
Abstract
Background
Alternative splicing of the Pkm gene product generates the PKM1 and PKM2 isoforms of the glycolytic enzyme pyruvate kinase. PKM2 expression is associated with embryogenesis, tissue regeneration, and cancer. PKM2 is also the pyruvate kinase isoform expressed in most wild-type adult tissues, with PKM1 restricted primarily to skeletal muscle, heart, and brain. To interrogate the functional requirement for PKM2 during tumor initiation in an autochthonous mouse model for soft tissue sarcoma (STS), we used a conditional Pkm2 allele (Pkm2fl) to abolish PKM2 expression.
Results
PKM2 deletion slowed tumor onset but did not abrogate eventual tumor outgrowth. PKM2-null sarcoma cells expressed PKM1 with tumors containing a high number of infiltrating PKM2 expressing stromal cells. End-stage PKM2-null tumors showed increased proliferation compared to tumors with a wild-type Pkm2 allele, and tumor metabolite analysis revealed metabolic changes associated with PKM2 loss.
Conclusions
While PKM2 is not required for soft tissue sarcoma growth, PKM2 expression may facilitate initiation of this tumor type. Because these data differ from what has been observed in other cancer models where PKM2 has been deleted, they argue that the consequences of PKM2 loss during tumor initiation are dependent on the tumor type.
Similar content being viewed by others

Background
Proliferating cells, including cancer cells, favor aerobic glycolysis and display increased glucose consumption and lactate production compared to many non-proliferating cells. Expression of the M2 isoform of pyruvate kinase (PKM2), the glycolytic enzyme that catalyzes the last step in glycolysis, has been implicated in promoting this metabolic state of cancer cells.
The pyruvate kinase gene, Pkm, encodes the PKM1 and PKM2 isoforms, which are the result of alternative splicing of mutually exclusive exons. The PKM1 isoform includes exon 9 but not exon 10 and shows a pattern of expression that is restricted in the adult largely to the heart, brain, and skeletal muscle. PKM2, on the other hand, includes exon 10 but not exon 9. Expression of the PKM2 isoform is widespread in adult epithelial tissues and has been associated with embryogenesis, tissue regeneration, and cancer [1, 2]. Importantly, expression of PKM2 impacts the fate of glucose, in part through regulated enzymatic activity. While PKM1 is a constitutively active enzyme, PKM2 enzymatic activity can be inhibited by a variety of cellular signaling events, many of which are associated with proliferation [3,4,5,6,7]. Consistent with this notion, activation of PKM2 enzymatic activity in cancer cells through either exogenous expression of PKM1 or use of small molecule activators leads to delayed xenograft tumor formation [8, 9].
PKM2 is highly expressed in many tumor types, and a number of metabolic and non-metabolic functions in cancer have been attributed to PKM2 [1, 5, 8, 10,11,12,13,14,15]. However, whether PKM2 is required for tumor formation remains a controversial question. Recent studies of PKM2 function in autochthonous mouse models of cancer have revealed a complicated picture of PKM2 function in cancer. For example, loss of PKM2 in a leukemia and breast cancer model led to contrasting results: while PKM2 loss limited progression in one case, it led to accelerated tumor growth in the other [16, 17]. PKM2 deletion also accelerated tumor growth in a mouse medulloblastoma model [18]. Loss of PKM2 in a colon cancer model driven by APC loss did not alter tumor initiation, growth, or progression [19]. Finally, germline loss of PKM2 led to late onset of hepatocellular carcinoma, at least in part through non-cell autonomous means [2]. In no cases was PKM2 absolutely required for tumor formation; however, the different response to PKM2 loss in various tumors might be indicative of distinct tissue metabolic requirements.
In this study, we used a conditional Pkm2 allele (M2fl) to assess the requirement for PKM2 in a mouse model of soft tissue sarcoma. In this model, Cre-mediated activation of oncogenic Kras and loss of p53 (KP) in the hind limb of mice leads to the formation of soft tissue sarcomas within 3 months [20]. Loss of PKM2 in this context led to delayed tumor initiation and decreased tumor penetrance. The tumors that formed in KP M2fl/fl mice expressed PKM1 instead of PKM2. Metabolite analysis of KP M2−/− sarcoma tissue showed changes in the pool sizes of several metabolites, arguing that metabolism in these tumors changes as a result of PKM2 deletion and consequent PKM1 expression.
Methods
Mouse strains and treatments
All animal work was conducted in accordance with a protocol approved by the MIT Committee on Animal Care. KP and M2 flox mouse strains have been described previously [17, 21]. All animals were maintained on a mixed C57BL/6J × 129SvJ genetic background.
Sarcoma tumorigenesis was initiated as previously described by intramuscular infection of mice with Ad5-CMV-Cre (University of Iowa Gene Transfer Core) [20, 22]. For histologic analysis, tissues were fixed in 4% formalin, embedded in paraffin, and sections were stained with hematoxylin and eosin (H&E).
