
Abstract
Combination therapy is increasingly becoming the cornerstone of current day antitumor therapy. Glioblastoma multiforme is an aggressive brain tumor with a dismal median survival post diagnosis and a high rate of disease recurrence. The poor prognosis can be attributed to unique treatment limitations, which include the infiltrative nature of tumor cells, failure of anti-glioma drugs to cross the blood–brain barrier, tumor heterogeneity and the highly metastatic and angiogenic nature of the tumor making cells resistant to chemotherapy. Combination therapy approach is being developed against glioblastoma with new innovative combination drug regimens being tested in preclinical and clinical trials. In this review, we discuss the pathophysiology of glioblastoma, diagnostic markers, therapeutic targeting strategies, current treatment limitations, novel combination therapies in the context of current treatment options and the ongoing clinical trials for glioblastoma therapy.
Similar content being viewed by others

Introduction
In recent years, a growing number of successful pre-clinical agents have failed to show reproducible effects on patient survival [1]. The “Basket trail” approach—testing the effect of one drug on a single mutation in a variety of tumor types is being used to address the irreproducibility issue [2]. Another approach being used to overcome this troubling trend is the use of combination of drugs that rely on complementary mechanisms of antitumor activity and can be combined into a therapeutic regimen [3, 4]. The success of combination therapy approach relies on three factors: (i) each component of the combination therapy regimen should have a single-agent activity without any cross-resistance, (ii) pre-clinical studies on the drug cocktail should indicate synergism, and (iii) each component should have a separate safety criterion [5]. Since, combination therapy works synergistically or in an additive manner, lower doses of individual drugs are administered reducing the problems of drug resistance to tumor cells and drug toxicity to healthy cells [6].
In cancer, combinations of two or more therapeutic treatments have been demonstrated to be more effective than monotherapy and chemotherapy [6]. Monotherapy non-selectively targets rapidly growing cells and chemotherapy leads to high toxicity burden and immunosuppression [6]. Glioblastoma multiforme (GBM) is the most frequent and aggressive brain tumor in adults with very poor prognosis [7]. In the past decade, despite novel therapeutic targets being discovered, monotherapy has failed in clinical trials [8, 9]. In this review, we discuss and summarize the current status of combination therapy in combating GBM.
Pathophysiology of Glioblastoma multiforme
Glioblastoma multiforme is the most aggressive primary malignant brain tumor, with a median survival of about 14.6 months post-diagnosis and is classified as Grade IV by the World Health Organization (WHO) [10]. It accounts for 45.2% of malignant primary brain and central nervous system (CNS) tumors, 54% of all gliomas and 16% of primary brain and CNS tumors [11]. The global incidence of GBM is 0.59–3.69 per 100,000 live births with an age-adjusted incidence rate of 3.97 cases per 100,000 for males and 2.53 cases per 100,000 for females [11, 12]. Glioblastoma is classified into two types: primary GBM, which is the predominant subtype (80% of cases) and is manifested at later age (mean age 62 years) whereas secondary GBM progresses from lower grade astrocytoma or oligodendroglioma and is prevalent in younger patients (mean age 45 years) [13,14,15,16]. A rare subtype of GBM, “with oligodendroglioma component” (GBMO) has been added to the WHO classification [10]. GBMO is defined as GBM with an area resembling anaplastic oligodendroglioma and necrosis with or without microvascular proliferation [10].
Molecular sub-classes of Glioblastoma multiforme
The histological homogeneity despite clinical heterogeneity of GBM sub-types that affect disease prognosis and therapeutic outcomes warrants a molecular profiling based classification [17]. The Cancer Genome Atlas (TCGA) summarized genomic alteration in 206 GBM patients. A total of 601 genes and sequencing data from 91 patients (a subset of the 206 GBM patients) were used to describe the mutational spectrum [18]. An independent set of gene expression profiling data from 260 GBM patients and 40 classifying genes have been used to cluster GBM into four clinically relevant subtypes; namely: Classical, Proneural, Mesenchymal and Neural (Table 1) [18].
Classical GBM
The classical subtype is associated with chromosome 7 amplification, loss of chromosome 10 (100% cases) and Epidermal growth factor receptor (EGFR) amplification (97% cases) [19]. Focal deletion of 9p21.3 harboring cyclin-dependent kinase Inhibitor 2A (CDKN2A) is associated (p < 0.01, two sided Student’s t test) with classical GBM, which co-occurs with EGFR amplification in 94% cases [18]. Homozygous deletion of 9p21.3 is almost mutually exclusive with retinoblastoma (RB) pathway genes (RB1, CDK4, CCDN2) [18]. Neural precursor and stem cell marker (NES) overexpression, mutation in phosphatase and tensin (PTEN), Notch (NOTCH3, JAG1, LFNG) and Sonic hedgehog (SMO, GAS1, GLI2) pathway activation are observed in this subtype [18].
Mesenchymal GBM
This subtype is associated with focal heterozygous deletion of 17q11.2 containing neurofibromin 1 (NF1) (p < 0.01, adjusted two-sided Student’s t test), mutation in PTEN, mutation in tumor protein p53 (TP53) and activation of genes in tumor necrosis factor (TNF) superfamily and NF-kB pathway [18]. Mesenchymal subtype is associated with mesenchymal markers: CHI3L1 and MET [18, 20].
Proneural GBM
This subtype is associated with platelet-derived growth factor receptor alpha (PDGFRA) amplification, mutation in isocitrate dehydrogenase 1 (IDH1), PIK3A/PIK3R1, TP53, CDKN2A and PTEN [18]. Several development genes (SOX, DCX, DLL3, ASCL1, TCF4) are overexpressed in the proneural subtype [20]. Interestingly, 90% of the IDH1 mutations in GBM are found in proneural subclass (p < 0.01, adjusted two sided Fisher’s exact test) [18].
