of a resected adrenal gland from a patient with congenital adrenal hyperplasia (CAH). Multinodular adrenal cortex (A, B) characterized by zona reticularis-like cells admixed with chronic lymphocytic infiltrates and multifocal myelolipomas. A myelolipoma (C) arising within the hyperplastic adrenal cortex. This patient carried a germline CYP21A2 mutation
Bilateral Adrenocortical Nodular Disease
The term “bilateral adrenocortical nodular disease” comprises cortisol-producing primary bilateral micronodular adrenal cortical disease (nodules < 10 mm) and primary bilateral macronodular adrenal cortical disease (nodules > 10 mm), of which the former can be sub-divided into primary pigmented micronodular adrenal cortical disease (PPNAD) and isolated micronodular adrenal cortical disease (i-MAD). Irrespectively of entity, these patients regularly exhibit a germline variant in a susceptibility gene, which is then followed by a “second hit” occurring on the trans allele. PPNAD is coupled to Carney complex, a syndrome of cardiac myxomas, spotty pigmentation and endocrine overactivity, including adrenal cortical lesions [86]. When occurring in the context of Carney complex (“c-PPNAD”), mutations in genes regulating the PKA pathway are predominant, such as PRKAR1A (most common alteration in c-PPNAD and isolated PPNAD (i-PPNAD) followed by PRKACA gene aberrancies and CNC2 locus alterations [5, 81]. In a similar manner, germline variants in PKA pathway related genes PDE11A and PDE8B may cause i-MAD [4, 15, 229, 230].
Primary bilateral macronodular adrenal cortical disease (formerly known primary bilateral macronodular adrenal cortical hyperplasia) is composed of independent clonal expansions in which the patients usually carry germline mutations in ARMC5, MEN1, FH, APC, GNAS, PDE8B or PDE11A [4, 5, 7,8,9,10,11,12,13,14, 92, 172]. ARMC5 mutations are most commonly seen (occurring in 25–55% of cases), with additional somatic alterations found in the trans allele in each independent nodule—strongly arguing for multiclonal tumor development rather than a “hyperplastic” phenomenon. Interestingly, patients with primary bilateral macronodular adrenal cortical disease carrying ARMC5 mutations more often present with larger adrenals and a more pronounced hypercortisolism than those without ARMC5 mutations [9].
Adrenocortical Adenoma and Carcinoma
Genetic syndromes predisposing for adrenal cortical tumors are summarized in Fig. 42. Adrenal cortical adenomas are reported as unusual manifestations of the Li-Fraumeni, MEN1, FAP, Beckwith Wiedemann and Carney complex syndromes. Moreover, in terms of aldosterone-producing adenomas, subsets of patients may present with a family history of primary aldosteronism in an autosomal dominant pattern with several affected first-degree relatives. These syndromes (familial hyperaldosteronism, FHA) include FHA type 1 (germline CYP11B1/CYP11B2 chimeric fusions) [231], FHA type II (germline CLCN2 mutations) [232], FHA type III (germline KCNJ5 mutations) [186] and FHA type IV (germline CACNA1H mutations) [191]. Moreover, germline CACNA1D mutations may also cause early onset primary aldosteronism with various neurological abnormalities; also known as the “PASNA” syndrome [189].

Hereditary syndromes coupled to adrenal cortical neoplasia. Schematic overview of the most common syndromes associated to the development of adrenal cortical adenoma (left) and adrenal cortical carcinoma (right). FHA, familial hyperaldosteronism syndrome; PASNA, primary aldosteronism, seizures, and neurologic abnormalities. Created with BioRender.com
Hereditary conditions in which ACCs are overrepresented include Li-Fraumeni, Lynch, MEN1, Beckwith Wiedemann, Carney complex and NF1 syndromes [4]. Overall, approximately 10% of ACCs are expected to arise in a hereditary setting [233]. The most common syndromic manifestation is Li-Fraumeni syndrome, which may be evident in 50–80% of children that present with ACCs, in addition to 3–5% of adults. Furthermore, approximately 3% of adults developing this disease are found be Lynch syndrome patients carrying inactivating germline mutations in one out of the well-known mismatch repair genes MSH2, MSH6, MLH1, and PSM2 [180]. Since mutations in these genes are coupled to absent immunoreactivity for the corresponding proteins, MSH2, MSH6, MLH1 and PMS2 immunohistochemistry may therefore be an efficient way to identify cases in need of genetic counseling [1, 179]. Moreover, germline SDHx variants have been coupled to the development of ACCs, but these findings need additional verification [184]. The role of ancillary biomarkers with respect to germline screening are discussed in Question 10; however, careful assessment of the non-tumorous adrenal cortex may also help to distinguish c-PPNAD-related ACCs [234]. Given the overall rarity of ACCs, genetic counseling may be considered for all patients irrespective of the age of the patient [233].
References
Hodgson A, Pakbaz S, Mete O (2019) A Diagnostic Approach to Adrenocortical Tumors. Surg Pathol Clin 12:967–995 . https://doi.org/10.1016/j.path.2019.08.005
Mete O, Asa SL (2013) Precursor lesions of endocrine system neoplasms. Pathology 45:316–330 . https://doi.org/10.1097/PAT.0b013e32835f45c5
van Nederveen FH, de Krijger RR (2007) Precursor lesions of the adrenal gland. Pathobiology 74:285–290 . https://doi.org/10.1159/000105811
Juhlin CC, Bertherat J, Giordano TJ, Hammer GD, Sasano H, Mete O (2021) What Did We Learn from the Molecular Biology of Adrenal Cortical Neoplasia? From Histopathology to Translational Genomics. Endocr Pathol 32:102–133 . https://doi.org/10.1007/s12022-021-09667-0
Mete O, Duan K (2018) The Many Faces of Primary Aldosteronism and Cushing Syndrome: A Reflection of Adrenocortical Tumor Heterogeneity. Front Med (Lausanne) 5:54 . https://doi.org/10.3389/fmed.2018.00054
Kamilaris CDC, Hannah-Shmouni F, Stratakis CA (2020) Adrenocortical tumorigenesis: Lessons from genetics. Best Pract Res Clin Endocrinol Metab 34:101428 . https://doi.org/10.1016/j.beem.2020.101428
Vaduva P, Bonnet F, Bertherat J (2020) Molecular Basis of Primary Aldosteronism and Adrenal Cushing Syndrome. J Endocr Soc 4:bvaa075 . https://doi.org/10.1210/jendso/bvaa075
Hannah-Shmouni F, Faucz FR, Stratakis CA (2016) Alterations of Phosphodiesterases in Adrenocortical Tumors. Front Endocrinol (Lausanne) 7:111 . https://doi.org/10.3389/fendo.2016.00111
Espiard S, Drougat L, Libé R, Assié G, Perlemoine K, Guignat L, Barrande G, Brucker-Davis F, Doullay F, Lopez S, Sonnet E, Torremocha F, Pinsard D, Chabbert-Buffet N, Raffin-Sanson M-L, Groussin L, Borson-Chazot F, Coste J, Bertagna X, Stratakis CA, Beuschlein F, Ragazzon B, Bertherat J (2015) ARMC5 Mutations in a Large Cohort of Primary Macronodular Adrenal Hyperplasia: Clinical and Functional Consequences. J Clin Endocrinol Metab 100:E926-935 . https://doi.org/10.1210/jc.2014-4204
Assié G, Libé R, Espiard S, Rizk-Rabin M, Guimier A, Luscap W, Barreau O, Lefèvre L, Sibony M, Guignat L, Rodriguez S, Perlemoine K, René-Corail F, Letourneur F, Trabulsi B, Poussier A, Chabbert-Buffet N, Borson-Chazot F, Groussin L, Bertagna X, Stratakis CA, Ragazzon B, Bertherat J (2013) ARMC5 mutations in macronodular adrenal hyperplasia with Cushing’s syndrome. N Engl J Med 369:2105–2114 . https://doi.org/10.1056/NEJMoa1304603
Gaujoux S, Pinson S, Gimenez-Roqueplo A-P, Amar L, Ragazzon B, Launay P, Meatchi T, Libé R, Bertagna X, Audebourg A, Zucman-Rossi J, Tissier F, Bertherat J (2010) Inactivation of the APC gene is constant in adrenocortical tumors from patients with familial adenomatous polyposis but not frequent in sporadic adrenocortical cancers. Clin Cancer Res 16:5133–5141 . https://doi.org/10.1158/1078-0432.CCR-10-1497
Hsiao H-P, Kirschner LS, Bourdeau I, Keil MF, Boikos SA, Verma S, Robinson-White AJ, Nesterova M, Lacroix A, Stratakis CA (2009) Clinical and genetic heterogeneity, overlap with other tumor syndromes, and atypical glucocorticoid hormone secretion in adrenocorticotropin-independent macronodular adrenal hyperplasia compared with other adrenocortical tumors. J Clin Endocrinol Metab 94:2930–2937 . https://doi.org/10.1210/jc.2009-0516
Ventura M, Melo M, Carrilho F (2019) Outcome and long-term follow-up of adrenal lesions in multiple endocrine neoplasia type 1. Arch Endocrinol Metab 63:516–523 . https://doi.org/10.20945/2359-3997000000170
Villares Fragoso MCB, Wanichi IQ, Cavalcante IP, Mariani BM de P (2016) The Role of gsp Mutations on the Development of Adrenocortical Tumors and Adrenal Hyperplasia. Front Endocrinol (Lausanne) 7:104 . https://doi.org/10.3389/fendo.2016.00104
Duan K, Gomez Hernandez K, Mete O (2015) Clinicopathological correlates of adrenal Cushing’s syndrome. J Clin Pathol 68:175–186 . https://doi.org/10.1136/jclinpath-2014-202612
Williams TA, Gomez-Sanchez CE, Rainey WE, Giordano TJ, Lam AK, Marker A, Mete O, Yamazaki Y, Zerbini MCN, Beuschlein F, Satoh F, Burrello J, Schneider H, Lenders JWM, Mulatero P, Castellano I, Knösel T, Papotti M, Saeger W, Sasano H, Reincke M (2021) International Histopathology Consensus for Unilateral Primary Aldosteronism. J Clin Endocrinol Metab 106:42–54 . https://doi.org/10.1210/clinem/dgaa484
