
Abstract
Background
A wide variety of pathogens could be maintained and transmitted by Haemaphysalis longicornis. The aim of this study is to systematically examine the variety of pathogens carried by Haemaphysalis longicornis, an importnatn vector, in tick-borne diseases epidemic area, and to estimate the risk of human infection imposed by tick bites.
Methods
Adult questing ticks were collected in Xinyang, central China. Genomic DNA and RNA were extracted from 144 H. longicornis ticks individually, and sequenced respectively as the templates for high-throughput sequencing. Clean reads were compared against the database of NCBI nucleotide collection and specific PCR was performed to confirm the presence of pathogen. Phylogenetic analysis was performed to explore the evolutionary status of pathogens.
Results
The assignment of reads to taxa based on BLASTN results revealed the existence of several potential pathogens, including Anaplasma spp., Rickettsia spp., Babesia sp., as well as severe fever with thrombocytopenia syndrome bunyavirus (SFTSV). Comfirmantory PCR assays revealed the existence of Anaplasma bovis (13/144, 9.03%), Anaplasma centrale (2/144, 1.39%), Rickettsia heilongjiangensis (3/144, 2.08%), Rickettsia sp. LON-13 (1/144, 0.69%), Rickettsia raoultii (5/144, 3.47%), Babesia sp. (1/144, 0.69%). SFTSV accounted for the highest detected pathogen with a positive rate of 18.75% (27/144). Three of the ticks (2.08%) were co-infected with SFTSV and A. bovis.
Conclusion
Our study provided a broadened list of microorganism that harbored by H. longicornis. In previously unrecognized endemic regions, prokaryotic and eukaryotic infection including Anaplasma spp., Rickettsiae spp., and Babesia spp. should be considered, along with the well-known SFTSV for patients with tick bites history. A novel Babesia species was identified in local natural foci, which needs further investigation in the future.
Similar content being viewed by others

Multilingual abstracts
Please see Additional file 1 for translations of the abstract into the five official working languages of the United Nations.
Background
Haemaphysalis longicornis is one of the most important tick species that imposes the risks of tick-bonre disease infection, which is widely distributed in the Asia-Pacific region, including Korea, Japan, Australia, the Pacific Islands, and New Zealand [1,2,3,4,5]. Common hosts of H. longicornis include goats, cattle, sheep, Bos mutus, donkeys, pigs, Cervus elaphus, cats, Rattus norregicus, Mus culus, Erinaceus europaeus, Mustela sibirica, Trichosurus vulpecula, and some birds, along with human beings, which is commonly considered as the definitive host [5,6,7]. A wide variety of pathogens can be maintained and transmitted by H.longicornis. A remarkable example is the novel phlebovirus in the Bunyaviridae family (severe fever with thrombocytopenia syndrome virus, SFTSV), which was newly identified to be a causal agent of severe fever with thrombocytopenia syndrome (SFTS) in China and other neighbouring countries [8,9,10]. SFTS is largely considered to be a tick-associated disease, as high proportion of patients had tick exposure before disease onset [8]. H. longicornis, the most prevelant tick species that infests human in SFTS-endemic areas, was determined to be a competent vector of SFTSV by an experimental maintenance and transmission study [11]. In this context, we performed a metagenomic analysis to provide an inventory of predicted and unexpected pathogenic agents carried by H. longicornis ticks, captured in Xinyang Administrative Area, Henan Province in central China.
Xinyang is the region mostly heavily inflicted by SFTSV, reporting 48% of SFTS cases in China [12]. The region has a humid subtropical climate with annual precipitation of around 1100 mm. The southern part of Xinyang, stretching across the Dabie Mountain range, is an important habitat for H. longicornis ticks in China [13]. Xinyang is also reported to be epidemic area of many tick-borne diseases such as human granulocytic anaplasmosis, spotted fever and typhus fever [14, 15]. These findings indicated that H. longicornis might be potential vector of causative agents. Therefore we expect the metagenomic analysis in this region might provide a broad list of the pathogens carried by this important vector, thereby offering the potential humans infection risk imposed by tick bites.