Immunofluorescence
For immunofluorescence analysis, mice were perfused with PBS, followed by perfusion with 2% paraformaldehyde (PFA). After an overnight fixation in 4% PFA at 4 °C, tissues were placed in 30% sucrose overnight at 4 °C with shaking. Tissue was allowed to equilibrate in OCT:PBS (1:1) solution and embedded in OCT (Tissue-Tek). Frozen sections (10 μm thick) were cut on a CryoStar NX70 (Thermo Scientific). PKM2 antibody (1:500, CST 4053) was used.
Immunohistochemistry (IHC)
IHC was performed on 4-μm-thick sections using ABC Vectastain kit (Vector Labs) with antibodies to PKM1 (1:500, CST 7067), PKM2 (1:500, CST 4053), and phospho-Histone H3 (1:200, CST 9701). The staining was visualized with DAB (Vector Labs, SK-4100), and the slides were counterstained with hematoxylin. For dual staining, DAB and ImmPACT VIP (Vector Labs, SK-4605) were used. Hematoxylin and eosin (H&E) staining was performed on a Varistain Gemini automated slide stainer (Thermo Shandon). Soft tissue sarcoma human tissue array, SO2081, was purchased from US Biomax Inc. (Rockville, MD USA). The TMA was scored for PKM1 and PKM2 intensity. Tumors that showed no positive staining were given a score of 0, those with weak staining were given a score of 1, those with strong positive staining were given a score of 2, and those with very strong staining were given a score of 3.
ImageJ software was used for quantification of pHH3 staining, and four to five 10× fields were counted per tumor.
RNA isolation and qRT-PCR
RNA was isolated from flash frozen crushed sarcoma tissue following manufacturer’s instructions for Trizol (Invitrogen). One to 2 μg of RNA was reverse transcribed following manufacturer’s instructions for High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). KAPA SYBR Fast mix (Kapa Biosystems, KK4604) was used for quantitative PCR with primers listed below. Expression levels were calculated relative to β-actin and normalized to WT samples.
Target gene | Forward | Reverse |
-actin | GGCATAGAGGTCTTTACGGATGTC | TATTGGCAACGAGCGGTTCC |
Pkm | TGACACCTTCCTGGAACACA | TTCAGCATCTCCACAGATCG |
Pkm1 | GTCTGGAGAAACAGCCAAGG | TCTTCAAACAGCAGACGGTG |
Pkm2 | GTCTGGAGAAACAGCCAAGG | CGGAGTTCCTCGAATAGCTG |
Immunoblotting
Sarcoma tissue lysates were generated from flash frozen ground whole tumors following lysis with ice cold RIPA buffer, supplemented with HALT phosphatase and protease inhibitors (Thermo-Scientific, PI-78420 and 87786). The following antibodies were used for immunoblotting: anti-Hsp90 (1:1000; CST 4877), anti-PKM1 (1:5000; Cell Signaling, 7067), and anti-PKM2 (1:5000; Cell signaling, 4053).
Metabolite extractions and LC/MS analysis
Metabolites were extracted from ground flash frozen whole tumors by methanol extraction. For polar metabolite analysis, dried metabolite samples were resuspended in 100 mL of water, centrifuged at 15,000×g at 4° C for 10 min, and 1 mL of supernatant was injected for LC/MS analysis using QExactive Orbitrap mass spectrometer as previously described [23, 24].
EdU incorporation
Mice were given one pulse of intraperitoneally injected EdU, and tissue was harvested after 3 h. EdU was visualized using Click-iT EDU AlexFluor-647 kit for IF (Invitrogen C10340). For EdU incorporation in cell lines, cells were grown on a monolayer and treated with EdU for 1 h.
M2/M1 expression-associated survival analyses in human sarcoma patients
RNA-seq gene expression profiles (n = 265) and relevant clinical data (n = 261) for sarcoma (SARC) patients were obtained from the Cancer Genome Atlas data portal (TCGA; cancergenome.nih.gov/). Patients with primary tumor samples and associated survival data (n = 259) were ranked by the ratio of the RPKM (reads per Kb per million mapped reads) values of PKM2 and PKM1 isoform-specific exons. Values greater than one indicate higher expression of the M2 isoform compared to the M1 isoform, with higher values indicating greater M2 isoform expression. Kaplan–Meier survival analysis was conducted to compare patients with high M2/M1 expression ratio (top 20th percentile) to those in the bottom 20th percentile (n = 51 within each group). The log-rank test was used to assess significance. Cox proportional hazards analysis was conducted across all patients in the TCGA SARC cohort (n = 261) to assess the prognostic significance of the M2/M1 expression ratio. Univariate Cox regression analysis for individual characteristics was performed to benchmark single-variable hazard ratios. Subsequently, a multivariable model was implemented to estimate the prognostic value of the M2/M1 ratio while controlling for other patient characteristics (age, gender). Hazard ratio proportionality assumptions for the Cox regression model fit were validated by testing for all interactions simultaneously (p = 0.0856). Interaction between significant covariates (M2/M1 ratio and age) was tested using a likelihood ratio test (LRT) to contrast a model consisting of both covariates with another model consisting of both covariates and an interaction term. All survival analyses were conducted within a 5-year survival timeframe using the survival package in R (www.r-project.org).