Volpe C, Hamberger B, Zedenius J, Juhlin CC (2020) Impact of immunohistochemistry on the diagnosis and management of primary aldosteronism: An important tool for improved patient follow-up. Scand J Surg 109:133–142 . https://doi.org/10.1177/1457496918822622
Mete O, Asa SL, Giordano TJ, Papotti M, Sasano H, Volante M (2018) Immunohistochemical Biomarkers of Adrenal Cortical Neoplasms. Endocr Pathol 29:137–149 . https://doi.org/10.1007/s12022-018-9525-8
Nakamura Y, Maekawa T, Felizola SJA, Satoh F, Qi X, Velarde-Miranda C, Plonczynski MW, Ise K, Kikuchi K, Rainey WE, Gomez-Sanchez EP, Gomez-Sanchez CE, Sasano H (2014) Adrenal CYP11B1/2 expression in primary aldosteronism: immunohistochemical analysis using novel monoclonal antibodies. Mol Cell Endocrinol 392:73–79 . https://doi.org/10.1016/j.mce.2014.05.002
Nanba K, Tsuiki M, Sawai K, Mukai K, Nishimoto K, Usui T, Tagami T, Okuno H, Yamamoto T, Shimatsu A, Katabami T, Okumura A, Kawa G, Tanabe A, Naruse M (2013) Histopathological diagnosis of primary aldosteronism using CYP11B2 immunohistochemistry. J Clin Endocrinol Metab 98:1567–1574 . https://doi.org/10.1210/jc.2012-3726
Volpe C, Höög A, Ogishima T, Mukai K, Lu M, Thorén M, Hamberger B (2013) Immunohistochemistry improves histopathologic diagnosis in primary aldosteronism. J Clin Pathol 66:351–354 . https://doi.org/10.1136/jclinpath-2012-201287
Falco EC, Daniele L, Metovic J, Bollito E, De Rosa G, Volante M, Papotti M (2021) Adrenal Rests in the Uro-genital Tract of an Adult Population. Endocr Pathol 32:375–384 . https://doi.org/10.1007/s12022-021-09685-y
Engels M, Span PN, van Herwaarden AE, Sweep FCGJ, Stikkelbroeck NMML, Claahsen-van der Grinten HL (2019) Testicular Adrenal Rest Tumors: Current Insights on Prevalence, Characteristics, Origin, and Treatment. Endocr Rev 40:973–987 . https://doi.org/10.1210/er.2018-00258
Lolis E, Juhlin CC, Nordenström A, Falhammar H (2018) Extensive Bilateral Adrenal Rest Testicular Tumors in a Patient With 3β-Hydroxysteroid Dehydrogenase Type 2 Deficiency. J Endocr Soc 2:513–517 . https://doi.org/10.1210/js.2018-00082
Ma L, Xia Y, Wang L, Liu R, Huang X, Ye T, Zhang L, Zhu Q, Li J, Jiang Y (2019) Sonographic features of the testicular adrenal rests tumors in patients with congenital adrenal hyperplasia: a single-center experience and literature review. Orphanet J Rare Dis 14:242 . https://doi.org/10.1186/s13023-019-1231-1
Corica D, Bottari A, Aversa T, Caudo D, Galletta K, Micalizzi MF, Pajno GB, Wasniewska M, Ascenti G (2019) An unusual epididymal localization of Testicular Adrenal Rest Tumor in an adolescent with congenital adrenal hyperplasia. Endocrine 66:695–698 . https://doi.org/10.1007/s12020-019-01986-x
Bouzidi L, Triki M, Charfi S, Ameur HB, Dhaou MB, Bouaziz T, Boudawara T (2021) Incidental Finding of Bilateral Ovarian Adrenal Rest Tumor in a Patient With Congenital Adrenal Hyperplasia: A Case Report and Brief Review. Pediatr Dev Pathol 24:137–141 . https://doi.org/10.1177/1093526620980614
Burman P, Falhammar H, Waldenström E, Sundin A, Bitzén U (2021) 11C-Metomidate PET/CT Detected Multiple Ectopic Adrenal Rest Tumors in a Woman With Congenital Adrenal Hyperplasia. J Clin Endocrinol Metab 106:e675–e679 . https://doi.org/10.1210/clinem/dgaa870
Uyanıkoglu H, Ozer G, Kahraman S (2020) A spontaneous pregnancy and live birth in a woman with primary infertility following the excision of an ovarian adrenal rest tumor: A rare case. Clin Exp Reprod Med 47:319–322 . https://doi.org/10.5653/cerm.2020.03692
Sisto JM, Liu FW, Geffner ME, Berman ML (2018) Para-ovarian adrenal rest tumors: gynecologic manifestations of untreated congenital adrenal hyperplasia. Gynecol Endocrinol 34:644–646 . https://doi.org/10.1080/09513590.2018.1441399
Chew KT, Abu MA, Arifuddin Y, Mohamed Ismail NA, Nasir NAM, Mohammed F, Nur Azurah AG (2017) Ectopic adrenal tissue associated with borderline mucinous cystadenoma of ovary: a case report with review of the literature. Horm Mol Biol Clin Investig 32:/j/hmbci.2017.32.issue-3/hmbci-2017–0021/hmbci-2017–0021.xml . https://doi.org/10.1515/hmbci-2017-0021
Chen H-D, Huang L-E, Zhong Z-H, Su Z, Jiang H, Zeng J, Liu J-C (2017) Ovarian Adrenal Rest Tumors Undetected by Imaging Studies and Identified at Surgery in Three Females with Congenital Adrenal Hyperplasia Unresponsive to Increased Hormone Therapy Dosage. Endocr Pathol 28:146–151 . https://doi.org/10.1007/s12022-016-9461-4
Tiosano D, Vlodavsky E, Filmar S, Weiner Z, Goldsher D, Bar-Shalom R (2010) Ovarian adrenal rest tumor in a congenital adrenal hyperplasia patient with adrenocorticotropin hypersecretion following adrenalectomy. Horm Res Paediatr 74:223–228 . https://doi.org/10.1159/000295722
Tingi E, Ogah J (2018) Ectopic adrenal rest cells of the fallopian tube: a case report and review of the literature. J Obstet Gynaecol 38:578–579 . https://doi.org/10.1080/01443615.2017.1379063
Khandakar B, Dey S, Ray PS, Sarkar R, Bhattacharyya P (2015) Ectopic Paratubal Adrenal Cell Rest Associated with Mucinous Cystadenoma of Ovary. J Clin Diagn Res 9:ED13–14 . https://doi.org/10.7860/JCDR/2015/15411.6638
N Schaumann K Hussein 2020 Hepatic and Adrenocortical Choristomas in the Placenta Fetal Pediatr Pathol 1–10 . https://doi.org/10.1080/15513815.2020.1792594
Kasajima A, Nakamura Y, Adachi Y, Takahashi Y, Fujishima F, Chiba Y, Uehara S, Watanabe M, Sasano H (2014) Oncocytic adrenocortical neoplasm arising from adrenal rest in the broad ligament of the uterus. Pathol Int 64:183–188 . https://doi.org/10.1111/pin.12154
Sasano H, Sato S, Yajima A, Akama J, Nagura H (1997) Adrenal rest tumor of the broad ligament: case report with immunohistochemical study of steroidogenic enzymes. Pathol Int 47:493–496 . https://doi.org/10.1111/j.1440-1827.1997.tb04529.x
Yokoyama H, Adachi T, Tsubouchi K, Tanaka M, Sasano H (2013) Non-functioning adrenocortical carcinoma arising in an adrenal rest: immunohistochemical study of an adult patient. Tohoku J Exp Med 229:267–270 . https://doi.org/10.1620/tjem.229.267
Akishima-Fukasawa Y, Yoshihara A, Ishikawa Y, Watanabe N, Hiroi N, Akasaka Y, Sasano H, Ishii T, Yoshino G (2011) Malignant adrenal rest tumor of the retroperitoneum producing adrenocortical steroids. Endocr Pathol 22:112–117 . https://doi.org/10.1007/s12022-011-9152-0
Cornejo KM, Afari HA, Sadow PM (2017) Adrenocortical Carcinoma Arising in an Adrenal Rest: a Case Report and Review of the Literature. Endocr Pathol 28:165–170 . https://doi.org/10.1007/s12022-017-9472-9
Senescende L, Bitolog PL, Auberger E, Zarzavadjian Le Bian A, Cesaretti M (2016) Adrenal ectopy of adult groin region: a systematic review of an unexpected anatomopathologic diagnosis. Hernia 20:879–885 . https://doi.org/10.1007/s10029-016-1535-1
Ketata S, Ketata H, Sahnoun A, FakhFakh H, Bahloul A, Mhiri MN (2008) Ectopic adrenal cortex tissue: an incidental finding during inguinoscrotal operations in pediatric patients. Urol Int 81:316–319 . https://doi.org/10.1159/000151411
Drut R, Quijano G, Altamirano ME, Jones MC, Maffessoli OB (2006) Vascular malformation and choroid plexus adrenal heterotopia: new findings in Beckwith-Wiedemann syndrome? Fetal Pediatr Pathol 25:191–197 . https://doi.org/10.1080/15513810601015704
Giner J, Esteban I, Carceller F, Saceda J, Regojo RM (2017) Spinal adrenal cortical adenoma associated with Beckwith-Wiedemann syndrome: case report and review of the literature. Childs Nerv Syst 33:1009–1013 . https://doi.org/10.1007/s00381-017-3397-y
Makino K, Kojima R, Nakamura H, Morioka M, Iyama K, Shigematsu K, Kuratsu J (2010) Ectopic adrenal cortical adenoma in the spinal region: case report and review of the literature. Brain Tumor Pathol 27:121–125 . https://doi.org/10.1007/s10014-010-0270-z
Mitchell A, Scheithauer BW, Sasano H, Hubbard EW, Ebersold MJ (1993) Symptomatic intradural adrenal adenoma of the spinal nerve root: report of two cases. Neurosurgery 32:658–661; discussion 661–662 . https://doi.org/10.1227/00006123-199304000-00024
Marchevsky AM (1995) Lung tumors derived from ectopic tissues. Semin Diagn Pathol 12:172–184
Armin A, Castelli M (1984) Congenital adrenal tissue in the lung with adrenal cytomegaly. Case report and review of the literature. Am J Clin Pathol 82:225–228 . https://doi.org/10.1093/ajcp/82.2.225
Turan H, Tarçın G, Mete Ö, Sinoplu AB, Evliyaoğlu SO, Öz B, Ercan O (2021) SILENT CORTICOTROPH TUMOR WITH ADRENOCORTICAL CHORISTOMA IN AN ELEVEN-YEAR-OLD BOY. J Clin Res Pediatr Endocrinol. https://doi.org/10.4274/jcrpe.galenos.2021.2020.0258
Mete O, Ng T, Christie-David D, McMaster J, Asa SL (2013) Silent corticotroph adenoma with adrenal cortical choristoma: a rare but distinct morphological entity. Endocr Pathol 24:162–166 . https://doi.org/10.1007/s12022-013-9256-9
Uğur Kılınç AN, Bayramoğlu Z, Ünlü Y, Keçeli AM, Dönmez Mİ (2021) Incidental ectopic adrenal cortical tissue: Retrospective analysis of 16 patients. J Pediatr Urol 17:258.e1-258.e6 . https://doi.org/10.1016/j.jpurol.2020.12.027