Methods
Ticks collection and DNA/RNA extraction
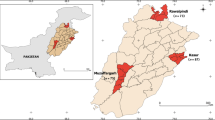
The questing ticks were captured from 10 sampling sites across Xinyang. Sample sites were selected from representative geographical areas where patients got tick bite in Dabie Mountain area and plain area. (Dabie Mountain area: 31.69 N, 115.45E; 31.59 N, 115.30E; 31.76 N, 115.27E; 31.70 N, 114.82E; 31.66 N, 115.01E; plain: 32.48 N, 115,31E; 32.40 N, 115.24E; 32.38 N, 115.45E; 32.25 N, 114.90E; 32.33 N, 115.11E), Henan Province, in May 2013. The ticks were collected by dragging over the vegetation layer during daytime. Morphologic features were used to identify the species and developmental stage of ticks by an entomologist (Sun Y) [16]. 10–15 H. longicornis from each site were randomly selected and totally 144 ticks were included in our study. The predominant tick species was confirmed to be H. longicornis. Live H. longicornis ticks were subsequently sterilised in 75% ethanol and then washed up with deionised water for 5 min each to remove environmental contaminants. DNA and RNA were extracted from single tick using TIANamp DNA/RNA extraction kit (Tiangen, Beijing, China) according to the manufacturer’s instructions.
Library preparation for high-throughput sequencing
The DNA/RNA extracted from 144 ticks were pooled respectively as the templates for library preparation. For prokaryotic pathogen screening, the pooled DNA was amplified in 50 μl reactions: 26.5 μl pure water, 10 μl 5 × phusion HF buffer (Thermo scientific, Hudson, NH, USA), 1 μl 10 mmol/L dNTPs (Thermo scientific), 2 μl of the 16S F and 16S R primers [17] (Invitrogen Corp., Carlsbad, CA), 0.5 μl Phusion High-Fidelity DNA polymerase (Thermo scientific), and 10 μl DNA(≈ 50 ng). The amplification was conducted according to a protocol involving initial denaturation for 30s at 98 °C and 35 cycles of 98 °C for 10 s, 55 °C for 30 s, 72 °C for 1 min, followed by a final extension at 72 °C for 7 min. Agarose gel with target fragments was purified using TIANgel Midi Purification Kit (Tiangen, Beijing, China) according to the manufacturer’s instruction. For eukaryotic pathogen screening, the pooled DNA was amplified as previously described using primers BTH-1F: cctgmgaracggctaccacatct and BTH-R: ttgcgaccatactccccca [18]. For virus screening, the pooled RNA was reverse transcripted with random hexmers to cDNA using Thermo Scientific Revert Aid First Strand cDNA Synthesis Kit (Thermo, Waltham, USA). The purified 16S and 18S PCR product, cDNA from RNA and DNA were sequenced with Ion Personal Genome Machine (PGM) System as described by Vogel and others [19]. Quality of the library was analysed using the Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA).
Bioinformatics analysis
After sequencing, the individual sequence reads were filtered within the PGM software to remove low quality sequences. Sequences matching the PGM 3′adaptor were also automatically trimmed. Sequences that were shorter than 100 bp were deleted with an in-house python script. Each clean read was compared against the NCBI nucleotide collection (non-redundant nt database) using Blastn with default parameters (−v 5, −b 5, −w 35). The hit with the highest “Max Score” for every query was picked up, and the resulting hits were grouped by species according to its GI number. The number of reads and the total matched length of each species were calculated.
Specific PCR for detection of three pathogens in ticks
Based on the results from the alignments, specific PCR was performed to confirm the presence of pathogens in the DNA and RNA of individual ticks. The genes used for phylogenetic analysis were as below: 16S rDNA for Anaplasma spp., gltA gene for Rickettsia spp., 18S rDNA for Babesia spp. and the S segment for SFTSV. Total RNA from each sample (0.1–1 μg) was used for reverse transcription using the SuperScript III First-Strand Synthesis System (Invitrogen). DNA extracted from each sample and cDNA reverse-transcripted from each sample were used as PCR template.