Results
PKM2 is expressed in mouse and human sarcomas
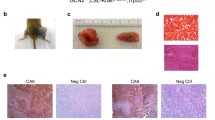
To inform our study of PKM isoform requirements in soft tissue sarcomas (STS), we examined the expression patterns of PKM1 and PKM2 in normal adult skeletal muscle and STS tissue. This was assessed by immunohistochemistry (IHC) staining using antibodies specific to either PKM1 or PKM2. IHC staining of both mouse and human skeletal muscle confirmed previous reports that PKM1 is the primary isoform expressed in this tissue (Fig. 1a, b, Additional file 1: Figure S1A) [2]. We next stained 48 primary human rhabdomyosarcomas including embryonal, spindle cell, alveolus, and pleomorphic rhabdomyosarcomas (undifferentiated sarcomas) for PKM1 and PKM2 (Fig. 1c). Consistent with the PKM expression pattern of their tissue of origin, the majority of the examined rhabdomyosarcomas showed positive staining for PKM1 (32/48, 67%). Less than half of the examined rhabdomyosarcomas showed positive staining for PKM2, arguing against an absolute requirement for PKM2 expression in this tumor type (Additional file 1: Figure S1B). PKM1 and PKM2 IHC intensity scoring revealed heterogeneity in the distribution of PKM2-expressing tumors according to rhabdomyosarcoma subtypes (Fig. 1d). In contrast to the other three examined subtypes, pleomorphic rhabdomyosarcomas contained a high percentage of PKM2-positive tumors (10/12, 83%). The majority of these tumors also expressed PKM1 (10/12, 83%).

Human and mouse soft tissue sarcomas express PKM2. Representative images of IHC for PKM1 and PKM2 in human (a) and mouse (b) skeletal muscle and human rhabdomyosarcoma (c). Corresponding H&E images are shown. Scale bars, 20 μm. d Quantification of PKM1 and PKM2 staining intensities in human rhabdomyosarcoma tumors relative to rhabdomyosarcoma subtype (embryonal, spindle cells, alveolus, pleomorphic). Score 0 = no staining, score 1 = weak, score 2 = positive, or score 3 = strong
In addition to our IHC analysis of PKM1 and PKM2 expression in primary human sarcomas, we also analyzed human sarcoma gene expression data from The Cancer Genome Atlas (TCGA) to determine whether the expression ratio of PKM2 to PKM1 could predict patient outcome [25]. Across > 200 sarcoma patients within TCGA, those within the highest quintile for PKM2/PKM1 expression ratio had a significantly shorter median 5-year survival when compared to either those within the lowest quintile for PKM2/PKM1 expression ratio, or when compared with all other patients combined (Fig. 2a, Additional file 2: Figure S2). Furthermore, Cox regression analysis using this TCGA sarcoma patient cohort showed that a high PKM2/PKM1 ratio was independently prognostic of shorter survival when controlling for age and gender (Fig. 2b).

A high PKM2/PKM1 expression ratio predicts poor prognosis. a Kaplan–Meier 5-year survival analysis comparing patients in the top quintile of PKM2/PKM1 expression ratio (n = 51; red) and those in the bottom quintile (n = 51; blue). Log-rank test p value is shown. b Results of univariate and multivariable Cox proportional hazards model on overall survival in the TCGA sarcoma cohort (all patients). Increasing PKM2/PKM1 expression ratio shows a significant association with poor survival after controlling for other characteristics
PKM2 loss constrains sarcoma tumor initiation
We were intrigued by the disparate PKM expression patterns in normal and transformed skeletal muscle tissue. While normal skeletal muscle exclusively expressed PKM1, a sizeable number of human primary tumors expressed both PKM1 and PKM2. This is in contrast to breast cancer, leukemia, and colon cancer where the normal and tumor tissue both express PKM2 [16, 17, 19]. This, in addition to the prognostic value of PKM2/PKM1 revealed by our analysis of the TCGA sarcoma cohort, led us to postulate that expression of PKM2 may play a role in malignant transformation of skeletal muscle tissue to form STS.
To directly test this hypothesis in a well-defined genetic context, we used a genetically engineered mouse model of soft tissue sarcoma. Conditional activation of oncogenic Kras and inactivation of p53 in a KrasLSL-G12D/+; p53fl/fl (KP) mouse model by viral delivery of Cre recombinase to skeletal muscle tissue initiates the development of high-grade sarcomas with myofibroblastic differentiation [20]. Cross-species gene expression analysis has shown that tumors from the KP model closely resemble human undifferentiated pleomorphic sarcomas [26] and our IHC analysis of human pleomorphic sarcomas showed that this subtype of STS is enriched for PKM2-expressing tumors (Fig. 1d). Importantly, IHC for PKM1 and PKM2 on STS derived from the KP model showed that most cells in all of the examined tumors expressed both PKM1 and PKM2 (Additional file 1: Figure S1C).