Yousif MQ, Salih ZT, DeYoung BR, Qasem SA (2018) Differentiating Intrarenal Ectopic Adrenal Tissue From Renal Cell Carcinoma in the Kidney. Int J Surg Pathol 26:588–592 . https://doi.org/10.1177/1066896918779449
Shigematsu K, Toriyama K, Kawai K, Takahara O (2007) Ectopic adrenal tissue in the thorax: a case report with in situ hybridization and immunohistochemical studies. Pathol Res Pract 203:543–548 . https://doi.org/10.1016/j.prp.2007.03.006
Yu H, He Y (2020) Hepatic adrenal adenoma-rare tumor on right lobe of liver: a case report and literature review. BMC Surg 20:128 . https://doi.org/10.1186/s12893-020-00780-1
Ren P-T, Fu H, He X-W (2013) Ectopic adrenal cortical adenoma in the gastric wall: case report. World J Gastroenterol 19:778–780 . https://doi.org/10.3748/wjg.v19.i5.778
Caliò A, Warfel KA, Eble JN (2019) Papillary Adenomas and Other Small Epithelial Tumors in the Kidney: An Autopsy Study. Am J Surg Pathol 43:277–287 . https://doi.org/10.1097/PAS.0000000000001189
Lottrup G, Nielsen JE, Skakkebæk NE, Juul A, Rajpert-De Meyts E (2015) Abundance of DLK1, differential expression of CYP11B1, CYP21A2 and MC2R, and lack of INSL3 distinguish testicular adrenal rest tumours from Leydig cell tumours. Eur J Endocrinol 172:491–499 . https://doi.org/10.1530/EJE-14-0810
Baba Y, Beppu T, Imai K, Masuda T, Iyama K-I, Sasano H, Baba H (2008) A case of adrenal rest tumor of the liver: Radiological imaging and immunohistochemical study of steroidogenic enzymes. Hepatol Res 38:1154–1158 . https://doi.org/10.1111/j.1872-034X.2008.00360.x
Arai K, Muro H, Suzuki M, Oba N, Ito K, Sasano H (2000) Adrenal rest tumor of the liver: a case report with immunohistochemical investigation of steroidogenesis. Pathol Int 50:244–248 . https://doi.org/10.1046/j.1440-1827.2000.01029.x
Issam R, Amine MM, Meriam T, Ahmed C, Mehdi B, Mourad HS (2021) Lymphangiomatous endothelial cyst of the adrenal gland: A case report. Urol Case Rep 39:101859 . https://doi.org/10.1016/j.eucr.2021.101859
Gubbiotti MA, Livolsi V, Montone K, Baloch Z (2021) A Cyst-ematic Analysis of the Adrenal Gland. Am J Clin Pathol aqab156 . https://doi.org/10.1093/ajcp/aqab156
Zheng W, Fung K-M, Cheng L, Osunkoya AO (2018) Benign vascular tumors, cysts, and pseudocysts of the adrenal gland: a contemporary multi-institutional clinicopathological analysis of 55 cases. Hum Pathol 82:95–102 . https://doi.org/10.1016/j.humpath.2018.07.013
Cavallaro G, Crocetti D, Paliotta A, De Gori A, Tarallo MR, Letizia C, De Toma G (2015) Cystic adrenal lesions: clinical and surgical management. The experience of a referral centre. Int J Surg 13:23–26 . https://doi.org/10.1016/j.ijsu.2014.11.023
Erickson LA, Lloyd RV, Hartman R, Thompson G (2004) Cystic adrenal neoplasms. Cancer 101:1537–1544 . https://doi.org/10.1002/cncr.20555
Mete O, Asa SL (2016) Endocrine pathology. Cambridge University Press, Cambridge
Chien H-P, Chang Y-S, Hsu P-S, Lin J-D, Wu Y-C, Chang H-L, Chuang C-K, Tsuei K-H, Hsueh C (2008) Adrenal cystic lesions: a clinicopathological analysis of 25 cases with proposed histogenesis and review of the literature. Endocr Pathol 19:274–281 . https://doi.org/10.1007/s12022-008-9046-y
Bouchaala H, Mejdoub I, Mseddi MA, Kammoun O, Rebai N, Slimen MH (2021) Giant primary hydatid cyst of the adrenal gland: A rare case report. Urol Case Rep 36:101580 . https://doi.org/10.1016/j.eucr.2021.101580
Olowu AA, Alzehairy AA (2021) A huge haemorrhagic suprarenal pseudocyst: an unusual presentation of a rare condition. BMJ Case Rep 14:e235158 . https://doi.org/10.1136/bcr-2020-235158
Zouari S, Marouene C, Bibani H, Saadi A, Sellami A, Haj Kacem L, Blel A, Bouzouita A, Derouiche A, Ben Slama R, Rammeh S, Ayed H, Chebil M (2020) Primary Hydatid Cyst of the adrenal gland: A case report and a review of the literature. Int J Surg Case Rep 70:154–158 . https://doi.org/10.1016/j.ijscr.2020.04.073
Akbulut S (2016) Incidentally detected hydatid cyst of the adrenal gland: A case report. World J Clin Cases 4:269–272 . https://doi.org/10.12998/wjcc.v4.i9.269
Suh J, Heimann A, Cohen H (2008) True adrenal mesothelial cyst in a patient with flank pain and hematuria: a case report. Endocr Pathol 19:203–205 . https://doi.org/10.1007/s12022-008-9026-2
Goel D, Enny L, Rana C, Ramakant P, Singh K, Babu S, Mishra A (2021) Cystic adrenal lesions: A report of five cases. Cancer Rep (Hoboken) 4:e1314 . https://doi.org/10.1002/cnr2.1314
Sebastiano C, Zhao X, Deng F-M, Das K (2013) Cystic lesions of the adrenal gland: our experience over the last 20 years. Hum Pathol 44:1797–1803 . https://doi.org/10.1016/j.humpath.2013.02.002
Romero-Rojas A, Bella-Cueto MR, Meza-Cabrera IA, Cabezuelo-Hernández A, García-Rojo D, Vargas-Uricoechea H, Cameselle-Teijeiro J (2013) Ectopic thyroid tissue in the adrenal gland: a report of two cases with pathogenetic implications. Thyroid 23:1644–1650 . https://doi.org/10.1089/thy.2013.0063
Tahri A, Abdellaoui W, Benyakhlef S, Boujtat K, Mahroug I, Kamaoui I, Barki A, Kettani F, Rouf S, Latrech H (2021) An Uncommon Presentation of Adrenal Cyst with Subclinical Cushing’s Syndrome: A Diagnosis Dilemma. Case Rep Endocrinol 2021:6662492 . https://doi.org/10.1155/2021/6662492
Tagawa H, Yamada T, Miyakawa T, Aida Y, Sekiguchi Z (2021) A collision between vascular adrenal cyst and adrenocortical adenoma. Radiol Case Rep 16:1294–1299 . https://doi.org/10.1016/j.radcr.2021.02.067
Babaya N, Okuda Y, Noso S, Hiromine Y, Taketomo Y, Niwano F, Ueda K, Tanaka Y, Yamazaki Y, Sasano H, Kawabata Y, Ohno Y, Ikegami H (2021) A Rare Case of Adrenal Cysts Associated With Bilateral Incidentalomas and Diffuse Hyperplasia of the Zona Glomerulosa. J Endocr Soc 5:bvaa184 . https://doi.org/10.1210/jendso/bvaa184
Sakaue T, Okuno Y, Mukai K, Fujita S, Kozawa J, Nishizawa H, Matsuoka T-A, Iwahashi H, Norikazu M, Yamazaki Y, Sasano H, Otsuki M, Shimomura I (2021) Coincidence of Large Adrenal Cyst and Prominent Hyporeninemic Hyperaldosteronism. Case Rep Endocrinol 2021:8860498 . https://doi.org/10.1155/2021/8860498
Sasano H, Miyazaki S, Sawai T, Sasano N, Nagura H, Funahashi H, Aiba M, Demura H (1992) Primary pigmented nodular adrenocortical disease (PPNAD): immunohistochemical and in situ hybridization analysis of steroidogenic enzymes in eight cases. Mod Pathol 5:23–29
Kamilaris CDC, Stratakis CA, Hannah-Shmouni F (2021) Molecular Genetic and Genomic Alterations in Cushing’s Syndrome and Primary Aldosteronism. Front Endocrinol (Lausanne) 12:632543 . https://doi.org/10.3389/fendo.2021.632543
Maillet M, Bourdeau I, Lacroix A (2020) Update on primary micronodular bilateral adrenocortical diseases. Curr Opin Endocrinol Diabetes Obes 27:132–139 . https://doi.org/10.1097/MED.0000000000000538
Espiard S, Vantyghem M-C, Assié G, Cardot-Bauters C, Raverot G, Brucker-Davis F, Archambeaud-Mouveroux F, Lefebvre H, Nunes M-L, Tabarin A, Lienhardt A, Chabre O, Houang M, Bottineau M, Stroër S, Groussin L, Guignat L, Cabanes L, Feydy A, Bonnet F, North MO, Dupin N, Grabar S, Duboc D, Bertherat J (2020) Frequency and Incidence of Carney Complex Manifestations: A Prospective Multicenter Study With a Three-Year Follow-Up. J Clin Endocrinol Metab 105:dgaa002 . https://doi.org/10.1210/clinem/dgaa002
Bertherat J, Horvath A, Groussin L, Grabar S, Boikos S, Cazabat L, Libe R, René-Corail F, Stergiopoulos S, Bourdeau I, Bei T, Clauser E, Calender A, Kirschner LS, Bertagna X, Carney JA, Stratakis CA (2009) Mutations in regulatory subunit type 1A of cyclic adenosine 5’-monophosphate-dependent protein kinase (PRKAR1A): phenotype analysis in 353 patients and 80 different genotypes. J Clin Endocrinol Metab 94:2085–2091 . https://doi.org/10.1210/jc.2008-2333
Vezzosi D, Tenenbaum F, Cazabat L, Tissier F, Bienvenu M, Carrasco CA, Laloi-Michelin M, Barrande G, Lefebvre H, Hiéronimus S, Tabarin A, Bertagna X, Legmann P, Vantyghem M-C, Bertherat J (2015) Hormonal, Radiological, NP-59 Scintigraphy, and Pathological Correlations in Patients With Cushing’s Syndrome Due to Primary Pigmented Nodular Adrenocortical Disease (PPNAD). J Clin Endocrinol Metab 100:4332–4338 . https://doi.org/10.1210/jc.2015-2174
Carney JA, Gordon H, Carpenter PC, Shenoy BV, Go VL (1985) The complex of myxomas, spotty pigmentation, and endocrine overactivity. Medicine (Baltimore) 64:270–283 . https://doi.org/10.1097/00005792-198507000-00007
Groussin L, Jullian E, Perlemoine K, Louvel A, Leheup B, Luton JP, Bertagna X, Bertherat J (2002) Mutations of the PRKAR1A gene in Cushing’s syndrome due to sporadic primary pigmented nodular adrenocortical disease. J Clin Endocrinol Metab 87:4324–4329 . https://doi.org/10.1210/jc.2002-020592
Bourdeau I, Parisien-La Salle S, Lacroix A (2020) Adrenocortical hyperplasia: A multifaceted disease. Best Pract Res Clin Endocrinol Metab 34:101386 . https://doi.org/10.1016/j.beem.2020.101386