The targeted genes were amplified from template in 30 μl PCR mixtures containing 120 mmol/L of each primer (Table 1) [20,21,22,23,24], 60 mmol/L of each dNTP, 3 μl of 10 × rTaq PCR buffer (Takara, Dalian, China), and 1.5 U of rTaq DNA polymerase (Takara, Dalian, China). Amplification cycling conditions were as follows: denaturation for 3 min at 94 °C and 35 cycles of 94 °C for 45 s, 56 °C for 35 s, 72 °C for 1 min, followed by a final extension at 72 °C for 15 min. Amplified products were visualized with SYBR® Safe (Thermo, Waltham, USA) after electrophoresis in 2% agarose gel. For nested PCR, the second round were performed using the same reaction and cycling conditions as described above, and 1 μl of the first-round PCR production were used as template.
The primers for amplification in this study are presented in Table 1. The PCR ampliconswere directly sequenced using an ABI 3730 machine (Applied Biosystems, Foster City, CA, USA). To reveal the evolutionary status of identified pathogens of interest, the phylogenetic analysis was performed using the Mega 5.0 software (http://www.megasoftware.net). The alignment was made under default parameters. Phylogenetic analysis was performed by the Maximum Likelihood method. All positions containing alignment gaps and missing data were deleted (complete-deletion).
Results
Taxonomic classification
The DNA and RNA extraction from a total of 144 adult H. longicornis ticks were respectively pooled into one sample, and subject to the high throughput sequencing. The fragments length of the constructed library ranged between 300 to 400 bp (Fig. 1). Totally 826 561 kb data were obtained, while 706 862 kb were of high quality (>Q20). A total of 6 926 470 reads were obtained with mean length of 233 bp (Fig. 2). The assignment of unassembled sequence reads to taxa based on BLASTN results revealed the existence of several pathogens, including Anaplasma spp., Rickettsia spp., Babesia spp., as well as SFTSV (Table 2).

The fragments’ distribution of sequencing library. Quality of the libraries were analysed using the Agilent 2100 Bioanalyzer. The fragments length of the constructed library mainly ranged between 300 and 400 bp

Run Report. Pseudo-colour image showing percent loading across the physical surface. This sequencing run had a 86% loading, which ensures a high ISP density. The number of total reads was up to 6 926 470 and 71% of the reads were usable. The median read length was 256 bp
Confirmation of the pathogens and phylogenetic analysis
The remaining DNA/RNA from the 144 H. longicornis ticks were individually detected for the presence of these pathogens by PCR or RT-PCR (Additional file 2: Figure S1). The sequence analysis revealed the existence of Anaplasma bovis (Ehrlichia bovis, 13/144, 9.03%), Anaplasma centrale (2/144, 1.39%), Rickettsia heilongjiangensis (3/144, 2.08%), Rickettsia sp. LON-13 (1/144, 0.69%), Rickettsia raoultii (5/144, 3.47%), Babesia sp. (0.69%, 1/144) (Table 3). Three ticks were co-infected with SFTSV and A. bovis.

Phylogenetic analysis of confirmed pathogens of interests. Sequences were aligned using the MEGA5 (Version5.1) software package. Phylogenetic analysis was performed by the Maximum Likelihood method. All positions containing alignment gaps and missing data were deleted (complete-deletion). Bars indicate the percentage of sequence divergence. All positions containing alignment gaps and missing data were deleted (Complete-deletion). a Phylogenetic tree of bacteria belonging to Anaplasma, inferred from comparison of the partial 16S rDNA gene sequences. b Phylogenetic tree of bacteria belonging to Rickettsia, inferred from comparison of the partial gltA gene sequences. c Phylogenetic tree of bacteria belonging to Babesia, inferred from comparison of the partial 18S rDNA gene sequences. d Phylogenetic tree of bacteria belonging to SFTSV, inferred from comparison of the partial S segment
By phylogenetic analysis, the Anaplasma spp. (KY007144-KY007156, KX817983, KX817984) identified in the H. longicornis ticks were shown to be clustered with A. centrale and A. bovis (Fig. 3a). Rickettsia, which is recognized as medically important arthropod-vectored pathogens, was found involved in symbiosis within 9 H. longicornis ticks, and clustered into three different branchs (KX817986-KX817988, KX965745- KX96575), R. heilongjiangensis, R. raoultii, and Rickettsia sp. LON-13 (Fig. 3b). One of the H. longicornis ticks was found carrying Babesia sp. (GenBank No. KX817985). The phylogenetic analysis showed the 18S rRNA gene was most close to Babesia sp. MA*361–1 (GenBank No.AB251610) and Babesia sp. SAP*091(GenBank No.AB251609) (Fig. 3c). The represented SFTSV sequences (KX817989, KX965742-KX965744) obtained from four H. longicornis ticks were clustered with sequences obtained from local SFTS patients [25] (Fig. 3d).