Neural GBM
This subtype is associated with neuronal marker expression such as NEFL, GABRA1, SYT1, SLC12A5 and EGFR amplification and overexpression. The gene ontology (GO) categories associated with this subtype include neuron projection, axon transmission and synaptic transmission. This subgroup shows strong association with oligodendrocytic and astrocytic differentiation. The neural subgroup also includes genes that are differentially expressed during neuronal differentiation [18].
In 2016, WHO updated the classification of CNS tumors [21]. The new update includes molecular features in addition to histopathology [21]. The nomenclature of the subtypes includes the histopathological name followed by genetic information [21]. Currently, GBM is classification into three major sub groups (Table 1) [21].
Glioblastoma, IDH1 wild type
About 90% of GBM are in this subtype, most cases are either primary or de novo glioblastoma [21]. Patients in this subtype are mostly 55 years or older [22]. The overall survival with surgery and radiotherapy is approximately 10 months. These tumors are located in supratentorial region of the brain and are associated with extensive necrosis, TERT promoter methylation (72% cases), TP53 mutations (27%), EGFR amplification (35%) and PTEN mutations (24%) [21]. A new variant, epithelioid glioblastoma, falls under IDH1-wild type and is characterized by the presence of large epithelioid cells, vesicular chromatin, rhabdoid cells and prominent nuclei [20, 21]. This subtype of GBM is found in patient ages ranging from 10 to 69 years and often harbors a v-raf murine sarcoma viral oncogenes homolog B1 (BRAF) V600E mutation [23]. This subtype lacks EGFR amplification and chromosome 10 loss [21].
Glioblastoma, IDH1 mutant
About 10% GBM cases form this subtype and are characterized as secondary glioblastoma associated with low-grade diffused glioma [21]. This subtype of GBM arises in younger patients (about 44 years) [22]. The median survival post surgery and radiotherapy is 24 months. Additional chemotherapy treatment extends the medial survival to 31 months. These tumors are located in frontal lobe of brain and are characterized by limited tumor necrosis, TERT promoter mutation (26%), TP53 mutations (81%) and ATRX mutations (71%) [21].
Glioblastoma, not otherwise specified (NOS)
This subtype includes GBM where full IDH evaluation cannot be performed [21].
There are two additional glioblastoma subtypes: (i) glioblastoma with primitive neural component and (ii) small cell glioblastoma and granular cell glioblastoma [21, 24, 25]. Both these subtypes show poor glioblastoma-like prognosis without microvascular proliferation and/or necrosis [21].
Disease prognosis in Glioblastoma multiforme and clinical correlation
Many signaling pathways, oncogene and transcription factors are altered in GBM [26]. Disease prognosis in GBM and patient response to therapy depends on the type genetic and epigenetic modifications [27]. The following section summarizes the factors that determine GBM prognosis and impact its clinical management.
Molecular marker based disease prognosis
Here we summarize the latest development in molecular marker based GBM prognosis and their clinical outcomes (Table 2).
O-6-methylguanine-DNAmethyltransferase (MGMT)
DNA repair enzyme MGMT removes alkyl groups from O6 position of guanine in DNA making cells resistant to the chemotherapeutic agent temozolomide (TMZ) [28]. Methylation of MGMT gene promoter leads to epigenetic silencing of MGMT in glioma cells. This in turn, leads to reduction in DNA repair function, increased genome instability and chemosensitivity to TMZ [29,30,31,32]. GBM patients with concurrent methylation of MGMT, TP53 and CDKN2A show better prognosis [33].
EGFR
EGFR mutation is mainly found in primary GBM [34]. EGFR variant III (EGFRvIII) is the most common mutation (40%) in GBM and its overexpression is highly associated with poor prognosis [35]. EGFR gene status and overexpression is a prognostic indicator in younger patients [35].
IDH mutation
Mutations in IDH1 or IDH2 genes, which encode isocitrate dehydrogenase enzyme involved in tricarboxylic acid cycle (TCA), are common in lower grade and anaplastic (II–III) glioma [36]. IDH1 mutated high grade gliomas arise from lower-grade glioma (secondary GBM) and shows specific radiographic, histologic and transcriptional features consistent with a less aggressive prognosis [37].
G-CIMP
The CpG island hypermethylation phenotype (G-CIMP) is strongly correlated with IDH1 mutation and the proneural sub-type of GBM [38, 39]. G-CIMP is rarely found in primary GBM (5–8%) [39]. GBM patients in this subgroup have the highest overall survival rates [18, 39, 40].
TP53 mutation
TP53 gene mutation is most commonly found in secondary GBM (60–70%) [41]. Mutation in this tumor suppressor is reported in younger patients, albeit the prognosis in this subtype is still unclear [42].
ATRX mutation
Mutation in alpha-thalassemia/mental retardation syndrome X-linked (ATRX) gene, involved in alternative telomere lengthening is most frequently found in secondary GBM (57%), less frequently in pediatric GBM (24%) and rarely in primary GBM (4%) [43, 44]. This mutation often clusters with IDH1 and TP53 mutations and is associated with poor patient prognosis [45, 46].
Loss of chromosome 10
Loss of chromosome 10, a part or the entire chromosome is found in 80–90% of GBM cases [41]. Mutation in PTEN, located at 10q23.3 is exclusively found in primary GBM and accounts for 20–40% of GBM [47].
1p19q co-deletion
GBMO results from 1p19q co-deletion [48]. As per the National Comprehensive Cancer Network (NCCN) treatment guidelines, 1p19q co-deletion is the only molecular biomarker for therapeutic use for GBMO subtype (NCCN, 2013).