Rockall AG, Babar SA, Sohaib SAA, Isidori AM, Diaz-Cano S, Monson JP, Grossman AB, Reznek RH (2004) CT and MR imaging of the adrenal glands in ACTH-independent cushing syndrome. Radiographics 24:435–452 . https://doi.org/10.1148/rg.242035092
Sasano H, Suzuki T, Nagura H (1994) ACTH-independent macronodular adrenocortical hyperplasia: immunohistochemical and in situ hybridization studies of steroidogenic enzymes. Mod Pathol 7:215–219
Burgess JR, Harle RA, Tucker P, Parameswaran V, Davies P, Greenaway TM, Shepherd JJ (1996) Adrenal lesions in a large kindred with multiple endocrine neoplasia type 1. Arch Surg 131:699–702 . https://doi.org/10.1001/archsurg.1996.01430190021006
Matyakhina L, Freedman RJ, Bourdeau I, Wei M-H, Stergiopoulos SG, Chidakel A, Walther M, Abu-Asab M, Tsokos M, Keil M, Toro J, Linehan WM, Stratakis CA (2005) Hereditary leiomyomatosis associated with bilateral, massive, macronodular adrenocortical disease and atypical cushing syndrome: a clinical and molecular genetic investigation. J Clin Endocrinol Metab 90:3773–3779 . https://doi.org/10.1210/jc.2004-2377
Zhu J, Cui L, Wang W, Hang X-Y, Xu A-X, Yang S-X, Dou J-T, Mu Y-M, Zhang X, Gao J-P (2013) Whole exome sequencing identifies mutation of EDNRA involved in ACTH-independent macronodular adrenal hyperplasia. Fam Cancer 12:657–667 . https://doi.org/10.1007/s10689-013-9642-y
Larose S, Bondaz L, Mermejo LM, Latour M, Prosmanne O, Bourdeau I, Lacroix A (2019) Coexistence of Myelolipoma and Primary Bilateral Macronodular Adrenal Hyperplasia With GIP-Dependent Cushing’s Syndrome. Front Endocrinol (Lausanne) 10:618 . https://doi.org/10.3389/fendo.2019.00618
Lacroix A, Bourdeau I, Lampron A, Mazzuco TL, Tremblay J, Hamet P (2010) Aberrant G-protein coupled receptor expression in relation to adrenocortical overfunction. Clin Endocrinol (Oxf) 73:1–15 . https://doi.org/10.1111/j.1365-2265.2009.03689.x
Mazzuco TL, Thomas M, Martinie M, Cherradi N, Sturm N, Feige J-J, Chabre O (2007) Cellular and molecular abnormalities of a macronodular adrenal hyperplasia causing beta-blocker-sensitive Cushing’s syndrome. Arq Bras Endocrinol Metabol 51:1452–1462 . https://doi.org/10.1590/s0004-27302007000900007
Lacroix A, Baldacchino V, Bourdeau I, Hamet P, Tremblay J (2004) Cushing’s syndrome variants secondary to aberrant hormone receptors. Trends Endocrinol Metab 15:375–382 . https://doi.org/10.1016/j.tem.2004.08.007
Lacroix A, N’Diaye N, Mircescu H, Hamet P, Tremblay J (1998) Abnormal expression and function of hormone receptors in adrenal Cushing’s syndrome. Endocr Res 24:835–843 . https://doi.org/10.3109/07435809809032694
Duan K, Mete O (2015) Clinicopathologic Correlates of Primary Aldosteronism. Arch Pathol Lab Med 139:948–954 . https://doi.org/10.5858/arpa.2014-0156-RS
Rossi GP, Bernini G, Caliumi C, Desideri G, Fabris B, Ferri C, Ganzaroli C, Giacchetti G, Letizia C, Maccario M, Mallamaci F, Mannelli M, Mattarello M-J, Moretti A, Palumbo G, Parenti G, Porteri E, Semplicini A, Rizzoni D, Rossi E, Boscaro M, Pessina AC, Mantero F, PAPY Study Investigators (2006) A prospective study of the prevalence of primary aldosteronism in 1,125 hypertensive patients. J Am Coll Cardiol 48:2293–2300 . https://doi.org/10.1016/j.jacc.2006.07.059
Mouat IC, Omata K, McDaniel AS, Hattangady NG, Talapatra D, Cani AK, Hovelson DH, Tomlins SA, Rainey WE, Hammer GD, Giordano TJ, Else T (2019) Somatic mutations in adrenocortical carcinoma with primary aldosteronism or hyperreninemic hyperaldosteronism. Endocr Relat Cancer 26:217–225 . https://doi.org/10.1530/ERC-18-0385
Griffin AC, Kelz R, LiVolsi VA (2014) Aldosterone-secreting adrenal cortical carcinoma. A case report and review of the literature. Endocr Pathol 25:344–349 . https://doi.org/10.1007/s12022-014-9307-x
Gomez-Sanchez CE, Qi X, Velarde-Miranda C, Plonczynski MW, Parker CR, Rainey W, Satoh F, Maekawa T, Nakamura Y, Sasano H, Gomez-Sanchez EP (2014) Development of monoclonal antibodies against human CYP11B1 and CYP11B2. Mol Cell Endocrinol 383:111–117 . https://doi.org/10.1016/j.mce.2013.11.022
Gao X, Yamazaki Y, Tezuka Y, Omata K, Ono Y, Morimoto R, Nakamura Y, Suzuki T, Satoh F, Sasano H (2021) Pathology of Aldosterone Biosynthesis and its Action. Tohoku J Exp Med 254:1–15 . https://doi.org/10.1620/tjem.254.1
Gao X, Yamazaki Y, Tezuka Y, Omata K, Ono Y, Morimoto R, Nakamura Y, Satoh F, Sasano H (2021) Gender differences in human adrenal cortex and its disorders. Mol Cell Endocrinol 526:111177 . https://doi.org/10.1016/j.mce.2021.111177
Yamazaki Y, Omata K, Tezuka Y, Ono Y, Morimoto R, Adachi Y, Ise K, Nakamura Y, Gomez-Sanchez CE, Shibahara Y, Kitamoto T, Nishikawa T, Ito S, Satoh F, Sasano H (2018) Tumor Cell Subtypes Based on the Intracellular Hormonal Activity in KCNJ5-Mutated Aldosterone-Producing Adenoma. Hypertension 72:632–640 . https://doi.org/10.1161/HYPERTENSIONAHA.118.10907
Yamazaki Y, Nakamura Y, Omata K, Ise K, Tezuka Y, Ono Y, Morimoto R, Nozawa Y, Gomez-Sanchez CE, Tomlins SA, Rainey WE, Ito S, Satoh F, Sasano H (2017) Histopathological Classification of Cross-Sectional Image-Negative Hyperaldosteronism. J Clin Endocrinol Metab 102:1182–1192 . https://doi.org/10.1210/jc.2016-2986
Inoue K, Yamazaki Y, Tsurutani Y, Suematsu S, Sugisawa C, Saito J, Omura M, Sasano H, Nishikawa T (2017) Evaluation of Cortisol Production in Aldosterone-Producing Adenoma. Horm Metab Res 49:847–853 . https://doi.org/10.1055/s-0043-119878
Perna V, Taylor NF, Dworakowska D, Schulte K-M, Aylwin S, Al-Hashimi F, Diaz-Cano SJ (2014) Adrenocortical adenomas with regression and myelolipomatous changes: urinary steroid profiling supports a distinctive benign neoplasm. Clin Endocrinol (Oxf) 81:343–349 . https://doi.org/10.1111/cen.12458
Duregon E, Volante M, Bollito E, Goia M, Buttigliero C, Zaggia B, Berruti A, Scagliotti GV, Papotti M (2015) Pitfalls in the diagnosis of adrenocortical tumors: a lesson from 300 consultation cases. Hum Pathol 46:1799–1807 . https://doi.org/10.1016/j.humpath.2015.08.012
Papotti M, Volante M, Duregon E, Delsedime L, Terzolo M, Berruti A, Rosai J (2010) Adrenocortical tumors with myxoid features: a distinct morphologic and phenotypical variant exhibiting malignant behavior. Am J Surg Pathol 34:973–983 . https://doi.org/10.1097/PAS.0b013e3181e2b726
Mete O, Asa SL (2012) Morphological distinction of cortisol-producing and aldosterone-producing adrenal cortical adenomas: not only possible but a critical clinical responsibility. Histopathology 60:1015–1016; author reply 1016–1017 . https://doi.org/10.1111/j.1365-2559.2011.04141.x
Nanba AT, Nanba K, Byrd JB, Shields JJ, Giordano TJ, Miller BS, Rainey WE, Auchus RJ, Turcu AF (2017) Discordance between imaging and immunohistochemistry in unilateral primary aldosteronism. Clin Endocrinol (Oxf) 87:665–672 . https://doi.org/10.1111/cen.13442
Wang H, Wang F, Zhang Y, Wen J, Dong D, Chang X, Sun H, Ma X, Cui Y, Chen S, Lu L, Ren W, Tong A, Li Y (2021) Surgical Outcomes of Aldosterone-Producing Adenoma on the Basis of the Histopathological Findings. Front Endocrinol (Lausanne) 12:663096 . https://doi.org/10.3389/fendo.2021.663096
Patel KA, Calomeni EP, Nadasdy T, Zynger DL (2014) Adrenal gland inclusions in patients treated with aldosterone antagonists (Spironolactone/Eplerenone): incidence, morphology, and ultrastructural findings. Diagn Pathol 9:147 . https://doi.org/10.1186/1746-1596-9-147
Del Gaudio AD, Del Gaudio GA (1993) Virilizing adrenocortical tumors in adult women. Report of 10 patients, 2 of whom each had a tumor secreting only testosterone. Cancer 72:1997–2003 . https://doi.org/10.1002/1097-0142(19930915)72:6<1997::aid-cncr2820720634>3.0.co;2-1
Allolio B, Fassnacht M (2006) Clinical review: Adrenocortical carcinoma: clinical update. J Clin Endocrinol Metab 91:2027–2037 . https://doi.org/10.1210/jc.2005-2639
Fassnacht M, Dekkers O, Else T, Baudin E, Berruti A, de Krijger R, Haak H, Mihai R, Assie G, Terzolo M (2018) European Society of Endocrinology Clinical Practice Guidelines on the management of adrenocortical carcinoma in adults, in collaboration with the European Network for the Study of Adrenal Tumors. Eur J Endocrinol 179:G1–G46 . https://doi.org/10.1530/EJE-18-0608
Moreno S, Guillermo M, Decoulx M, Dewailly D, Bresson R, Proye C (2006) Feminizing adreno-cortical carcinomas in male adults. A dire prognosis. Three cases in a series of 801 adrenalectomies and review of the literature. Ann Endocrinol (Paris) 67:32–38 . https://doi.org/10.1016/s0003-4266(06)72537-9
Mete O, Gucer H, Kefeli M, Asa SL (2018) Diagnostic and Prognostic Biomarkers of Adrenal Cortical Carcinoma. Am J Surg Pathol 42:201–213 . https://doi.org/10.1097/PAS.0000000000000943
Giordano TJ, Berney D, de Krijger RR, Erickson L, Fassnacht M, Mete O, Papathomas T, Papotti M, Sasano H, Thompson LDR, Volante M, Gill AJ (2020) Data set for reporting of carcinoma of the adrenal cortex: explanations and recommendations of the guidelines from the International Collaboration on Cancer Reporting. Hum Pathol. https://doi.org/10.1016/j.humpath.2020.10.001