Discussion
In recent years, due to intense research interest in SFTS, there has been an increasing number of pathogens that were detected from H. longicornis ticks in SFTS endemic region [26]. Our study, based on the Next Generation Sequencing (NGS) methods, provided a broadened list of the microorganism harboured by this tick species, including prokaryotic and eukaryotic pathogens and viruses. In addition to SFTSV, other tick-borne pathogens, A. bovis (13/144, 9.03%), A. centrale (2/144, 1.39%), R. heilongjiangensis (3/144, 2.08%), R. raoultii (5/144, 3.47%), were detected as well.
Members of the genus Anaplasma include A. phagocytophilum, A.marginale, A. bovis, A. ovis, A. platys and A. centrale, all of which are obligate intracellular bacteria that infect a variety of cell types [27]. In China, wild and domestic ruminants play active roles as Anaplasma.sppcarriers and reservoirs. The presence of A. bovis in sheep and goats has been reported in Northwest, Central and Southern China [28, 29]. A. centrale has been reported in goats and sheep from South-eastern China [30]. Our results indicate the epidemic of A. bovis and A. centrale in central China. In the study, spotted fever group Rickettsia, including R. heilongjiangensis, R. raoultii and Rickettsia sp. LON-13 was detected in H. longicornis ticks in the same region. R. heilongjiangensis can cause spotted fever in humans, which was detected in Dermacentor. silvarum and H. longicornis ticks in Heilongjiang Province and Zhejiang Province [31,32,33]. R. raoultii, the predominant Rickettsia found in Dermacentor silvarum ticks in China-Russia border areas [34], was also found in this area. According to phylogenetic analysis, Rickettsia sp. LON-13 was clustered in spotted fever group, and further investigation should be taken in livestock and human. Babesia sp. was detected in only one tick. According to phylogenetic analysis of 18S rRNA gene, Babesia from this tick was clustered between Babesia divergens and Babesia gibsoni, closing to Babesia sp. MA*361–1. This finding indicates the possible existence of a new Babesia species in local environment. Its prevalence in livestock and the risk to human beings requires to be investigated by an enhanced surveillance in the future.
Co-infection of A.bovis with SFTSV was identified, which is interesting while unexpected. Before the discovery of SFTSV, co-infection between Babesia microti (KJ715163) and Rickettsia sp. (KJ715194), Theileria luwenshuni (KJ715167) and Ehrlichia sp. (KJ715196), T. luwenshuni (KJ715168) and Anaplasma phagocytophilum (KJ715199) in H. longicornis ticks had been reported [26]. In addition, dual infection with A. phagocytophilum and B. microti in a Rattus norvegicus was found in this region [35]. However, the evidence of human dual infection was only recently reported from our previous research on the existence of CRT (Candidatus Rickettsia tarasevichiae) infection in clinical diagnosed SFTS patients [36]. Taken all of the findings together, we propose that in SFTS endemic areas bacterial infection including Anaplasma spp., Rickettsiae spp., Babesia spp., and co-infecitons of various tick-borne pathogens should be considered for patients after tick bite.