Metabolic profiling based prognosis
Protein expression analysis in GBM patients revealed two populations of glioblastoma stem-like cells (GSC) associated with different clinical outcomes: (i) proto-oncogene, tyrosine protein kinase and SRC activation is associated with the subgroup showing better prognosis (ii) ribosomal protein S6 (RPS6), an effector of the mTOR pathway is associated with poor patient prognosis [49].
Immunological profiling based prognosis
Analysis of tumor-associated stroma revealed that mesenchymal (MES) subtype of GBM is associated with the presence of tumor-associated glial cells and microglial cells [50]. Furthermore, genetic deactivation of NF1 is associated with increased macrophage/microglia in the tumor microenvironment [50]. Since there is a higher frequency of M2 macrophages and CD4+ T cells observed after radiotherapy, M2 macrophages are presumed to play a role in radioresistance [50].
Glioblastoma multiforme: treatment challenges
Despite the identification of well-defined molecular markers, therapeutic targeting and disease prognosis in GBM is poor. Multiple factors contribute to this disease complexity and are summarized below.
Glioblastoma stem cell (GSC) and therapeutic resistance
The poor prognosis of GBM can be attributed to its resistance to current therapeutic approach consisting of radiotherapy (RT) with concomitant and adjuvant TMZ therapy post surgery [51]. Although, GBM is most comprehensively characterized based on genetic (IDH1 mutation), epigenetic (MGMT promoter methylation) and transcriptional (classical, mesenchyme, proneural, neural) profiling, these mutations show therapeutic resistance and recurrence due to the self-renewing tumor cell type known as GSCs that escape chemo-RT and proliferate residual tumor cells following treatment [52,53,54]. GSCs are defined as tumor cells capable of self-renewal, high tumorigenic ability, and capacity for multipotent differentiation [55, 56]. GSCs can acquire resistance to chemo-RT either through innate properties of genetic heterogeneity of the tumor or through adaptive resistance pathways [57,58,59].
Intratumoral heterogeneity
Genomic landscape study of pre- and post treatment GBM patients pairs revealed a variable degree of genetically related clones to the original tumor (clonal evolution) and accumulation of new clones (sub clonal evolution)—giving rise to high degree of intratumoral heterogeneity: the primary cause for the poor prognosis and lack of effective therapeutic options in GBM [60, 61]. Intratumoral heterogeneity can be experimentally concluded from (i) single cell RNA-seq data showing the presence of heterogeneous mixture of cells in various GBM subgroups, and (ii) genetically distinct identity and differential drug resistance profiles in single cell derived GBM subclones [62, 63]. The proneural subgroup has the highest proportion of markers specific to other subgroups and reports the worst prognosis [62]. Altogether, intratumoral heterogeneity fuels therapy resistance and failure of effective therapeutic strategies.
Post-therapy resistance
A number of molecular mechanisms have been implicated in the post-therapy resistance in GBM: DNA damage checkpoints; tumor microenvironment such as hypoxia, acidic and metabolic stresses; oncogene and transcription factor activation such as Notch, NF-kB and EZH2 [54, 64,65,66,67,68,69,70,71]. Some examples of these mechanisms include (i) enrichment of CD133+ GSCs post chemo-RT results in post-therapy resistance and is mediated by the activation of DNA damage checkpoint kinases (ChK1 and ChK2) [54], (ii) MGMT plays an important role in TMZ resistance mediated by MMR pathway that acts by reversing the mutagenic DNA lesion O-6-lguanine (introduced by TMZ) back to guanine [28, 72], (iii) the Notch pathway inhibitor: γ-secretase inhibitor (GSI) sensitizes GSCs to radiation [67] and overexpression of Notch 1 or Notch 2 sensitizes GSC to RT by blocking the transition into endothelial progenitors [67], (iv) combinations of receptor tyrosine kinase (RTK) inhibitors and/or RNA interference show promising anticancer activity by inhibiting cell growth in PTEN-deficient glioma cells [73], (v) hypoxia favors GSCs maintenance and hypoxia inducible transcription factors (HIF-1 and HIF-2) are involved in tumor maintenance and angiogenesis [74], and (vi) GSCs suppress the adaptive immune system by recruiting microglia/macrophages to induce secretion of immunosuppressive cytokines interleukin-10 (IL-10) and TGF-β1, in turn promoting tumor growth [75].
Therapeutic targeting
The compromised responses to radio and chemotherapy in GBM results from therapeutic resistance and inefficient targeting of GSCs. Novel therapeutic approaches are being developed to overcome these treatment limitations [76,77,78,79,80].
Targeting GSCs by surface markers
Patients with CD133+ expression show poor clinical outcomes [81]. Silencing of CD133+ in GBM derived neurosphere impairs the self-renewal and tumorigenic capacity of neurosphere cells [82]. Selective targeting of CD133+ GBM cells by anti-CD133 monoclonal antibody selectively kills tumor cells while sparing normal cells [83]. Using CD133 antibody conjugated immune liposomes that encapsulate gemcitabine to target GSCs showed 15 times higher anti-tumor effect than that of free gemcitabine [84].
Targeting GSCs by signaling pathways
Signaling pathways including Notch, Sonic-hedgehog (Shh), VEGF, STAT3 and Bone morphogenetic proteins (BMPs) are critical in GSCs maintenance and targeting these pathways have promising therapeutic potential [85]. GSI mediated Notch inhibition leads to reduced neurosphere proliferation, reduced CD133+ cell fraction in vitro and decreased tumor growth in vivo [86]. STAT3 is a critical signaling node involved in GSC maintenance through regulation of Toll-like receptor TLR9 expression [87, 88]. A variant of BMP7 (BMP7v), member of the TGF-β superfamily, has been shown to decrease GSC proliferation and angiogenesis providing a novel approach to the treatment of GBM [89].