To investigate the role of PKM2 in sarcoma development in the KP model, we crossed mice with a conditional allele of Pkm2 (M2fl) [17] to KP mice and performed intramuscular infections of cohorts of KP M2fl/fl and KP M2+/+ mice with Ad-Cre (Fig. 3a). In contrast to similar experiments performed in other autochthonous mouse cancer models, we found that deletion of PKM2 concomitant with sarcoma initiation delayed tumor onset and decreased penetrance of the model (Fig. 3b) (p = 0.003 by log-rank test). Deletion of PKM2 delayed the date of tumor onset by approximately 8 days (56.45 days for KP M2+/+ versus 64.05 days for KP M2flfl) (Fig. 3c).

PKM2 loss delays KP soft tissue sarcoma initiation. KP M2+/+ or KP M2fl/fl mice were injected intramuscularly with Ad5-CMV-Cre (a) and the onset of palpable sarcomas was monitored, p value by log-rank test is 0.003 (b). The percentage of total mice (n) with sarcomas by 62 days (gray box) is indicated. c Time for palpable tumor formation, p value using unpaired t test is 0.04
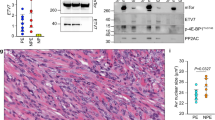
To characterize the PKM expression patterns of sarcomas arising in KP M2+/+ and KP M2fl/fl mice, we performed Western blot analysis of PKM1 and PKM2 expression in primary tumors. While KP M2+/+ tumors primarily expressed PKM2, tumors from KP M2fl/fl mice (KP M2−/− tumors) lost PKM2 expression and expressed PKM1 (Fig. 4a). Consistent with our protein analysis, qRT-PCR analysis of the same tumors showed very low transcript levels for Pkm2 accompanied by a dramatic increase in the transcript levels for Pkm1 in KP M2−/− tumors relative to KP M2+/+ tumors (Fig. 4b). Of note, the levels of Pkm1 in KP M2−/− tumors were lower than the levels of Pkm1 in wild-type untransformed skeletal muscle tissue.

PKM2-deficient sarcoma tumor cells express PKM1. a Western blot analysis for PKM1 and PKM2 on lysates from KP M2+/+ or KP M2−/− sarcoma tissue. Hsp90 was used as a loading control. b Expression of Pkm1, Pkm2, and total Pkm mRNA in KP M2+/+, KP M2−/−, or wild-type skeletal muscle tissue was assessed by qRT-PCR. Mean values ± SEM and p values using unpaired t test are shown, *p < 0.05; **p < 0.01; ****p < 0.0001. c Pyruvate kinase enzymatic activity of KP M2+/+, KP M2−/−, or wild-type skeletal muscle tissue, p value using unpaired t test is shown, *p < 0.05. d Representative images for PKM1 and PKM2 IHC in KP M2+/+ and KP M2−/− sarcoma tissue. Asterisks (*) indicate skeletal muscle fibers. Scale bars, 20 μm. e Representative images of IF for tdTomato and PKM2 on KP M2−/− sarcoma tissue
This quantitative assessment of mRNA abundance also revealed that the levels of total Pkm transcripts were reduced in KP M2−/− tumors compared to KP M2+/+ tumors. The observed decrease in total Pkm transcripts in KP M2−/− tumors was accompanied by a reduction in the maximum rate of reaction (Vmax) when pyruvate kinase enzyme activity was assessed in KP M2−/− tumor lysates compared to KP M2+/+ tumor lysates (Fig. 4c). Interestingly, normal skeletal muscle tissue showed a significantly higher Vmax for pyruvate kinase enzyme activity than tumors of either genotype. These findings are consistent with what has been described in adult tissue isolated from germline Pkm2 knockout mice [2] and suggest a common mechanism of decreased total Pkm expression to compensate for replacement of PKM2 by the more active PKM1 isozyme.
To further characterize the PKM expression patterns of sarcomas from KP; M2+/+ and KP; M2fl/fl mice at the cell and tissue level, we performed IHC for PKM1 and PKM2 (Fig. 4d). Consistent with Western blot analysis, KP M2+/+ tumors primarily and uniformly expressed PKM2. In contrast, KP M2−/− tumors consisted primarily of PKM1-expressing cells and also contained a number of cells that had high expression of PKM2. To determine whether the PKM2-expressing cells in KP M2−/− tumors were cancer cells, we performed immunofluorescence (IF) for PKM2 (Fig. 4e). We used KP mice for this study that carried a fluorescent Cre-reporter allele (Rosa26LSL-tdTomato), allowing us to distinguish tumor cells from stroma through tumor-specific expression of the red-fluorescent protein, tdTomato [27]. Consistent with efficient deletion of PKM2 and consequent expression of PKM1 in tumor cells, we found that PKM2 was expressed exclusively in the tdTomato-negative stromal cells. Collectively, these data argue that while loss of PKM2 concomitant with KP sarcoma initiation delays tumor onset, it does not fully abrogate the eventual outgrowth of tumors in most cases.