Bisceglia M, Ludovico O, Di Mattia A, Ben-Dor D, Sandbank J, Pasquinelli G, Lau SK, Weiss LM (2004) Adrenocortical oncocytic tumors: report of 10 cases and review of the literature. Int J Surg Pathol 12:231–243 . https://doi.org/10.1177/106689690401200304
Jeremie G, Lifante JC, Chazot FB, Sajous C, Raymond P, Decaussin-Petrucci M (2021) [Myxoid variant of adrenocortical carcinoma]. Ann Pathol 41:186–191 . https://doi.org/10.1016/j.annpat.2020.12.010
Hsieh M-S, Chen J-H, Lin L-W (2011) Myxoid adrenal cortical carcinoma presenting as primary hyperaldosteronism: case report and review of the literature. Int J Surg Pathol 19:803–807 . https://doi.org/10.1177/1066896909356925
de Krijger RR, Papathomas TG (2012) Adrenocortical neoplasia: evolving concepts in tumorigenesis with an emphasis on adrenal cortical carcinoma variants. Virchows Arch 460:9–18 . https://doi.org/10.1007/s00428-011-1166-y
Papathomas TG, Duregon E, Korpershoek E, Restuccia DF, van Marion R, Cappellesso R, Sturm N, Rossi G, Coli A, Zucchini N, Stoop H, Oosterhuis W, Ventura L, Volante M, Fassina A, Dinjens WNM, Papotti M, de Krijger RR (2016) Sarcomatoid adrenocortical carcinoma: a comprehensive pathological, immunohistochemical, and targeted next-generation sequencing analysis. Hum Pathol 58:113–122 . https://doi.org/10.1016/j.humpath.2016.08.006
Zheng S, Cherniack AD, Dewal N, Moffitt RA, Danilova L, Murray BA, Lerario AM, Else T, Knijnenburg TA, Ciriello G, Kim S, Assie G, Morozova O, Akbani R, Shih J, Hoadley KA, Choueiri TK, Waldmann J, Mete O, Robertson AG, Wu H-T, Raphael BJ, Shao L, Meyerson M, Demeure MJ, Beuschlein F, Gill AJ, Sidhu SB, Almeida MQ, Fragoso MCBV, Cope LM, Kebebew E, Habra MA, Whitsett TG, Bussey KJ, Rainey WE, Asa SL, Bertherat J, Fassnacht M, Wheeler DA, Cancer Genome Atlas Research Network, Hammer GD, Giordano TJ, Verhaak RGW (2016) Comprehensive Pan-Genomic Characterization of Adrenocortical Carcinoma. Cancer Cell 29:723–736 . https://doi.org/10.1016/j.ccell.2016.04.002
Weiss LM, Medeiros LJ, Vickery AL (1989) Pathologic features of prognostic significance in adrenocortical carcinoma. Am J Surg Pathol 13:202–206 . https://doi.org/10.1097/00000478-198903000-00004
Weiss LM (1984) Comparative histologic study of 43 metastasizing and nonmetastasizing adrenocortical tumors. Am J Surg Pathol 8:163–169 . https://doi.org/10.1097/00000478-198403000-00001
Duregon E, Fassina A, Volante M, Nesi G, Santi R, Gatti G, Cappellesso R, Dalino Ciaramella P, Ventura L, Gambacorta M, Dei Tos AP, Loli P, Mannelli M, Mantero F, Berruti A, Terzolo M, Papotti M (2013) The reticulin algorithm for adrenocortical tumor diagnosis: a multicentric validation study on 245 unpublished cases. Am J Surg Pathol 37:1433–1440 . https://doi.org/10.1097/PAS.0b013e31828d387b
Volante M, Bollito E, Sperone P, Tavaglione V, Daffara F, Porpiglia F, Terzolo M, Berruti A, Papotti M (2009) Clinicopathological study of a series of 92 adrenocortical carcinomas: from a proposal of simplified diagnostic algorithm to prognostic stratification. Histopathology 55:535–543 . https://doi.org/10.1111/j.1365-2559.2009.03423.x
Pennanen M, Heiskanen I, Sane T, Remes S, Mustonen H, Haglund C, Arola J (2015) Helsinki score-a novel model for prediction of metastases in adrenocortical carcinomas. Hum Pathol 46:404–410 . https://doi.org/10.1016/j.humpath.2014.11.015
Duregon E, Cappellesso R, Maffeis V, Zaggia B, Ventura L, Berruti A, Terzolo M, Fassina A, Volante M, Papotti M (2017) Validation of the prognostic role of the “Helsinki Score” in 225 cases of adrenocortical carcinoma. Hum Pathol 62:1–7 . https://doi.org/10.1016/j.humpath.2016.09.035
Fonseca D, Murthy SS, Tagore KR, Rao BV, Thamminedi SR, Raju KVVN, Sharma R, Challa S (2017) Diagnosis of Adrenocortical Tumors by Reticulin Algorithm. Indian J Endocrinol Metab 21:734–737 . https://doi.org/10.4103/ijem.IJEM_573_16
Lin BT, Bonsib SM, Mierau GW, Weiss LM, Medeiros LJ (1998) Oncocytic adrenocortical neoplasms: a report of seven cases and review of the literature. Am J Surg Pathol 22:603–614 . https://doi.org/10.1097/00000478-199805000-00012
Brondani VB, Lacombe AMF, Mariani BM de P, Montenegro L, Soares IC, Bezerra-Neto JE, Tanno FY, Srougi V, Chambo JL, Mendonca BB, Almeida MQ, Zerbini MCN, Fragoso MCBV (2021) Low Protein Expression of both ATRX and ZNRF3 as Novel Negative Prognostic Markers of Adult Adrenocortical Carcinoma. Int J Mol Sci 22:1238 . https://doi.org/10.3390/ijms22031238
Duan K, Gucer H, Kefeli M, Asa SL, Winer DA, Mete O (2020) Immunohistochemical Analysis of the Metabolic Phenotype of Adrenal Cortical Carcinoma. Endocr Pathol 31:231–238 . https://doi.org/10.1007/s12022-020-09624-3
Duregon E, Volante M, Giorcelli J, Terzolo M, Lalli E, Papotti M (2013) Diagnostic and prognostic role of steroidogenic factor 1 in adrenocortical carcinoma: a validation study focusing on clinical and pathologic correlates. Hum Pathol 44:822–828 . https://doi.org/10.1016/j.humpath.2012.07.025
Martins-Filho SN, Almeida MQ, Soares I, Wakamatsu A, Alves VAF, Fragoso MCBV, Zerbini MCN (2021) Clinical Impact of Pathological Features Including the Ki-67 Labeling Index on Diagnosis and Prognosis of Adult and Pediatric Adrenocortical Tumors. Endocr Pathol 32:288–300 . https://doi.org/10.1007/s12022-020-09654-x
Papathomas TG, Pucci E, Giordano TJ, Lu H, Duregon E, Volante M, Papotti M, Lloyd RV, Tischler AS, van Nederveen FH, Nose V, Erickson L, Mete O, Asa SL, Turchini J, Gill AJ, Matias-Guiu X, Skordilis K, Stephenson TJ, Tissier F, Feelders RA, Smid M, Nigg A, Korpershoek E, van der Spek PJ, Dinjens WNM, Stubbs AP, de Krijger RR (2016) An International Ki67 Reproducibility Study in Adrenal Cortical Carcinoma. Am J Surg Pathol 40:569–576 . https://doi.org/10.1097/PAS.0000000000000574
Beuschlein F, Weigel J, Saeger W, Kroiss M, Wild V, Daffara F, Libé R, Ardito A, Al Ghuzlan A, Quinkler M, Oßwald A, Ronchi CL, de Krijger R, Feelders RA, Waldmann J, Willenberg HS, Deutschbein T, Stell A, Reincke M, Papotti M, Baudin E, Tissier F, Haak HR, Loli P, Terzolo M, Allolio B, Müller H-H, Fassnacht M (2015) Major prognostic role of Ki67 in localized adrenocortical carcinoma after complete resection. J Clin Endocrinol Metab 100:841–849 . https://doi.org/10.1210/jc.2014-3182
Duregon E, Molinaro L, Volante M, Ventura L, Righi L, Bolla S, Terzolo M, Sapino A, Papotti MG (2014) Comparative diagnostic and prognostic performances of the hematoxylin-eosin and phospho-histone H3 mitotic count and Ki-67 index in adrenocortical carcinoma. Mod Pathol 27:1246–1254 . https://doi.org/10.1038/modpathol.2013.230
McAteer JP, Huaco JA, Gow KW (2013) Predictors of survival in pediatric adrenocortical carcinoma: a Surveillance, Epidemiology, and End Results (SEER) program study. J Pediatr Surg 48:1025–1031 . https://doi.org/10.1016/j.jpedsurg.2013.02.017
Pinto EM, Zambetti GP (2020) What 20 years of research has taught us about the TP53 p.R337H mutation. Cancer 126:4678–4686 . https://doi.org/10.1002/cncr.33143
Ferreira AM, Brondani VB, Helena VP, Charchar HLS, Zerbini MCN, Leite LAS, Hoff AO, Latronico AC, Mendonca BB, Diz MDPE, de Almeida MQ, Fragoso MCBV (2019) Clinical spectrum of Li-Fraumeni syndrome/Li-Fraumeni-like syndrome in Brazilian individuals with the TP53 p.R337H mutation. J Steroid Biochem Mol Biol 190:250–255 . https://doi.org/10.1016/j.jsbmb.2019.04.011
Pinto EM, Zambetti GP, Rodriguez-Galindo C (2020) Pediatric adrenocortical tumours. Best Pract Res Clin Endocrinol Metab 34:101448 . https://doi.org/10.1016/j.beem.2020.101448
Stratakis CA (2008) Cushing syndrome caused by adrenocortical tumors and hyperplasias (corticotropin- independent Cushing syndrome). Endocr Dev 13:117–132 . https://doi.org/10.1159/000134829
Wagner J, Portwine C, Rabin K, Leclerc JM, Narod SA, Malkin D (1994) High frequency of germline p53 mutations in childhood adrenocortical cancer. J Natl Cancer Inst 86:1707–1710 . https://doi.org/10.1093/jnci/86.22.1707
Amadou A, Achatz MIW, Hainaut P (2018) Revisiting tumor patterns and penetrance in germline TP53 mutation carriers: temporal phases of Li-Fraumeni syndrome. Curr Opin Oncol 30:23–29 . https://doi.org/10.1097/CCO.0000000000000423
Olivier M, Goldgar DE, Sodha N, Ohgaki H, Kleihues P, Hainaut P, Eeles RA (2003) Li-Fraumeni and related syndromes: correlation between tumor type, family structure, and TP53 genotype. Cancer Res 63:6643–6650
Else T (2012) Association of adrenocortical carcinoma with familial cancer susceptibility syndromes. Mol Cell Endocrinol 351:66–70 . https://doi.org/10.1016/j.mce.2011.12.008
Pinto EM, Chen X, Easton J, Finkelstein D, Liu Z, Pounds S, Rodriguez-Galindo C, Lund TC, Mardis ER, Wilson RK, Boggs K, Yergeau D, Cheng J, Mulder HL, Manne J, Jenkins J, Mastellaro MJ, Figueiredo BC, Dyer MA, Pappo A, Zhang J, Downing JR, Ribeiro RC, Zambetti GP (2015) Genomic landscape of paediatric adrenocortical tumours. Nat Commun 6:6302 . https://doi.org/10.1038/ncomms7302
Dehner LP, Hill DA (2009) Adrenal cortical neoplasms in children: why so many carcinomas and yet so many survivors? Pediatr Dev Pathol 12:284–291 . https://doi.org/10.2350/08-06-0489.1