Targeting tumor microenvironment
Tumor microenvironment plays a significant role in GSC stemness and thus targeting microenvironment for therapy has shown promising results [90]. Cancer stem cells (CSCs) orchestrate vascular niches that maintain the CSCs pool [74]. VEGF is involved in microvasculature formation and is an important mediator of angiogenesis in GBM [90]. Bevacizumab is a recombinant humanized monoclonal antibody that inhibits VEGF signaling pathway by inhibiting interaction with VEGF receptors [91]. Tumor-associated macrophages (TAMs) are enriched in GBMs and promote tumor progression [92]. GSCs secrete periostin (POSTN) to recruit TAMs and disrupted POSTN attenuate the tumor-supportive M2 type TAMs in xenografts [92].
Combination therapy in Glioblastoma multiforme: preclinical development
Characterization of molecular targets from in vitro and in vivo studies have led to the development of clinical trials in GBM. Experimental models and clinical trials are most rewarding when they combine multiple gene targets [93,94,95,96]. This approach, also referred to as combination therapy, shows synergistic effects in terms of drug efficacy and is by far the most effective way of managing aggressive tumors like GBM.
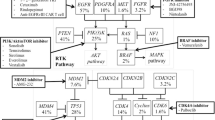
Maximum surgical resection of the tumor followed by focal chemotherapy with TMZ is the current standard of care for GBM [97]. TMZ, a second-generation imidazotetrazine, is a DNA-alkylating agent [98]. It has the ability to cross the blood–brain barrier (BBB) making it particularly effective in the treatment of brain tumors [98,99,100]. However, in addition to severe side effects of TMZ such as myelotoxicity, ulcers, nausea, vomiting, fatigue and toxic DNA damage, the resistance to this drug is common in GBM patients [101, 102]. A potential approach in the first-line treatment of GBM may be to explore a more effective combination regimen. In this section we summarize a comprehensive list of gene targets and small molecule inhibitors that are in various stages of preclinical development for GBM therapy and are being tested in combination with radio- and/or chemotherapy (Fig. 1).

Schematic representation of combination therapy in treatment of GBM. Targeted therapy approaches have the potential to improve patient outcome in the treatment of Glioblastoma. Small-molecule inhibitors in combination with chemo/radiotherapy can overcome chemo/radioresistance in patients improving treatment efficacy when compared to monotherapy. We summarize small molecule inhibitors and their gene targets that are in preclinical development for GBM therapy and are being tested in combination with radio- and/or chemotherapy
TMZ in combination therapy
TMZ and micellarized Cyp (MCyp)
Shh signaling pathway is up regulated in high-grade gliomas and inhibition of Shh leads to apoptosis and cell death [103, 104]. Cyclopamine (Cyp) is a Shh antagonist, which is selective and non-toxic to cell types not dependent on activation of the Shh pathway [105]. Combination of MCyp with TMZ has been shown to block the Shh pathway and eliminate neurosphere formation [105].
TMZ and morphine
The efflux of P-glycoprotein 1 (P-gp1) in endothelium cells of the BBB mediates TMZ resistance [106]. Morphine is an inhibitor of P-gp1 [106]. Combinatorial effect of morphine with lower dose of TMZ shows significant reduction in tumor growth [106]. Additionally, reducing the TMZ drug dose reduces chemoresistance, thus improving therapeutic response in the long term [106].
TMZ and nutlin3a
nutlin3a is a murine double minute 2 (MDM2) protein–protein interaction inhibitor [107]. It blocks the MDM2-p53 associated signaling pathways [107]. The combination of TMZ/nutlin3a synergistically decreases the growth of p53 wild type GBM cells and leads to significant increase in survival of mice with GBM10 intracranial tumor [107].
TMZ and SGT-53
Combining TMZ with exogenous delivery of TP53 via tumor-targeted nanocomplex (SGT-53) significantly chemosensitized GBM cells (U87 and U251) to chemotherapy and prolonged median survival [108].
TMZ and sulforaphane (SFN)
Coadministration of the transcriptional NF-kB inhibitor, SFN with TMZ in TMZ-resistant cell lines (U87-R and U373-R) lead to reversal of chemoresistance, suppression of cell growth and enhanced cell death in chemoresistant xenograft in nude mice [109]. Additionally, SFN has also been shown to inhibit miR-21 via Wnt/β-catenin/TCF4 signaling pathway, thereby enhancing chemosensitivity [110].
TMZ and XL765
The PI3K/mTOR pathway is dysregulated in many tumors. It acts by inhibiting Akt signaling and promotes resistance to EGFR inhibitors [111, 112]. XL765 is a novel PI3K/mTOR dual inhibitor [113]. XL765 in combination with TMZ shows additive cytotoxicity in genetically diverse GBM xenografts [113].
TMZ and JQ1
Delivery of transferring-functionized nanoparticle (Tf-NP) based dual drug combination of TMZ with bromodomain inhibitor JQ1 leads to increased DNA damage and apoptosis. This results in 1.5 to 2-fold decrease of tumor burden and increased survival when compared with free-drug dosing [114].
TMZ and nimotuzumab
The mutant form of EGFR, EGFRvIII confers therapeutic resistance and tumor growth. Nimotuzumab, an anti-EFGR antibody in combination with TMZ showed enhanced antitumor activity in nude mice bearing subcutaneous or intracerebral tumor expressing EGFRvIII [115].