Since the discovery of SFTS in China, enormous efforts have been applied to identify SFTSV infection in both human being and the predominant tick species. However, other tick-borne pathogens were largely neglected. Due to nonspecific clinical presentation and less access to confirmatory laboratory findings, it is rather difficult to make diagnosis. In addition, novel Rickettsia and Babesia species of undetermined pathogenicity continue to be detected from ticks, highly possible to cause human illness. The current findings might have important application in determining the etiological determination in SFTS endemic region with H. longicornis as the predominant tick species, the most important tick-borne infectious disease not only in China, but also in countries where SFTSV infection has been reported.
Conclusions
In the study, a broadened list of the microorganism harboured by H. longicornis was provided. In SFTS region with abundant H. longicornis, prokaryotic infection including Anaplasma spp., Rickettsiae spp., and Babesia spp. should also be considered. The possibility of their co-infection with tick-borne viral pathogens in H. longicornis ticks, and dual infection in human, should be acknowledged by the clinicians. Specially, a novel Babesia species was identified in local natural foci, which needs further investigation in the future.
Abbreviations
- SFTS:
-
Severe fever with thrombocytopenia syndrome
- SFTSV:
-
Severe fever with thrombocytopenia syndrome virus
References
Park SW, Song BG, Shin EH, Yun SM, Han MG, Park MY, et al. Prevalence of severe fever with thrombocytopenia syndrome virus in Haemaphysalis longicornis ticks in South Korea. Ticks Tick Borne Dis. 2014;5(6):975–7.
Tateno M, Sunahara A, Nakanishi N, Izawa M, Matsuo T, Setoguchi A, et al. Molecular survey of arthropod-borne pathogens in ticks obtained from Japanese wildcats. Ticks Tick Borne Dis. 2015;6(3):281–9.
Hammer JF, Emery D, Bogema DR, Jenkins C. Detection of Theileria orientalis genotypes in Haemaphysalis longicornis ticks from southern Australia. Parasit Vectors. 2015;8(1):229.
Mediannikov O, Davoust B, Cabre O, Rolain JM, Raoult D. Bartonellae in animals and vectors in New Caledonia. Comp Immunol Microbiol Infect Dis. 2011;34(6):497–501.
Heath A. Biology, ecology and distribution of the tick, Haemaphysalis longicornis Neumann (Acari: Ixodidae) in New Zealand. N Z Vet J. 2015;7:1–32.
Zhang YZ, Li J, Li JC, Hu XB, Liang DF, An DS. Preliminary investigation of ticks in Yigong, Tibet. Medical Journal of National Defending Forces in Southwest China. 2007;4:519–20. (in Chinese)
Deng GF, Jiang ZJ. Economic insect fauna of China. Fasc 39. Acarina: Ixodidae. Beijing: Science Press; 1991. p. 181–5.
Yu XJ, Liang MF, Zhang SY, Liu Y, Li JD, Sun YL, et al. Fever with thrombocytopenia associated with a novel bunyavirus in China. N Engl J Med. 2011;364(16):1523–32.
Takahashi T, Maeda K, Suzuki T, Ishido A, Shigeoka T, Tominaga T, et al. The first identification and retrospective study of severe fever with thrombocytopenia syndrome in Japan. J Infect Dis. 2014;209(6):816–27.
Yun SM, Lee WG, Ryou J, Yang SC, Park SW, Roh JY, et al. Severe fever with thrombocytopenia syndrome virus in ticks collected from humans, South Korea, 2013. Emerg Infect Dis. 2014;20(8):1358–61.
Luo LM, Zhao L, Wen HL, Zhang ZT, Liu JW, Fang LZ, et al. Haemaphysalis longicornis ticks as reservoir and vector of severe fever with thrombocytopenia syndrome virus in China. Emerg Infect Dis. 2015;21(10):1770–6.
Ding F, Zhang W, Wang L, Hu W, Soares Magalhaes RJ, sun H, et al. epidemiologic features of severe fever with thrombocytopenia syndrome in China, 2011-2012. Clin Infect Dis. 2013;56(11):1682–3.
Zhang YZ, Zhou DJ, Qin XC, Tian JH, Xiong Y, Wang JB, et al. The ecology, genetic diversity, and phylogeny of Huaiyangshan virus in China. J Virol. 2012;86(5):2864–8.