Wieneke JA, Thompson LDR, Heffess CS (2003) Adrenal cortical neoplasms in the pediatric population: a clinicopathologic and immunophenotypic analysis of 83 patients. Am J Surg Pathol 27:867–881 . https://doi.org/10.1097/00000478-200307000-00001
Picard C, Orbach D, Carton M, Brugieres L, Renaudin K, Aubert S, Berrebi D, Galmiche L, Dujardin F, Leblond P, Thomas-Teinturier C, Dijoud F (2019) Revisiting the role of the pathological grading in pediatric adrenal cortical tumors: results from a national cohort study with pathological review. Mod Pathol 32:546–559 . https://doi.org/10.1038/s41379-018-0174-8
Lam AK-Y (2021) Adrenocortical Carcinoma: Updates of Clinical and Pathological Features after Renewed World Health Organisation Classification and Pathology Staging. Biomedicines 9:175 . https://doi.org/10.3390/biomedicines9020175
Michalkiewicz E, Sandrini R, Figueiredo B, Miranda ECM, Caran E, Oliveira-Filho AG, Marques R, Pianovski M a. D, Lacerda L, Cristofani LM, Jenkins J, Rodriguez-Galindo C, Ribeiro RC (2004) Clinical and outcome characteristics of children with adrenocortical tumors: a report from the International Pediatric Adrenocortical Tumor Registry. J Clin Oncol 22:838–845 . https://doi.org/10.1200/JCO.2004.08.085
Sandrini R, Ribeiro RC, DeLacerda L (1997) Childhood adrenocortical tumors. J Clin Endocrinol Metab 82:2027–2031 . https://doi.org/10.1210/jcem.82.7.4057
Gupta N, Rivera M, Novotny P, Rodriguez V, Bancos I, Lteif A (2018) Adrenocortical Carcinoma in Children: A Clinicopathological Analysis of 41 Patients at the Mayo Clinic from 1950 to 2017. Horm Res Paediatr 90:8–18 . https://doi.org/10.1159/000488855
Papotti M, Duregon E, Volante M, McNicol AM (2014) Pathology of the adrenal cortex: a reappraisal of the past 25 years focusing on adrenal cortical tumors. Endocr Pathol 25:35–48 . https://doi.org/10.1007/s12022-013-9291-6
van Slooten H, Schaberg A, Smeenk D, Moolenaar AJ (1985) Morphologic characteristics of benign and malignant adrenocortical tumors. Cancer 55:766–773 . https://doi.org/10.1002/1097-0142(19850215)55:4<766::aid-cncr2820550414>3.0.co;2-7
Hough AJ, Hollifield JW, Page DL, Hartmann WH (1979) Prognostic factors in adrenal cortical tumors. A mathematical analysis of clinical and morphologic data. Am J Clin Pathol 72:390–399 . https://doi.org/10.1093/ajcp/72.3.390
Giordano TJ (2011) The argument for mitotic rate-based grading for the prognostication of adrenocortical carcinoma. Am J Surg Pathol 35:471–473 . https://doi.org/10.1097/PAS.0b013e31820bcf21
Mete O, Asa SL, Baker TP, Berman MA, Davis J, Erickson L, Giordano TJ, Rudzinski ER, Tischler A, Thompson LDR, Weiss LM (2021) College of American Pathologists (CAP) Protocol for the examination of specimens from patients with carcinoma of the adrenal gland. Version:4.2.1.0. Protocol Posting Date: November 2021. https://www.cap.org/protocols-and-guidelines/cancer-reporting-tools/cancer-protocol-templates
Stojadinovic A, Brennan MF, Hoos A, Omeroglu A, Leung DHY, Dudas ME, Nissan A, Cordon-Cardo C, Ghossein RA (2003) Adrenocortical adenoma and carcinoma: histopathological and molecular comparative analysis. Mod Pathol 16:742–751 . https://doi.org/10.1097/01.MP.0000081730.72305.81
Arola J, Salmenkivi K, Liu J, Kahri AI, Heikkilä P (2000) p53 and Ki67 in adrenocortical tumors. Endocr Res 26:861–865 . https://doi.org/10.3109/07435800009048609
Soon PSH, Gill AJ, Benn DE, Clarkson A, Robinson BG, McDonald KL, Sidhu SB (2009) Microarray gene expression and immunohistochemistry analyses of adrenocortical tumors identify IGF2 and Ki-67 as useful in differentiating carcinomas from adenomas. Endocr Relat Cancer 16:573–583 . https://doi.org/10.1677/ERC-08-0237
Wachenfeld C, Beuschlein F, Zwermann O, Mora P, Fassnacht M, Allolio B, Reincke M (2001) Discerning malignancy in adrenocortical tumors: are molecular markers useful? Eur J Endocrinol 145:335–341 . https://doi.org/10.1530/eje.0.1450335
Elhassan YS, Altieri B, Berhane S, Cosentini D, Calabrese A, Haissaguerre M, Kastelan D, Fragoso MCBV, Bertherat J, Al Ghuzlan A, Haak H, Boudina M, Canu L, Loli P, Sherlock M, Kimpel O, Laganà M, Sitch AJ, Kroiss M, Arlt W, Terzolo M, Berruti A, Deeks JJ, Libé R, Fassnacht M, Ronchi CL (2021) S-GRAS score for prognostic classification of adrenocortical carcinoma: an international, multicenter ENSAT study. Eur J Endocrinol 186:25–36 . https://doi.org/10.1530/EJE-21-0510
Cree IA (2022) From Counting Mitoses to Ki67 Assessment: Technical Pitfalls in the New WHO Classification of Endocrine and Neuroendocrine Tumors. Endocr Pathol. https://doi.org/10.1007/s12022-021-09701-1
Cree IA, Tan PH, Travis WD, Wesseling P, Yagi Y, White VA, Lokuhetty D, Scolyer RA (2021) Counting mitoses: SI(ze) matters! Mod Pathol 34:1651–1657 . https://doi.org/10.1038/s41379-021-00825-7
Faillot S, Foulonneau T, Néou M, Espiard S, Garinet S, Vaczlavik A, Jouinot A, Rondof W, Septier A, Drougat L, Hécale-Perlemoine K, Ragazzon B, Rizk-Rabin M, Sibony M, Bonnet-Serrano F, Guibourdenche J, Libé R, Groussin L, Dousset B, de Reyniès A, Bertherat J, Assié G (2021) Genomic classification of benign adrenocortical lesions. Endocr Relat Cancer 28:79–95 . https://doi.org/10.1530/ERC-20-0128
Mohan DR, Lerario AM, Else T, Mukherjee B, Almeida MQ, Vinco M, Rege J, Mariani BMP, Zerbini MCN, Mendonca BB, Latronico AC, Marie SKN, Rainey WE, Giordano TJ, Fragoso MCBV, Hammer GD (2019) Targeted Assessment of G0S2 Methylation Identifies a Rapidly Recurrent, Routinely Fatal Molecular Subtype of Adrenocortical Carcinoma. Clin Cancer Res 25:3276–3288 . https://doi.org/10.1158/1078-0432.CCR-18-2693
Giordano TJ, Kuick R, Else T, Gauger PG, Vinco M, Bauersfeld J, Sanders D, Thomas DG, Doherty G, Hammer G (2009) Molecular classification and prognostication of adrenocortical tumors by transcriptome profiling. Clin Cancer Res 15:668–676 . https://doi.org/10.1158/1078-0432.CCR-08-1067
de Reyniès A, Assié G, Rickman DS, Tissier F, Groussin L, René-Corail F, Dousset B, Bertagna X, Clauser E, Bertherat J (2009) Gene expression profiling reveals a new classification of adrenocortical tumors and identifies molecular predictors of malignancy and survival. J Clin Oncol 27:1108–1115 . https://doi.org/10.1200/JCO.2008.18.5678
Mete O, Pakbaz S, Lerario AM, Giordano TJ, Asa SL (2021) Significance of Alpha-inhibin Expression in Pheochromocytomas and Paragangliomas. Am J Surg Pathol 45:1264–1273 . https://doi.org/10.1097/PAS.0000000000001715
Mete O, Asa SL (2020) Structure, Function, and Morphology in the Classification of Pituitary Neuroendocrine Tumors: the Importance of Routine Analysis of Pituitary Transcription Factors. Endocr Pathol 31:330–336 . https://doi.org/10.1007/s12022-020-09646-x
Schmitt A, Saremaslani P, Schmid S, Rousson V, Montani M, Schmid DM, Heitz PU, Komminoth P, Perren A (2006) IGFII and MIB1 immunohistochemistry is helpful for the differentiation of benign from malignant adrenocortical tumours. Histopathology 49:298–307 . https://doi.org/10.1111/j.1365-2559.2006.02505.x
Domènech M, Grau E, Solanes A, Izquierdo A, Del Valle J, Carrato C, Pineda M, Dueñas N, Pujol M, Lázaro C, Capellà G, Brunet J, Navarro M (2021) Characteristics of Adrenocortical Carcinoma Associated With Lynch Syndrome. J Clin Endocrinol Metab 106:318–325 . https://doi.org/10.1210/clinem/dgaa833
Raymond VM, Everett JN, Furtado LV, Gustafson SL, Jungbluth CR, Gruber SB, Hammer GD, Stoffel EM, Greenson JK, Giordano TJ, Else T (2013) Adrenocortical carcinoma is a lynch syndrome-associated cancer. J Clin Oncol 31:3012–3018 . https://doi.org/10.1200/JCO.2012.48.0988
Pozdeyev N, Fishbein L, Gay LM, Sokol ES, Hartmaier R, Ross JS, Darabi S, Demeure MJ, Kar A, Foust LJ, Koc K, Bowles DW, Leong S, Wierman ME, Kiseljak-Vassiliades K (2021) Targeted genomic analysis of 364 adrenocortical carcinomas. Endocr Relat Cancer 28:671–681 . https://doi.org/10.1530/ERC-21-0040
D’Avolio A, De Francia S, Basile V, Cusato J, De Martino F, Pirro E, Piccione F, Ardito A, Zaggia B, Volante M, Di Perri G, Terzolo M (2013) Influence of the CYP2B6 polymorphism on the pharmacokinetics of mitotane. Pharmacogenet Genomics 23:293–300 . https://doi.org/10.1097/FPC.0b013e3283606cb2
Volante M, Terzolo M, Fassnacht M, Rapa I, Germano A, Sbiera S, Daffara F, Sperone P, Scagliotti G, Allolio B, Papotti M, Berruti A (2012) Ribonucleotide reductase large subunit (RRM1) gene expression may predict efficacy of adjuvant mitotane in adrenocortical cancer. Clin Cancer Res 18:3452–3461 . https://doi.org/10.1158/1078-0432.CCR-11-2692
Else T, Lerario AM, Everett J, Haymon L, Wham D, Mullane M, Wilson TL, Rainville I, Rana H, Worth AJ, Snyder NW, Blair IA, McKay R, Kilbridge K, Hammer G, Barletta J, Vaidya A (2017) Adrenocortical carcinoma and succinate dehydrogenase gene mutations: an observational case series. Eur J Endocrinol 177:439–444 . https://doi.org/10.1530/EJE-17-0358
Williams TA, Monticone S, Schack VR, Stindl J, Burrello J, Buffolo F, Annaratone L, Castellano I, Beuschlein F, Reincke M, Lucatello B, Ronconi V, Fallo F, Bernini G, Maccario M, Giacchetti G, Veglio F, Warth R, Vilsen B, Mulatero P (2014) Somatic ATP1A1, ATP2B3, and KCNJ5 mutations in aldosterone-producing adenomas. Hypertension 63:188–195 . https://doi.org/10.1161/HYPERTENSIONAHA.113.01733