RIST/aRIST in combination therapy
The RIST (rapamycin, irinotecan, sunitinib, temozolomide) and the variant aRIST (alternative to rapamycin, GDC-0941) combination regimen inhibit GBM cell growth in primary patient culture via up-regulation of apoptotic pathways [116]. Rapamycin is an mTOR inhibitor, irinotecan is a topoisomerase-I inhibitor and sunitinib inhibits RTKs [116]. These individual components have shown partial success, but while in combinations, they significantly reduce cell viability [116]. Additionally, in the combinatorial regimen all inhibitors are administered at relatively lower doses thereby reducing toxicity and other side effects [116].
Radiotherapy in combination therapy
RT and poly (ADP ribose) polymerase (PARP) inhibitor
Inhibition of PARP proteins radiosensitizes glioma cells by inhibiting DNA repair [93,94,95]. Studies show that PARP inhibitors (PARPi) decreased colony formation in MGMT unmethylated GBM patient derived xenografts. This suggests PARP inhibition as a new therapeutic approach in GBM [117]. RT in GBM patients’ lead to upregulation of PARP1 mediated repair of DNA damage in glioblastoma CSCs. Preclinical study of the PARPi, talazoparib (BMN-673; Pfizer), in combination with RT showed prolonged G2/M block and a significant reduction in GSC proliferation [118].
RT and palbociclib
RB pathway (Cycline-dependent kinase (CDK4/6), RB tumor suppressor and the E2F-family of transcription factors are important for cell cycle regulation [119]. Amplification of CDK6 and deletion of CDK inhibitor 2A/B (CDKN2A/B) genes are frequently found in GBM patients (~ 86%), specifically in classical and mesenchymal subtypes [119]. Such alterations cause constitutive expression of E2F transcription factors leading to cell cycle acceleration, DNA replication and mitotic progress [119]. Palbociclib (PD0332991; Pfizer) is a selective inhibitor of CDK4/6 kinase in RB proficient cells and can promote cell cycle arrest and apoptosis both in vitro and in vivo [120]. Palbociclib when coadministered with RT, showed a survival advantage in mice [121].
Tyrosine kinase receptor inhibitors in combination therapy
Activation of RTKs, including EGFR, PDGFRα, PDGFRβ and MET is frequently observed in GBM. Anti-EGFR therapy or targeted therapy against a particular RTK is not effective in suppressing the downstream PI3K pathway leading to therapeutic resistance. Combination of RTK inhibitors (erlotinib, SU11274, imatinib) show improved cell survival and anchorage-independent growth in PTEN-deficient glioma cells [73].
Immunotherapy in combination therapy
Immunosuppression is one of the primary reason for poor prognosis in GBM [122]. Reduction in T cell mediated immune response is due to co-inhibitory receptors on T-cells known as immune checkpoint molecules [122]. CTLA-4 and PD-1 are two such immune checkpoint molecules, blocking these two molecules induces tumor regression and promotes long-term survival [123, 124]. Combination of anti-PD-1 antibodies and RT doubled median survival and enhanced long-term survival in 15–40% GBM mice [125]. Studies in GBM models and human samples have shown that the accumulation of myeloid-derived suppressor cells (MDSCs) in the tumor microenvironment induces immunosuppression [126, 127]. Inhibiting MDSC accumulation with PD-1 or CTLA-4 (that acts early in T-cell activation) enhances efficacy of immune-simulated gene therapy [128].
Antisense oligo-based therapy in combination therapy
Immune-suppression in the tumor microenvironment leads to down-regulation of TGFβ-2 in GBM cells, which inhibits T and B cell activation and proliferation. Nanoparticle mediated delivery of antisense oligonucleotides (AON) to GBM xenografts in rats showed better delivery of AON to target cells and increased rates of activation of CD25 + T cells leading to immunostimulation [129, 130].
Combination therapy in GBM: ongoing clinical trials
Molecular profiling of GBM and preclinical research has led to the discovery of several targets for GBM therapy. The success of poly (ADP ribose) polymerase inhibitors (PARPi) in the treatment of breast and ovarian cancer in recent years has led to an intensive focus on exploiting the underlying combination therapy approach for both discerning the mechanisms of oncogenesis as well as for developing better treatment regimens. Depending upon the GBM sub-type, treatment modalities that combine two or more chemotherapeutic agents are being tested in ongoing clinical trials. In this section we summarize a list of the ongoing clinical trials for various GBM targets that are being tested in combination with radio-and/or chemotherapy (Table 3).
Future perspectives
Recent therapeutic approaches in cancer are based on the major advances made in areas of molecular biology, cellular biology and genomics. Administering drug combinations to patients that provide better clinical outcomes than individual agents do, is one such advance that has been successfully translated into therapy. PARP inhibitors, which utilize the combination therapy approach, have shown excellent results in the treatment of breast and ovarian cancer changing the diagnostic and therapeutic landscape of the disease. Since combination drugs target multiple pathways, they rely on smaller drug doses, which help minimize drug resistance, a pitfall of adjuvant therapy in the clinic. However, it is important to consider some limitations to the use combination therapy in GBM. First, a better understanding of the signaling networks and molecular players in GBM is crucial for the development of combination regimens and the success of this approach. Second, some drug combinations are effective and act synergistically for therapeutic benefits; some drug interactions may produce unwanted side effects on the patient’s health. Tumor heterogeneity and the unique immunological milieu of CNS is a major challenge in designing effective therapy regimens for GBM. One approach to overcome these limitations is to build mathematical models of synergism/antagonism of drugs and pathways that are effective in predicting drug combinations for multifactorial disorders like GBM. Mathematical models are effective in visualizing drug combinations in a dose response matrix that can be subsequently validated by in vitro and/or in vivo experiments [131]. Quantification of synergism (when the result of combining two or more chemical compounds produces an effect that is greater than additive effects of individual compounds) or antagonism (when the result of combining two or more chemical compounds produces an effect that is less than the additive effects of the individual compounds) is assessed based on the deviation of the observed combination response from the expected combination response calculated using a reference model [132]. The most commonly used reference models for calculating combination drug response include the Highest single agent (HSA) model, Loewe additivity model, Bliss independence model, and the Zero interaction potency (ZIP) model [133,134,135,136]. Combination therapy and drug synergism for targeted heterogeneous tumors and the interacting tumor microenvironment thus holds promise. Future research should focus on identifying synergistic interactions between chemotherapy, radiotherapy and immunotherapy in order to maximize the antitumor potential of individual treatment approaches.