Expression of PKM1 in late-stage soft tissue sarcomas does not limit proliferation
Expression of PKM1 in tumor cells has been shown to limit proliferation and progression in some mouse models of cancer [28]. Furthermore, in breast cancers with PKM2 deletion, PKM1 expression was only observed in the non-proliferating population of tumor cells [17]. To ask whether the proliferating cells in KP M2−/− sarcomas expressed PKM1, we performed two-color IHC for PKM1 or PKM2 and the proliferative marker, phospho-histone H3 (pHH3) (Fig. 5a). Contrary to the conclusions from breast cancer, we found that a large portion of PKM1-expressing tumor cells in KP M2−/− sarcomas also expressed pHH3. Thus, once these tumors have been initiated, PKM1 expression does not appear to abrogate tumor cell proliferation. Analysis of 5-ethynyl-2′-deoxyuridine (EdU) incorporation in tdTomato-positive tumor cells was also consistent with this conclusion (Fig. 5b), as was an analysis of proliferating cells in cell lines derived from primary tumors in KP M2+/+ and KP M2fl/fl mice (Additional file 3: Figure S3A). Furthermore, expression of PKM1 in sarcoma cell lines derived from KP M2−/− tumors did not affect the rates of in vitro proliferation as measured through cumulative populations doublings, which were variable across cell lines but did not correlate with Pkm genotype (Additional file 3: Figure S3B).

PKM2-deficient sarcoma tumors have increased proliferation and an altered metabolic profile. a Representative images of dual-color IHC for PKM1 or PKM2 (brown) and pHH3 (purple) on KP M2+/+ and KP M2−/− sarcoma tissue. Scale bars, 20 μm. b Representative images of IF for tdTomato, PKM1, and EdU on KP M2−/− sarcoma tissue. c Quantification of pHH3+ cells in KP M2+/+ and KP M2−/− sarcomas. Representative images of the IHC for pHH3 are shown. Scale bars, 20 μm, n = 6 tumors per genotype. p value using unpaired t test is shown, *p < 0.05. d Heatmap showing measured metabolites that were significantly different between KP M2+/+ and KP M2−/− sarcoma tissue. A full list of the measured metabolites is included in Additional file 4: Table S1
Despite the longer time to palpable tumor formation and the decreased tumor penetrance in KP M2fl/fl mice, there was no significant difference in the tumor weight at time of death of KP M2fl/fl mice relative to KP M2+/+ mice (Additional file 3: Figure S3C). To determine whether KP M2−/− tumors had higher rates of proliferation than KP M2+/+ tumors, we quantified pHH3-positive cells in tumors collected at the time of death from mice of each genotype. Unexpectedly, we found that KP M2−/− tumors had more pHH3-positive cells compared to KP M2+/+ tumors, consistent with the notion that PKM1 expression in late-stage KP sarcomas does not abrogate tumor proliferation or progression (Fig. 5c). These findings are also consistent with PKM2 loss promoting proliferation in some other tissues [2, 17, 18].
PKM2 loss in soft tissue sarcomas leads to metabolic changes
The fact that deletion of PKM2 in our KP model of soft tissue sarcoma led to delayed tumor onset and decreased penetrance but did not appear to limit proliferation or progression of late-stage tumors raised the possibility that KP M2−/− tumors had arisen through an adaptive process that might modulate the effects of PKM2 loss and consequent expression of PKM1. Therefore, we hypothesized that the adaptive process that had allowed for KP M2−/− tumor outgrowth might involve changes in glucose metabolism that could be apparent as changes in metabolite pool sizes. Therefore, we conducted targeted metabolomics analysis of primary tumors derived from KP M2+/+ and KP M2fl/fl mice. The metabolite profiles of primary tumor tissue revealed significant differences in levels of five metabolites in KP M2−/− tumors compared to KP M2+/+ tumors: FAD, NADP, and allantoin, which were increased, and aspartate and fumarate, which were decreased (Fig. 5d, Additional file 4: Table S1). Although it is unclear how these metabolic changes are related to PKM2 loss, it is notable that allantoin is a purine catabolite and aspartate plays an important role in purine biosynthesis.