Choi M, Scholl UI, Yue P, Björklund P, Zhao B, Nelson-Williams C, Ji W, Cho Y, Patel A, Men CJ, Lolis E, Wisgerhof MV, Geller DS, Mane S, Hellman P, Westin G, Åkerström G, Wang W, Carling T, Lifton RP (2011) K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science 331:768–772 . https://doi.org/10.1126/science.1198785
Nanba K, Yamazaki Y, Bick N, Onodera K, Tezuka Y, Omata K, Ono Y, Blinder AR, Tomlins SA, Rainey WE, Satoh F, Sasano H (2020) Prevalence of Somatic Mutations in Aldosterone-Producing Adenomas in Japanese Patients. J Clin Endocrinol Metab 105. https://doi.org/10.1210/clinem/dgaa595
Azizan EAB, Poulsen H, Tuluc P, Zhou J, Clausen MV, Lieb A, Maniero C, Garg S, Bochukova EG, Zhao W, Shaikh LH, Brighton CA, Teo AED, Davenport AP, Dekkers T, Tops B, Küsters B, Ceral J, Yeo GSH, Neogi SG, McFarlane I, Rosenfeld N, Marass F, Hadfield J, Margas W, Chaggar K, Solar M, Deinum J, Dolphin AC, Farooqi IS, Striessnig J, Nissen P, Brown MJ (2013) Somatic mutations in ATP1A1 and CACNA1D underlie a common subtype of adrenal hypertension. Nat Genet 45:1055–1060 . https://doi.org/10.1038/ng.2716
Scholl UI, Goh G, Stölting G, de Oliveira RC, Choi M, Overton JD, Fonseca AL, Korah R, Starker LF, Kunstman JW, Prasad ML, Hartung EA, Mauras N, Benson MR, Brady T, Shapiro JR, Loring E, Nelson-Williams C, Libutti SK, Mane S, Hellman P, Westin G, Åkerström G, Björklund P, Carling T, Fahlke C, Hidalgo P, Lifton RP (2013) Somatic and germline CACNA1D calcium channel mutations in aldosterone-producing adenomas and primary aldosteronism. Nat Genet 45:1050–1054 . https://doi.org/10.1038/ng.2695
Nanba K, Blinder AR, Rege J, Hattangady NG, Else T, Liu C-J, Tomlins SA, Vats P, Kumar-Sinha C, Giordano TJ, Rainey WE (2020) Somatic CACNA1H Mutation As a Cause of Aldosterone-Producing Adenoma. Hypertension 75:645–649 . https://doi.org/10.1161/HYPERTENSIONAHA.119.14349
Scholl UI, Stölting G, Nelson-Williams C, Vichot AA, Choi M, Loring E, Prasad ML, Goh G, Carling T, Juhlin CC, Quack I, Rump LC, Thiel A, Lande M, Frazier BG, Rasoulpour M, Bowlin DL, Sethna CB, Trachtman H, Fahlke C, Lifton RP (2015) Recurrent gain of function mutation in calcium channel CACNA1H causes early-onset hypertension with primary aldosteronism. Elife 4:e06315 . https://doi.org/10.7554/eLife.06315
Beuschlein F, Boulkroun S, Osswald A, Wieland T, Nielsen HN, Lichtenauer UD, Penton D, Schack VR, Amar L, Fischer E, Walther A, Tauber P, Schwarzmayr T, Diener S, Graf E, Allolio B, Samson-Couterie B, Benecke A, Quinkler M, Fallo F, Plouin P-F, Mantero F, Meitinger T, Mulatero P, Jeunemaitre X, Warth R, Vilsen B, Zennaro M-C, Strom TM, Reincke M (2013) Somatic mutations in ATP1A1 and ATP2B3 lead to aldosterone-producing adenomas and secondary hypertension. Nat Genet 45:440–444, 444e1–2 . https://doi.org/10.1038/ng.2550
Wannachalee T, Zhao L, Nanba K, Nanba AT, Shields JJ, Rainey WE, Auchus RJ, Turcu AF (2019) Three Discrete Patterns of Primary Aldosteronism Lateralization in Response to Cosyntropin During Adrenal Vein Sampling. J Clin Endocrinol Metab 104:5867–5876 . https://doi.org/10.1210/jc.2019-01182
Lenzini L, Rossitto G, Maiolino G, Letizia C, Funder JW, Rossi GP (2015) A Meta-Analysis of Somatic KCNJ5 K(+) Channel Mutations In 1636 Patients With an Aldosterone-Producing Adenoma. J Clin Endocrinol Metab 100:E1089-1095 . https://doi.org/10.1210/jc.2015-2149
Azizan EAB, Lam BYH, Newhouse SJ, Zhou J, Kuc RE, Clarke J, Happerfield L, Marker A, Hoffman GJ, Brown MJ (2012) Microarray, qPCR, and KCNJ5 sequencing of aldosterone-producing adenomas reveal differences in genotype and phenotype between zona glomerulosa- and zona fasciculata-like tumors. J Clin Endocrinol Metab 97:E819-829 . https://doi.org/10.1210/jc.2011-2965
Nanba K, Omata K, Gomez-Sanchez CE, Stratakis CA, Demidowich AP, Suzuki M, Thompson LDR, Cohen DL, Luther JM, Gellert L, Vaidya A, Barletta JA, Else T, Giordano TJ, Tomlins SA, Rainey WE (2019) Genetic Characteristics of Aldosterone-Producing Adenomas in Blacks. Hypertension 73:885–892 . https://doi.org/10.1161/HYPERTENSIONAHA.118.12070
Nanba K, Omata K, Else T, Beck PCC, Nanba AT, Turcu AF, Miller BS, Giordano TJ, Tomlins SA, Rainey WE (2018) Targeted Molecular Characterization of Aldosterone-Producing Adenomas in White Americans. J Clin Endocrinol Metab 103:3869–3876 . https://doi.org/10.1210/jc.2018-01004
Omata K, Anand SK, Hovelson DH, Liu C-J, Yamazaki Y, Nakamura Y, Ito S, Satoh F, Sasano H, Rainey WE, Tomlins SA (2017) Aldosterone-Producing Cell Clusters Frequently Harbor Somatic Mutations and Accumulate With Age in Normal Adrenals. J Endocr Soc 1:787–799 . https://doi.org/10.1210/js.2017-00134
Fernandes-Rosa FL, Giscos-Douriez I, Amar L, Gomez-Sanchez CE, Meatchi T, Boulkroun S, Zennaro M-C (2015) Different Somatic Mutations in Multinodular Adrenals With Aldosterone-Producing Adenoma. Hypertension 66:1014–1022 . https://doi.org/10.1161/HYPERTENSIONAHA.115.05993
Wu V-C, Wang S-M, Chueh S-CJ, Yang S-Y, Huang K-H, Lin Y-H, Wang J-J, Connolly R, Hu Y-H, Gomez-Sanchez CE, Peng K-Y, Wu K-D (2017) The prevalence of CTNNB1 mutations in primary aldosteronism and consequences for clinical outcomes. Sci Rep 7:39121 . https://doi.org/10.1038/srep39121
Åkerström T, Maharjan R, Sven Willenberg H, Cupisti K, Ip J, Moser A, Stålberg P, Robinson B, Alexander Iwen K, Dralle H, Walz MK, Lehnert H, Sidhu S, Gomez-Sanchez C, Hellman P, Björklund P (2016) Activating mutations in CTNNB1 in aldosterone producing adenomas. Sci Rep 6:19546 . https://doi.org/10.1038/srep19546
Juhlin CC, Goh G, Healy JM, Fonseca AL, Scholl UI, Stenman A, Kunstman JW, Brown TC, Overton JD, Mane SM, Nelson-Williams C, Bäckdahl M, Suttorp A-C, Haase M, Choi M, Schlessinger J, Rimm DL, Höög A, Prasad ML, Korah R, Larsson C, Lifton RP, Carling T (2015) Whole-exome sequencing characterizes the landscape of somatic mutations and copy number alterations in adrenocortical carcinoma. J Clin Endocrinol Metab 100:E493-502 . https://doi.org/10.1210/jc.2014-3282
Bonnet S, Gaujoux S, Launay P, Baudry C, Chokri I, Ragazzon B, Libé R, René-Corail F, Audebourg A, Vacher-Lavenu M-C, Groussin L, Bertagna X, Dousset B, Bertherat J, Tissier F (2011) Wnt/β-catenin pathway activation in adrenocortical adenomas is frequently due to somatic CTNNB1-activating mutations, which are associated with larger and nonsecreting tumors: a study in cortisol-secreting and -nonsecreting tumors. J Clin Endocrinol Metab 96:E419-426 . https://doi.org/10.1210/jc.2010-1885
Tadjine M, Lampron A, Ouadi L, Bourdeau I (2008) Frequent mutations of beta-catenin gene in sporadic secreting adrenocortical adenomas. Clin Endocrinol (Oxf) 68:264–270 . https://doi.org/10.1111/j.1365-2265.2007.03033.x
Berthon A, Drelon C, Ragazzon B, Boulkroun S, Tissier F, Amar L, Samson-Couterie B, Zennaro M-C, Plouin P-F, Skah S, Plateroti M, Lefèbvre H, Sahut-Barnola I, Batisse-Lignier M, Assié G, Lefrançois-Martinez A-M, Bertherat J, Martinez A, Val P (2014) WNT/β-catenin signalling is activated in aldosterone-producing adenomas and controls aldosterone production. Hum Mol Genet 23:889–905 . https://doi.org/10.1093/hmg/ddt484
Espiard S, Knape MJ, Bathon K, Assié G, Rizk-Rabin M, Faillot S, Luscap-Rondof W, Abid D, Guignat L, Calebiro D, Herberg FW, Stratakis CA, Bertherat J (2018) Activating PRKACB somatic mutation in cortisol-producing adenomas. JCI Insight 3. https://doi.org/10.1172/jci.insight.98296
Thiel A, Reis A-C, Haase M, Goh G, Schott M, Willenberg HS, Scholl UI (2015) PRKACA mutations in cortisol-producing adenomas and adrenal hyperplasia: a single-center study of 60 cases. Eur J Endocrinol 172:677–685 . https://doi.org/10.1530/EJE-14-1113
Di Dalmazi G, Kisker C, Calebiro D, Mannelli M, Canu L, Arnaldi G, Quinkler M, Rayes N, Tabarin A, Laure Jullié M, Mantero F, Rubin B, Waldmann J, Bartsch DK, Pasquali R, Lohse M, Allolio B, Fassnacht M, Beuschlein F, Reincke M (2014) Novel somatic mutations in the catalytic subunit of the protein kinase A as a cause of adrenal Cushing’s syndrome: a European multicentric study. J Clin Endocrinol Metab 99:E2093-2100 . https://doi.org/10.1210/jc.2014-2152
Sato Y, Maekawa S, Ishii R, Sanada M, Morikawa T, Shiraishi Y, Yoshida K, Nagata Y, Sato-Otsubo A, Yoshizato T, Suzuki H, Shiozawa Y, Kataoka K, Kon A, Aoki K, Chiba K, Tanaka H, Kume H, Miyano S, Fukayama M, Nureki O, Homma Y, Ogawa S (2014) Recurrent somatic mutations underlie corticotropin-independent Cushing’s syndrome. Science 344:917–920 . https://doi.org/10.1126/science.1252328
Goh G, Scholl UI, Healy JM, Choi M, Prasad ML, Nelson-Williams C, Kunstman JW, Kuntsman JW, Korah R, Suttorp A-C, Dietrich D, Haase M, Willenberg HS, Stålberg P, Hellman P, Akerström G, Björklund P, Carling T, Lifton RP (2014) Recurrent activating mutation in PRKACA in cortisol-producing adrenal tumors. Nat Genet 46:613–617 . https://doi.org/10.1038/ng.2956