Abbreviations
- ATRX:
-
alpha-thalassemia/mental retardation syndrome X-linked
- aRIST:
-
alternative to rapamycin, GDC-0941
- AONs:
-
antisense oligonucleotides
- BMP:
-
bone morphogenetic protein
- CSCs:
-
cancer stem cells
- CNS:
-
central nervous system
- G-CIMP:
-
CpG island hypermethylation phenotype
- CDK:
-
cycline-dependent kinase
- Cyp:
-
cyclopamine
- EGFR:
-
epidermal growth factor receptor
- GSI:
-
γ-secretase inhibitor
- GBM:
-
glioblastoma multiforme
- GBM-O:
-
glioblastoma multiforme with oligodendroglial component
- GSC:
-
glioblastoma stem-like cells
- HIF:
-
hypoxia inducible transcription factors
- IL:
-
interleukin
- IDH1:
-
isocitrate dehydrogenase 1
- mCyp:
-
micellarized Cyp
- MMR:
-
mismatch repair
- MDM2:
-
murine double minute 2
- MDSCs:
-
myeloid-derived suppressor cells
- NF1:
-
neurofibromin 1
- MGMT:
-
O(6)-methylguanine-DNAmethyltransferase
- PARPi:
-
PARP inhibitors
- POSTN:
-
periostin
- PTEN:
-
phosphatase and tensin
- PI3K:
-
phosphatidylinositol 3-kinase
- PDGFRA:
-
platelet-derived growth factor receptor alpha
- RT:
-
radiotherapy
- RIST:
-
rapamycin, irinotecan, sunitinib, temozolomide
- Shh:
-
Sonic-hedgehog
- TMZ:
-
temozolomide
- TCGA:
-
The Cancer Genome Atlas
- Tf-NP:
-
transferring-functionized nanoparticle
- TAMs:
-
tumor-associated macrophages
- TNF:
-
tumor necrosis factor
- TP53:
-
tumor protein p53
- VEGF:
-
vascular endothelial growth factor
References
Manolagas SC, Kronenberg HM (2014) Reproducibility of results in preclinical studies: a perspective from the bone field. J Bone Miner Res 29(10):2131–2140
Cunanan KM et al (2017) Basket trials in oncology: a trade-off between complexity and efficiency. J Clin Oncol 35(3):271–273
Reardon DA et al (2015) Immunotherapy for neuro-oncology: the critical rationale for combinatorial therapy. Neuro Oncol 17(Suppl 7):vii32–vii40
Coleman CN et al (2016) Improving the predictive value of preclinical studies in support of radiotherapy clinical trials. Clin Cancer Res 22(13):3138–3147
Miles D, von Minckwitz G, Seidman AD (2002) Combination versus sequential single-agent therapy in metastatic breast cancer. Oncologist 7(Suppl 6):13–19
Bayat Mokhtari R et al (2017) Combination therapy in combating cancer. Oncotarget 8(23):38022–38043
Paolillo M, Boselli C, Schinelli S (2018) Glioblastoma under siege: an overview of current therapeutic strategies. Brain Sci 8(1):15
Malkki H (2016) Trial Watch: glioblastoma vaccine therapy disappointment in Phase III trial. Nat Rev Neurol 12(4):190
Polivka J Jr et al (2017) Advances in experimental targeted therapy and immunotherapy for patients with glioblastoma multiforme. Anticancer Res 37(1):21–33
Louis DN et al (2007) The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 114(2):97–109
Ostrom QT et al (2013) CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2006–2010. Neuro Oncol 15(Suppl 2):ii1–ii56
Koshy M et al (2012) Improved survival time trends for glioblastoma using the SEER 17 population-based registries. J Neurooncol 107(1):207–212
Kleihues P, Ohgaki H (1999) Primary and secondary glioblastomas: from concept to clinical diagnosis. Neuro Oncol 1(1):44–51
Ohgaki H, Kleihues P (2009) Genetic alterations and signaling pathways in the evolution of gliomas. Cancer Sci 100(12):2235–2241
Ohgaki H et al (2004) Genetic pathways to glioblastoma: a population-based study. Cancer Res 64(19):6892–6899
Watanabe T et al (2009) IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. Am J Pathol 174(4):1149–1153
Ohgaki H, Kleihues P (2005) Epidemiology and etiology of gliomas. Acta Neuropathol 109(1):93–108
Verhaak RG et al (2010) Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17(1):98–110
Olar A, Aldape KD (2014) Using the molecular classification of glioblastoma to inform personalized treatment. J Pathol 232(2):165–177
Phillips HS et al (2006) Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 9(3):157–173
Louis DN et al (2016) The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol 131(6):803–820
Ohgaki H, Kleihues P (2013) The definition of primary and secondary glioblastoma. Clin Cancer Res 19(4):764–772
Kleinschmidt-DeMasters BK et al (2013) Epithelioid GBMs show a high percentage of BRAF V600E mutation. Am J Surg Pathol 37(5):685–698
Song XY et al (2011) Glioblastoma with PNET-like components has a higher frequency of isocitrate dehydrogenase 1 (IDH1) mutation and likely a better prognosis than primary glioblastoma. Int J Clin Exp Pathol 4(7):651–660
Schittenhelm J, Psaras T (2010) Glioblastoma with granular cell astrocytoma features: a case report and literature review. Clin Neuropathol 29(5):323–329
Parsons DW et al (2008) An integrated genomic analysis of human glioblastoma multiforme. Science 321(5897):1807–1812
Chen J, McKay RM, Parada LF (2012) Malignant glioma: lessons from genomics, mouse models, and stem cells. Cell 149(1):36–47
Hegi ME et al (2005) MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 352(10):997–1003
Nakagawachi T et al (2003) Silencing effect of CpG island hypermethylation and histone modifications on O6-methylguanine-DNA methyltransferase (MGMT) gene expression in human cancer. Oncogene 22(55):8835–8844