Discussion
Here, we show that PKM2 loss concomitant with tumor initiation leads to delayed sarcoma tumor onset, which is ultimately overcome and accompanied by changes in metabolism. The delay in tumor initiation that we observe upon loss of PKM2 suggests that tumor initiation in this model is partially dependent on the activation of PKM2 expression in tumor-initiating cells. While the exact cell of origin for soft tissue sarcoma in this mouse model is not well defined, several studies suggest that a diverse array of skeletal muscle cell types, including mature skeletal muscle cells and skeletal muscle progenitor cells, can give rise to sarcomas with comparable frequency in this model [29, 30]. The heterogeneity in potential cells of origin for sarcomas raises the possibility that the consequences of PKM2 loss in this cancer type are dependent on the specific cell type that gives rise to the tumor. We have found that while isolated Pax7+ and Sca1+ skeletal muscle progenitor cells express appreciable levels of Pkm1 mRNA, they also express high levels of Pkm2 mRNA (data not shown).
The fact that KP M2−/− sarcomas were ultimately comparable in size to KP M2+/+ sarcomas shows that the consequences of PKM2 loss can be overcome. Moreover, the metabolic changes that we observe in KP M2−/− tumors might be indicative of a compensatory change in the metabolic program of these cells compared to KP M2+/+ tumor cells. It is interesting to note that while PKM1 expression in PKM2-null breast tumors was shown to be limited to a subpopulation of non-proliferating cells [17], PKM1 was uniformly expressed in KP M2−/− sarcomas and did not abrogate proliferation in these cells. This may represent a better tolerance for PKM1 expression in non-epithelial cancers. Consistent with these in vivo data, sarcoma cell lines derived from KP M2−/− tumors also expressed PKM1, and this did not affect their rates of in vitro proliferation. The fact that the in vitro proliferation of KP M2+/+ and KP M2−/− cell lines were not significantly different even though end-stage KP M2−/− tumors contained a higher number of proliferating cells than end-stage KP M2+/+ tumors may point to a role for the microenvironment in allowing end-stage KP M2−/− tumors to catch up to their KP M2+/+ counterparts. Further investigation is needed to delineate the exact mechanism by which KP M2−/− sarcoma tumors overcome PKM2 loss.
This study adds to a growing body of evidence that the importance of PKM2 expression in tumor cells varies across tumor types and might be indicative of different tumor-specific metabolic requirements [16, 17, 19]. Importantly, these results have general implications for the use of therapeutic strategies that target PKM2. The success of PKM2-targeted therapies is likely to depend on both the tumor type and the stage at which a tumor is treated.
Conclusions
Our study found that PKM2 loss at the time of tumor initiation in a mouse model of STS driven by activation of oncogenic Kras and loss of p53 leads to delayed tumor onset and decreased tumor penetrance. Nonetheless, PKM2 was not required for eventual tumor formation or outgrowth. KP M2−/− tumors express PKM1 and contain infiltrating stromal cells that are PKM2-positive. PKM1 expression in these tumors did not abrogate tumor cell proliferation and, in fact, end-stage KP M2−/− tumors contain more cancer cells that stain positive for proliferation markers than end-stage KP M2+/+ tumors. We show that KP M2−/− tumors have an altered metabolite profile, suggesting metabolic changes accompany loss of PKM2 expression in these tumors.
References
Mazurek S. Pyruvate kinase type M2: a key regulator of the metabolic budget system in tumor cells. Int J Biochem Cell Biol. 2011;43:9 69–980.
Dayton TL, Gocheva V, Miller KM, Israelsen WJ, Bhutkar A, Clish CB, Davidson SM, Luengo A, Bronson RT, Jacks T, Vander Heiden MG. Germline loss of PKM2 promotes metabolic distress and hepatocellular carcinoma. Genes Dev. 2016;30:1020–33.
Macintyre AN, Rathmell JC. PKM2 and the tricky balance of growth and energy in cancer. Mol Cell. 2011;42:713–4.
Hitosugi T, Kang S, Vander Heiden MG, Chung T-W, Elf S, Lythgoe K, Dong S, Lonial S, Wang X, Chen GZ, Xie J, Gu T-L, Polakiewicz RD, Roesel JL, Boggon TJ, Khuri FR, Gilliland DG, Cantley LC, Kaufman J, Chen J. Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth. Sci Signal. 2009;2:ra73.
Lv L, Li D, Di Zhao LR, Chu Y, Zhang H, Zha Z, Liu Y, Li Z, Xu Y, Wang G, Huang Y, Xiong Y, Guan KL, Lei QY. Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone-mediated autophagy and promotes tumor growth. Mol Cell. 2011;42:719–30.
Christofk HR, Vander Heiden MG, Wu N, Asara JM, Cantley LC. Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nat Publ Group. 2008;452:181–6.
Anastasiou D, Poulogiannis G, Asara JM, Boxer MB, Jiang J-K, Shen M, Bellinger G, Sasaki AT, Locasale JW, Auld DS, Thomas CJ, Vander Heiden MG, Cantley LC. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science. 2011;334:1278–83.
Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL, Cantley LC. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nat Publ Group. 2008;452:230–3.