Beuschlein F, Fassnacht M, Assié G, Calebiro D, Stratakis CA, Osswald A, Ronchi CL, Wieland T, Sbiera S, Faucz FR, Schaak K, Schmittfull A, Schwarzmayr T, Barreau O, Vezzosi D, Rizk-Rabin M, Zabel U, Szarek E, Salpea P, Forlino A, Vetro A, Zuffardi O, Kisker C, Diener S, Meitinger T, Lohse MJ, Reincke M, Bertherat J, Strom TM, Allolio B (2014) Constitutive activation of PKA catalytic subunit in adrenal Cushing’s syndrome. N Engl J Med 370:1019–1028 . https://doi.org/10.1056/NEJMoa1310359
Bertherat J, Groussin L, Sandrini F, Matyakhina L, Bei T, Stergiopoulos S, Papageorgiou T, Bourdeau I, Kirschner LS, Vincent-Dejean C, Perlemoine K, Gicquel C, Bertagna X, Stratakis CA (2003) Molecular and functional analysis of PRKAR1A and its locus (17q22-24) in sporadic adrenocortical tumors: 17q losses, somatic mutations, and protein kinase A expression and activity. Cancer Res 63:5308–5319
Sheekey E, Narita M (2021) p53 in senescence - it’s a marathon not a sprint. FEBS J. https://doi.org/10.1111/febs.16325
Malkin D, Li FP, Strong LC, Fraumeni JF, Nelson CE, Kim DH, Kassel J, Gryka MA, Bischoff FZ, Tainsky MA (1990) Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 250:1233–1238 . https://doi.org/10.1126/science.1978757
Assié G, Letouzé E, Fassnacht M, Jouinot A, Luscap W, Barreau O, Omeiri H, Rodriguez S, Perlemoine K, René-Corail F, Elarouci N, Sbiera S, Kroiss M, Allolio B, Waldmann J, Quinkler M, Mannelli M, Mantero F, Papathomas T, De Krijger R, Tabarin A, Kerlan V, Baudin E, Tissier F, Dousset B, Groussin L, Amar L, Clauser E, Bertagna X, Ragazzon B, Beuschlein F, Libé R, de Reyniès A, Bertherat J (2014) Integrated genomic characterization of adrenocortical carcinoma. Nat Genet 46:607–612 . https://doi.org/10.1038/ng.2953
de Krijger RE, Bertherat J (2016) 5th International ACC Symposium: Classification of Adrenocortical Cancers from Pathology to Integrated Genomics: Real Advances or Lost in Translation? Horm Cancer 7:3–8 . https://doi.org/10.1007/s12672-015-0242-1
Gicquel C, Bertagna X, Schneid H, Francillard-Leblond M, Luton JP, Girard F, Le Bouc Y (1994) Rearrangements at the 11p15 locus and overexpression of insulin-like growth factor-II gene in sporadic adrenocortical tumors. J Clin Endocrinol Metab 78:1444–1453 . https://doi.org/10.1210/jcem.78.6.7911125
Henry I, Jeanpierre M, Couillin P, Barichard F, Serre JL, Journel H, Lamouroux A, Turleau C, de Grouchy J, Junien C (1989) Molecular definition of the 11p15.5 region involved in Beckwith-Wiedemann syndrome and probably in predisposition to adrenocortical carcinoma. Hum Genet 81:273–277 . https://doi.org/10.1007/BF00279003
Sasaki H, Ishihara K, Kato R (2000) Mechanisms of Igf2/H19 imprinting: DNA methylation, chromatin and long-distance gene regulation. J Biochem 127:711–715 . https://doi.org/10.1093/oxfordjournals.jbchem.a022661
Barreau O, Assié G, Wilmot-Roussel H, Ragazzon B, Baudry C, Perlemoine K, René-Corail F, Bertagna X, Dousset B, Hamzaoui N, Tissier F, de Reynies A, Bertherat J (2013) Identification of a CpG island methylator phenotype in adrenocortical carcinomas. J Clin Endocrinol Metab 98:E174-184 . https://doi.org/10.1210/jc.2012-2993
de Fraipont F, El Atifi M, Cherradi N, Le Moigne G, Defaye G, Houlgatte R, Bertherat J, Bertagna X, Plouin P-F, Baudin E, Berger F, Gicquel C, Chabre O, Feige J-J (2005) Gene expression profiling of human adrenocortical tumors using complementary deoxyribonucleic Acid microarrays identifies several candidate genes as markers of malignancy. J Clin Endocrinol Metab 90:1819–1829 . https://doi.org/10.1210/jc.2004-1075
Giordano TJ, Thomas DG, Kuick R, Lizyness M, Misek DE, Smith AL, Sanders D, Aljundi RT, Gauger PG, Thompson NW, Taylor JMG, Hanash SM (2003) Distinct transcriptional profiles of adrenocortical tumors uncovered by DNA microarray analysis. Am J Pathol 162:521–531 . https://doi.org/10.1016/S0002-9440(10)63846-1
Assié G, Jouinot A, Fassnacht M, Libé R, Garinet S, Jacob L, Hamzaoui N, Neou M, Sakat J, de La Villéon B, Perlemoine K, Ragazzon B, Sibony M, Tissier F, Gaujoux S, Dousset B, Sbiera S, Ronchi CL, Kroiss M, Korpershoek E, De Krijger R, Waldmann J, Quinkler M, Haissaguerre M, Tabarin A, Chabre O, Luconi M, Mannelli M, Groussin L, Bertagna X, Baudin E, Amar L, Coste J, Beuschlein F, Bertherat J (2019) Value of Molecular Classification for Prognostic Assessment of Adrenocortical Carcinoma. JAMA Oncol 5:1440–1447 . https://doi.org/10.1001/jamaoncol.2019.1558
Claahsen-van der Grinten HL, Speiser PW, Ahmed SF, Arlt W, Auchus RJ, Falhammar H, Flück CE, Guasti L, Huebner A, Kortmann BBM, Krone N, Merke DP, Miller WL, Nordenström A, Reisch N, Sandberg DE, Stikkelbroeck NMML, Touraine P, Utari A, Wudy SA, White PC (2022) Congenital Adrenal Hyperplasia-Current Insights in Pathophysiology, Diagnostics, and Management. Endocr Rev 43:91–159 . https://doi.org/10.1210/endrev/bnab016
Speiser PW, Azziz R, Baskin LS, Ghizzoni L, Hensle TW, Merke DP, Meyer-Bahlburg HFL, Miller WL, Montori VM, Oberfield SE, Ritzen M, White PC (2010) A Summary of the Endocrine Society Clinical Practice Guidelines on Congenital Adrenal Hyperplasia due to Steroid 21-Hydroxylase Deficiency. Int J Pediatr Endocrinol 2010:494173 . https://doi.org/10.1155/2010/494173
Falhammar H, Wedell A, Nordenström A (2015) Biochemical and genetic diagnosis of 21-hydroxylase deficiency. Endocrine 50:306–314 . https://doi.org/10.1007/s12020-015-0731-6
228. Calissendorff J, Juhlin CC, Sundin A, Bancos I, Falhammar H (2021) Adrenal myelolipomas. Lancet Diabetes Endocrinol 9:767–775 . https://doi.org/10.1016/S2213-8587(21)00178-9
Nermoen I, Falhammar H (2020) Prevalence and Characteristics of Adrenal Tumors and Myelolipomas in Congenital Adrenal Hyperplasia: A Systematic Review and Meta-Analysis. Endocr Pract 26:1351–1365 . https://doi.org/10.4158/EP-2020-0058
Horvath A, Giatzakis C, Tsang K, Greene E, Osorio P, Boikos S, Libè R, Patronas Y, Robinson-White A, Remmers E, Bertherat J, Nesterova M, Stratakis CA (2008) A cAMP-specific phosphodiesterase (PDE8B) that is mutated in adrenal hyperplasia is expressed widely in human and mouse tissues: a novel PDE8B isoform in human adrenal cortex. Eur J Hum Genet 16:1245–1253 . https://doi.org/10.1038/ejhg.2008.85
Horvath A, Giatzakis C, Robinson-White A, Boikos S, Levine E, Griffin K, Stein E, Kamvissi V, Soni P, Bossis I, de Herder W, Carney JA, Bertherat J, Gregersen PK, Remmers EF, Stratakis CA (2006) Adrenal hyperplasia and adenomas are associated with inhibition of phosphodiesterase 11A in carriers of PDE11A sequence variants that are frequent in the population. Cancer Res 66:11571–11575 . https://doi.org/10.1158/0008-5472.CAN-06-2914
Ise T, Shimoda A, Takakuwa H, Kato T, Izumiya Y, Shimizu K, Suzuki T, Sasano H, Yokoyama H, Kobayashi K (2001) A chimeric CYP11B1/CYP11B2 gene in glucocorticoid-insuppressible familial hyperaldosteronism. Clin Endocrinol (Oxf) 55:131–134 . https://doi.org/10.1046/j.1365-2265.2001.01192.x
Scholl UI, Stölting G, Schewe J, Thiel A, Tan H, Nelson-Williams C, Vichot AA, Jin SC, Loring E, Untiet V, Yoo T, Choi J, Xu S, Wu A, Kirchner M, Mertins P, Rump LC, Onder AM, Gamble C, McKenney D, Lash RW, Jones DP, Chune G, Gagliardi P, Choi M, Gordon R, Stowasser M, Fahlke C, Lifton RP (2018) CLCN2 chloride channel mutations in familial hyperaldosteronism type II. Nat Genet 50:349–354 . https://doi.org/10.1038/s41588-018-0048-5
Else T, Rodriguez-Galindo C (2016) 5th International ACC Symposium: Hereditary Predisposition to Childhood ACC and the Associated Molecular Phenotype: 5th International ACC Symposium Session: Not Just for Kids! Horm Cancer 7:36–39 . https://doi.org/10.1007/s12672-015-0244-z
Morin E, Mete O, Wasserman JD, Joshua AM, Asa SL, Ezzat S (2012) Carney complex with adrenal cortical carcinoma. J Clin Endocrinol Metab 97:E202-206 . https://doi.org/10.1210/jc.2011-2321
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics Approval
Not applicable.
Consent for Publication
All authors consent to publication.
Conflict of Interest
Dr. Ozgur Mete is the Editor-in-Chief of Endocrine Pathology. Dr. Lori A. Erickson is the Senior Associate Editor of Endocrine Pathology. This review article was handled by an independent editor and peer-reviewed as per the journal standards. The remaining authors declare that they have no competing interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Mete, O., Erickson, L.A., Juhlin, C.C. et al. Overview of the 2022 WHO Classification of Adrenal Cortical Tumors. Endocr Pathol 33, 155–196 (2022). https://doi.org/10.1007/s12022-022-09710-8
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12022-022-09710-8
Keywords
- Adrenal cortical adenoma
- Adrenal cortical carcinoma
- Adrenal cortical hyperplasia
- Adrenal cortical nodular disease
- Primary bilateral macronodular adrenal cortical hyperplasia
- IGF2
- Primary aldosteronism
- Cushing syndrome
- Virilization and feminization
- Adrenal incidentaloma
- Reticulin algorithm
- Reticulin histochemistry
- Lynch syndrome
- WHO classification
- Biomarkers