Wick W et al (2014) MGMT testing—the challenges for biomarker-based glioma treatment. Nat Rev Neurol 10(7):372–385
Bhattacharjee S et al (2013) MHF1-2/CENP-S-X performs distinct roles in centromere metabolism and genetic recombination. Open Biol 3(9):130102
Castel SE et al (2014) Dicer promotes transcription termination at sites of replication stress to maintain genome stability. Cell 159(3):572–583
Manuel JM et al (2016) Role of concurrent methylation pattern of MGMT, TP53 and CDKN2A genes in the prognosis of high grade glioma. J Carcinog Mutagen 7(1):2
Heimberger AB et al (2005) The natural history of EGFR and EGFRvIII in glioblastoma patients. J Transl Med 3:38
Shinojima N et al (2003) Prognostic value of epidermal growth factor receptor in patients with glioblastoma multiforme. Cancer Res 63(20):6962–6970
Cohen AL, Holmen SL, Colman H (2013) IDH1 and IDH2 mutations in gliomas. Curr Neurol Neurosci Rep 13(5):345
Lai A et al (2011) Phase II study of bevacizumab plus temozolomide during and after radiation therapy for patients with newly diagnosed glioblastoma multiforme. J Clin Oncol 29(2):142–148
Malta TM et al (2018) Glioma CpG island methylator phenotype (G-CIMP): biological and clinical implications. Neuro Oncol 20(5):608–620
Noushmehr H et al (2010) Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 17(5):510–522
Brennan CW et al (2013) The somatic genomic landscape of glioblastoma. Cell 155(2):462–477
Ohgaki H, Kleihues P (2007) Genetic pathways to primary and secondary glioblastoma. Am J Pathol 170(5):1445–1453
Hill C, Hunter SB, Brat DJ (2003) Genetic markers in glioblastoma: prognostic significance and future therapeutic implications—Commentary. Adv Anat Pathol 10(4):212–217
Nandakumar P, Mansouri A, Das S (2017) The role of ATRX in glioma biology. Front Oncol 7:236
Koschmann C, Lowenstein PR, Castro MG (2016) ATRX mutations and glioblastoma: impaired DNA damage repair, alternative lengthening of telomeres, and genetic instability. Mol Cell Oncol 3(3):e1167158
Wiestler B et al (2013) ATRX loss refines the classification of anaplastic gliomas and identifies a subgroup of IDH mutant astrocytic tumors with better prognosis. Acta Neuropathol 126(3):443–451
Jiao Y et al (2012) Frequent ATRX, CIC, FUBP1 and IDH1 mutations refine the classification of malignant gliomas. Oncotarget 3(7):709–722
Knobbe CB, Merlo A, Reifenberger G (2002) Pten signaling in gliomas. Neuro Oncol 4(3):196–211
Hu N, Richards R, Jensen R (2016) Role of chromosomal 1p/19q co-deletion on the prognosis of oligodendrogliomas: a systematic review and meta-analysis. Interdiscip Neurosurg 5:58–63
Marziali G et al (2016) Metabolic/proteomic signature defines two glioblastoma subtypes with different clinical outcome. Sci Rep 6:21557
Wang Q et al (2017) Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell 32(1):42–56.e6
Stupp R et al (2009) Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol 10(5):459–466
Singh SK et al (2004) Identification of human brain tumour initiating cells. Nature 432(7015):396–401
Liu G et al (2006) Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol Cancer 5:67
Bao S et al (2006) Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444(7120):756–760
Lathia JD et al (2015) Cancer stem cells in glioblastoma. Genes Dev 29(12):1203–1217
Hadjipanayis CG, Van Meir EG (2009) Brain cancer propagating cells: biology, genetics and targeted therapies. Trends Mol Med 15(11):519–530
Qazi MA et al (2017) Intratumoral heterogeneity: pathways to treatment resistance and relapse in human glioblastoma. Ann Oncol 28(7):1448–1456
Osuka S et al (2013) IGF1 receptor signaling regulates adaptive radioprotection in glioma stem cells. Stem Cells 31(4):627–640
Chen J et al (2012) A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 488(7412):522–526
Sottoriva A et al (2013) Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc Natl Acad Sci USA 110(10):4009–4014
Inda MD et al (2010) Tumor heterogeneity is an active process maintained by a mutant EGFR-induced cytokine circuit in glioblastoma. Genes Dev 24(16):1731–1745
Patel AP et al (2014) Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344(6190):1396–1401
Reinartz R et al (2017) Functional subclone profiling for prediction of treatment-induced intratumor population shifts and discovery of rational drug combinations in human glioblastoma. Clin Cancer Res 23(2):562–574
Venere M et al (2015) The mitotic kinesin KIF11 is a driver of invasion, proliferation, and self-renewal in glioblastoma. Sci Transl Med. 7(304):304ra143
Kim H et al (2015) Whole-genome and multisector exome sequencing of primary and post-treatment glioblastoma reveals patterns of tumor evolution. Genome Res 25(3):316–327
Bhat KPL et al (2013) Mesenchymal differentiation mediated by NF-kappaB promotes radiation resistance in glioblastoma. Cancer Cell 24(3):331–346
Wang R et al (2010) Glioblastoma stem-like cells give rise to tumour endothelium. Nature 468(7325):829–833