Ge XC. Epidemic analysisi of human granulocytic anaplasmosis in Shihe, Xinyang. Chinese Journal of Public Health Management. 2012;28(3):304–5. (in Chinese)
Zheng HJ. The investigation on scrub typhus group, Typhi group and Spored fever Group in Xinyang district of Henan. Zhengzhou University. 2013:23–4. (in Chinese)
Yamaguti N, Tipton VJ, Keegan HL. S. T. Ticks of Japan, Korea and the Ryukyu islands. Brighan young university Sci Bull. 1971;15:1–226.
Zhuang L, Wang CY, Tong YG, Tang F, Yang H, Liu W, et al. Discovery of Rickettsia species in Dermacentor niveus Neumann ticks by investigating the diversity of bacterial communities. Ticks Tick Borne Dis. 2014;5(5):564–8.
Criado-Fornelio A, Martinez-Marcos A, Buling-Sarana A, Barba-Carretero JC. Molecular studies on Babesia, Theileria and Hepatozoon in southern Europe Part I. Vet Parasitol. 2003;113(3–4):189–201.
Vogel U, Szczepanowski R, Claus H, Junemann S, Prior K, Harmsen D. Ion torrent personal genome machine sequencing for genomic typing of Neisseria meningitidis for rapid determination of multiple layers of typing information. J Clin Microbiol. 2012;50(6):1889–94.
Li H, Jiang JF, Liu W, Zheng YC, Huo QB, Tang K, et al. Human infection with Candidatus Neoehrlichia mikurensis, China. Emerg Infect Dis. 2012;18(10):1636–9.
Wen B, Jian R, Zhang Y, Chen R. Simultaneous detection of Anaplasma marginale and a new Ehrlichia species closely related to Ehrlichia chaffeensis by sequence analyses of 16S ribosomal DNA in Boophilus microplus ticks from Tibet. J Clin Microbiol. 2002;40(9):3286–90.
Kawabuchi T, Tsuji M, Sado A, Matoba Y, Asakawa M, Ishihara C. Babesia microti-like parasites detected in feral raccoons (Procyon lotor) captured in Hokkaido, Japan. J Vet Med Sci. 2005;67(8):825–7.
Das DP, Malik SV, Rawool DB, Das S, Shoukat S, Gandham RK, et al. Isolation of Coxiella burnetii from bovines with history of reproductive disorders in India and phylogenetic inference based on the partial sequencing of IS1111 element. Infect Genet Evol. 2014;22:67–71.
Zhang YZ, He YW, Dai YA, Xiong Y, Zheng H, Zhou DJ, et al. Hemorrhagic fever caused by a novel Bunyavirus in China: pathogenesis and correlates of fatal outcome. Clin Infect Dis. 2012;54(4):527–33.
Liu W, Lu QB, Cui N, Li H, Wang LY, Liu K, et al. Case-fatality ratio and effectiveness of ribavirin therapy among hospitalized patients in China who had severe fever with thrombocytopenia syndrome. Clin Infect Dis. 2013;57(9):1292–9.
Chen Z, Liu Q, Liu JQ, Xu BL, Lv S, Xia S, et al. Tick-borne pathogens and associated co-infections in ticks collected from domestic animals in Central China. Parasit Vectors. 2014;7:237.
Dumler JS, Barbet AF, Bekker CP, Dasch GA, Palmer GH, Ray SC, et al. Reorganization of genera in the families Rickettsiaceae and Anaplasmataceae in the order Rickettsiales: unification of some species of Ehrlichia with Anaplasma, Cowdria with Ehrlichia and Ehrlichia with Neorickettsia, descriptions of six new species combinations and designation of Ehrlichia equi and 'HGE agent' as subjective synonyms of Ehrlichia phagocytophila. Int J Syst Evol Microbiol. 2001;51(6):2145–65.
Liu Z, Ma M, Wang Z, Wang J, Peng Y, Li Y, et al. Molecular survey and genetic identification of Anaplasma species in goats from central and southern China. Appl Environ Microbiol. 2012;78(2):464–70.