Anastasiou D, Yu Y, Israelsen WJ, Jiang J-K, Boxer MB, Hong BS, Tempel W, Dimov S, Shen M, Jha A, Yang H, Mattaini KR, Metallo CM, Fiske BP, Courtney KD, Malstrom S, Khan TM, Kung C, Skoumbourdis AP, Veith H, Southall N, Walsh MJ, Brimacombe KR, Leister W, Lunt SY, Johnson ZR, Yen KE, Kunii K, Davidson SM, Christofk HR, et al. Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis. Nat Chem Biol. 2012;8:839–47.
Dayton TL, Jacks T, Vander Heiden MG. PKM2, cancer metabolism, and the road ahead. EMBO Rep. 2016;17:1721–30.
Lu Z. Nonmetabolic functions of pyruvate kinase isoform M2 in controlling cell cycle progression and tumorigenesis. Chin J Cancer. 2013;32:5–7.
Yang W, Zheng Y, Xia Y, Ji H, Chen X, Guo F, Lyssiotis CA, Aldape K, Cantley LC, Lu Z. ERK1/2-dependent phosphorylation and nuclear translocation of PKM2 promotes the Warburg effect. Nat Cell Biol. 2012;14:1295–304.
Yang W, Xia Y, Ji H, Zheng Y, Liang J, Huang W, Gao X, Aldape K, Lu Z. Nuclear PKM2 regulates b-catenin transactivation upon EGFR activation. Nat Publ Group. 2011;480:118–22.
Yang W, Xia Y, Hawke D, Li X, Liang J, Xing D, Aldape K, Hunter T, Yung WKA, Lu Z. PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell. 2012;150:685–96.
Gao X, Wang H, Yang JJ, Liu X, Liu Z-R. Pyruvate kinase M2 regulates gene transcription by acting as a protein kinase. Mol Cell. 2012;45:598–609.
Wang Y-H, Israelsen WJ, Lee D, Yu VWC, Jeanson NT, Clish CB, Cantley LC, Vander Heiden MG, Scadden DT. Cell-state-specific metabolic dependency in hematopoiesis and leukemogenesis. Cell. 2014;158:1309–23.
Israelsen WJ, Dayton TL, Davidson SM, Fiske BP, Hosios AM, Bellinger G, Li J, Yu Y, Sasaki M, Horner JW, Burga LN, Xie J, Jurczak MJ, DePinho RA, Clish CB, Jacks T, Kibbey RG, Wulf GM, Di Vizio D, Mills GB, Cantley LC, Vander Heiden MG. PKM2 isoform-specific deletion reveals a differential requirement for pyruvate kinase in tumor cells. Cell. 2013;155:397–409.
Tech K, Tikunov AP, Farooq H, Morrissy AS, Meidinger J, Fish T, Green SC, Liu H, Li Y, Mungall AJ, Moore RA, Ma Y, Jones SJM, Marra MA, Vander Heiden MG, Taylor MD, Macdonald JM, Gershon TR. Pyruvate kinase inhibits proliferation during postnatal cerebellar neurogenesis and suppresses medulloblastoma formation. Cancer Res. 2017;77:3217–30.
Lau AN, Israelsen WJ, Roper J, Sinnamon MJ, Georgeon L, Dayton TL, Hillis AL, Yilmaz ÖH, Di Vizio D, Hung KE, Vander Heiden MG. PKM2 is not required for colon cancer initiated by APC loss. Cancer Metab. 2017;5:10.
Kirsch DG, Dinulescu DM, Miller JB, Grimm J, Santiago PM, Young NP, Nielsen GP, Quade BJ, Chaber CJ, Schultz CP, Takeuchi O, Bronson RT, Crowley D, Korsmeyer SJ, Yoon SS, Hornicek FJ, Weissleder R, Jacks T. A spatially and temporally restricted mouse model of soft tissue sarcoma. Nat Med. 2007;13:992–7.
Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, Jacks T, Tuveson DA. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15:3243–8.
DuPage M, Mazumdar C, Schmidt LM, Cheung AF, Jacks T. Expression of tumour-specific antigens underlies cancer immunoediting. Nature. 2012;482:405–9.
Sullivan LB, Gui DY, Hosios AM, Bush LN, Freinkman E, Vander Heiden MG. Supporting aspartate biosynthesis is an essential function of respiration in proliferating cells. Cell. 2015;162:552–63.
Birsoy K, Wang T, Chen WW, Freinkman E, Abu-Remaileh M, Sabatini DM. An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell. 2015;162:540–51.
Cancer genome atlas research network. Electronic address: elizabeth.demicco@sinaihealthsystem.ca, Cancer genome atlas research network. Comprehensive and integrated genomic characterization of adult soft tissue sarcomas. Cell. 2017;171:950–965.e28.
Mito JK, Riedel RF, Dodd L, Lahat G, Lazar AJ, Dodd RD, Stangenberg L, Eward WC, Hornicek FJ, Yoon SS, Brigman BE, Jacks T, Lev D, Mukherjee S, Kirsch DG. Cross species genomic analysis identifies a mouse model as undifferentiated pleomorphic sarcoma/malignant fibrous histiocytoma. PLoS One. 2009;4:e8075.
Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, Lein ES, Zeng H. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 2009;13:133–40.
Israelsen WJ, Vander Heiden MG. Pyruvate kinase: function, regulation and role in cancer. Semin Cell Dev Biol. 2015;43:43–51.
Hettmer S, Liu J, Miller CM, Lindsay MC, Sparks CA, Guertin DA, Bronson RT, Langenau DM, Wagers AJ. Sarcomas induced in discrete subsets of prospectively isolated skeletal muscle cells. Proc Natl Acad Sci. 2011;108:20002–7.
Blum JM, Añó L, Li Z, Van Mater D, Bennett BD, Sachdeva M, Lagutina I, Zhang M, Mito JK, Dodd LG, Cardona DM, Dodd RD, Williams N, Ma Y, Lepper C, Linardic CM, Mukherjee S, Grosveld GC, Fan C-M, Kirsch DG. Distinct and overlapping sarcoma subtypes initiated from muscle stem and progenitor cells. CellReports. 2013;5:933–40.
Acknowledgements
We thank Kim Mercer for technical assistance; Elizaveta Freinkman and the Whitehead Metabolomics Core for the metabolite analysis. We also thank David Feldser and the entire Jacks and Vander Heiden labs for helpful discussions and experimental assistance. We thank the Koch Institute Swanson Biotechnology Center (SBC) for the technical support, specifically Denise Crowley and Kathleen Cormier in the Hope Babette Tang (1983) Histology Facility. The authors wish to dedicate this paper to the memory of Officer Sean Collier for his caring service to the MIT community and for his sacrifice.
Funding
This work was partially supported by the Cancer Center Support Grant P30CA14051 from the National Cancer Institute and by grants from the Howard Hughes Medical Institute (T.J., M.G.V.H.), the Burroughs Wellcome Fund (M.G.V.H.), the Ludwig Center at MIT (M.G.V.H.), Stand-up to Cancer (M.G.V.H.), the MIT Center for Precision Cancer Medicine, and the Department of Health and Human Services R01CA168653 (M.G.V.H.). T.L.D. was partially supported by a National Defense Science and Engineering Graduate (NDSEG) Fellowship Award. V.G. was supported by a Jane Coffin Childs Memorial Fund Postdoctoral Fellowship. T.J. is a Howard Hughes Investigator, the David H. Koch Professor of Biology, and a Daniel K. Ludwig Scholar at MIT.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Author information
Authors and Affiliations
Contributions
TLD, VG, CAL, and KMM performed and/or supervised experiments. RTB and AB were involved in the data analysis. VG and CAL gave constructive input on the experimental design and data analysis. TLD, TJ, and MGVH conceived and designed the study and were involved in the interpretation of datasets and writing the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval
All animal work was conducted in accordance with a protocol approved by the MIT Committee on Animal Care.
Competing interests
M.G.V.H. is a consultant and scientific advisory board member for Agios Pharmaceuticals and Aeglea Biotherapeutics. The other authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file1:
Figure S1. Quantification of PKM1 and PKM2 staining intensities shown as percent of tissue cores scored in 16 normal human skeletal muscle samples (A) and 48 primary human rhabdomyosarcomas (B). Score 0 = no staining, Score 1 = weak, Score 2 = positive, or Score 3 = strong. (C) Representative images of IHC for PKM1 and PKM2 in KP mouse sarcoma tissue. Corresponding H&E images are shown. Scale bars, 20 μm. (JPG 1314 kb)
Additional file 2:
Figure S2. (A) Kaplan–Meier 5-year survival analysis comparing patients in the top quintile of PKM2/PKM1 expression ratio (n = 51; red) and all other patients combined (n = 208; blue). Log-rank test p value is shown. (JPG 683 kb)
Additional file 3:
Figure S3. (A) Representative images of IF for tdTomato, PKM1 or PKM2, and EdU on KP M2+/+ or KP M2−/− sarcoma cell lines. (B) Cumulative populaion doublings of KP M2+/+ or KP M2−/− sarcoma cell lines, n = 3 KP M2+/+ cell lines and n = 4 KP M2−/− cell lines. (C) Sarcoma weight in grams. (JPG 2694 kb)
Additional file 4:
Table S1. Raw data for LC-MS metabolomics on KP M2+/+ and KP M2-/- sarcomas. Related to Fig. 5. This table contains all the LC-MS metabolomics data. Includes normalization and statistical analysis for each measured metabolite. (XLSX 140 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

{kind=link}
{kind=link}
{kind=link}
Cite this article
Dayton, T.L., Gocheva, V., Miller, K.M. et al. Isoform-specific deletion of PKM2 constrains tumor initiation in a mouse model of soft tissue sarcoma. Cancer Metab 6, 6 (2018). https://doi.org/10.1186/s40170-018-0179-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40170-018-0179-2