Yang J, Li Y, Liu Z, Liu J, Niu Q, Ren Q, et al. Molecular detection and characterization of Anaplasma spp. in sheep and cattle from Xinjiang, Northwest China. Parasit Vectors. 2015;8:108.
Ge Y, Yin H, Rikihisa Y, Pan W, Yin H. Molecular detection of tick-borne rickettsiales in goats and sheep from southeastern China. Vector Borne Zoonotic Dis. 2016;16(5):309–16.
Fournier PE, Dumler JS, Greub G, Zhang J, Wu Y, Raoult D. Gene sequence-based criteria for identification of new rickettsia isolates and description of Rickettsia heilongjiangensis sp. nov. J Clin Microbiol. 2003;41(12):5456–65.
Jiao Y, Wen B, Chen M, Niu D, Zhang J, Qiu L. Analysis of immunoprotectivity of the recombinant OmpA of Rickettsia heilongjiangensis. Ann N Y Acad Sci. 2005;1063:261–5.
Sun J, Lin J, Gong Z, Chang Y, Ye X, Gu S, et al. Detection of spotted fever group Rickettsiae in ticks from Zhejiang Province, China. Exp Appl Acarol. 2015;65(3):403–11.
Wen J, Jiao D, Wang JH, Yao DH, Liu ZX, Zhao G, et al. Rickettsia raoultii, the predominant Rickettsia found in Dermacentor silvarum ticks in China-Russia border areas. Exp Appl Acarol. 2014;63(4):579–85.
Zhao XG, Li H, Sun Y, Zhang YY, Jiang JF, Liu W, et al. Dual infection with Anaplasma phagocytophilum and Babesia microti in a Rattus norvegicus, China. Ticks Tick Borne Dis. 2013;4(5):399–402.
Liu W, Li H, Lu QB, Cui N, Yang ZD, Hu JG, et al. Candidatus rickettsia tarasevichiae infection in eastern Central China: a case series. Ann Intern Med. 2016;164(10):641–8.
Acknowledgements
We would like to thank all of the study participants for their commitment. We also specifically thank Mr. Hang Fan, Mr. Wei Wang, and other staff members at State Key Laboratory of Pathogen and Biosecurity, Beijing Institute of Microbiology and Epidemiology, for their efforts in data mining.
Funding
This study was funded by the Natural Science Foundation of China (81621005, 81472005, 81473023).
Availability of data and materials
The datasets collected and/or analysed during the current study are available from the corresponding author upon reasonable request. Please contact author for data requests.
Author information
Authors and Affiliations
Contributions
WL, ZCF and LZ conceived the study and led the writing of this article. LZ conducted the experiments, analysed the results, and wrote the manuscript. JD conducted the experiments and wrote the manuscript. XMC conducted the experiments. HL, FT, PHZ and JGH collected the ticks in the field. YGT led the data analysis. All authors reviewed and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional files
Additional file 1:
Multilingual abstracts in the five official working languages of the United Nations. (PDF 532 kb)
Additional file 2:
Figure S1. Experimental confirmation of predicted pathogens of interest predicted by bioinformatics. A), PCR amplification of the H. longicornis ticks to confirm the predicted Anaplasma spp.. S1 represented PCR amplification with primer Out1, Out2; S2 represented PCR amplification with primer Out2F, 317Pan. B), The second run of nested PCR (with primer RpCS877F, RpCS1258R) amplification of H.longicornis ticks to confirm the predicted Rickettsia spp.. C), The second run of nested PCR (with primer Piro1F, Piro5.5R) amplification of H. longicornis ticks to confirm the predicted Babesia spp.. D), The PCR (with primer BNYS1-F, BNYS1-R) amplification of the H. longicornis ticks to confirm the predicted SFTSV. (JPEG 80 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

{kind=link}
Cite this article
Zhuang, L., Du, J., Cui, XM. et al. Identification of tick-borne pathogen diversity by metagenomic analysis in Haemaphysalis longicornis from Xinyang, China. Infect Dis Poverty 7, 45 (2018). https://doi.org/10.1186/s40249-018-0417-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40249-018-0417-4