Heddleston JM et al (2011) Glioma stem cell maintenance: the role of the microenvironment. Curr Pharm Des 17(23):2386–2401
Li Z et al (2009) Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell 15(6):501–513
Hjelmeland AB et al (2011) Acidic stress promotes a glioma stem cell phenotype. Cell Death Differ 18(5):829–840
Flavahan WA et al (2016) Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 529(7584):110–114
Hegi ME et al (2008) Correlation of O6-methylguanine methyltransferase (MGMT) promoter methylation with clinical outcomes in glioblastoma and clinical strategies to modulate MGMT activity. J Clin Oncol 26(25):4189–4199
Stommel JM et al (2007) Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science 318(5848):287–290
Calabrese C et al (2007) A perivascular niche for brain tumor stem cells. Cancer Cell 11(1):69–82
Wu A et al (2010) Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro Oncol 12(11):1113–1125
Anjum K et al (2017) Current status and future therapeutic perspectives of glioblastoma multiforme (GBM) therapy: a review. Biomed Pharmacother 92:681–689
Arcella A et al (2018) Brain targeting by liposome-biomolecular corona boosts anticancer efficacy of temozolomide in glioblastoma cells. ACS Chem Neurosci. https://doi.org/10.1021/acschemneuro.8b00339
Chowdhury FA et al (2018) Therapeutic potential of thymoquinone in glioblastoma treatment: targeting major gliomagenesis signaling pathways. Biomed Res Int 2018:4010629
Jiang Y et al (2018) MicroRNA-599 suppresses glioma progression by targeting RAB27B. Oncol Lett 16(1):1243–1252
Zhang Q et al (2018) Current status and potential challenges of mesenchymal stem cell-based therapy for malignant gliomas. Stem Cell Res Ther 9(1):228
Shibahara I et al (2013) The expression status of CD133 is associated with the pattern and timing of primary glioblastoma recurrence. Neuro Oncol 15(9):1151–1159
Brescia P et al (2013) CD133 is essential for glioblastoma stem cell maintenance. Stem Cells 31(5):857–869
Wang CH et al (2011) Photothermolysis of glioblastoma stem-like cells targeted by carbon nanotubes conjugated with CD133 monoclonal antibody. Nanomedicine 7(1):69–79
Shin DH et al (2015) Synergistic effect of immunoliposomal gemcitabine and bevacizumab in glioblastoma stem cell-targeted therapy. J Biomed Nanotechnol 11(11):1989–2002
Safa AR et al (2015) Emerging targets for glioblastoma stem cell therapy. J Biomed Res 30:19
Fan X et al (2010) NOTCH pathway blockade depletes CD133-positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem Cells 28(1):5–16
Herrmann A et al (2014) TLR9 is critical for glioma stem cell maintenance and targeting. Cancer Res 74(18):5218–5228
Guryanova OA et al (2011) Nonreceptor tyrosine kinase BMX maintains self-renewal and tumorigenic potential of glioblastoma stem cells by activating STAT3. Cancer Cell 19(4):498–511
Tate CM et al (2012) A BMP7 variant inhibits the tumorigenic potential of glioblastoma stem-like cells. Cell Death Differ 19(10):1644–1654
Chung AS, Ferrara N (2011) Developmental and pathological angiogenesis. Annu Rev Cell Dev Biol 27:563–584
Kong DH et al (2017) A review of anti-angiogenic targets for monoclonal antibody cancer therapy. Int J Mol Sci 18(8):1786
Zhou W et al (2015) Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat Cell Biol 17(2):170–182
Bhattacharjee S, Nandi S (2017) Synthetic lethality in DNA repair network: a novel avenue in targeted cancer therapy and combination therapeutics. IUBMB Life 69(12):929–937
Bhattacharjee S, Nandi S (2017) DNA damage response and cancer therapeutics through the lens of the Fanconi Anemia DNA repair pathway. Cell Commun Signal 15(1):41
Bhattacharjee S, Nandi S (2016) Choices have consequences: the nexus between DNA repair pathways and genomic instability in cancer. Clin Transl Med 5(1):45
Bhattacharjee S, Nandi S (2018) Rare genetic diseases with defects in DNA repair: opportunities and challenges in orphan drug development for targeted cancer therapy. Cancers (Basel) 10(9):298
Cachia D et al (2015) Primary and secondary gliosarcomas: clinical, molecular and survival characteristics. J Neurooncol 125(2):401–410
Weller M, Steinbach JP, Wick W (2005) Temozolomide: a milestone in the pharmacotherapy of brain tumors. Future Oncol 1(6):747–754
Sanderson BJ, Shield AJ (1996) Mutagenic damage to mammalian cells by therapeutic alkylating agents. Mutat Res 355(1–2):41–57
Goldwirt L et al (2013) Development of a new UPLC-MSMS method for the determination of temozolomide in mice: application to plasma pharmacokinetics and brain distribution study. Biomed Chromatogr 27(7):889–893
Trinh VA, Patel SP, Hwu WJ (2009) The safety of temozolomide in the treatment of malignancies. Expert Opin Drug Saf 8(4):493–499
Choi JS, Kim CS, Berdis A (2018) Inhibition of translesion DNA synthesis as a novel therapeutic strategy to treat brain cancer. Cancer Res 78(4):1083–1096
Becher OJ et al (2008) Gli activity correlates with tumor grade in platelet-derived growth factor-induced gliomas. Cancer Res 68(7):2241–2249
Singec I et al (2006) Defining the actual sensitivity and specificity of the neurosphere assay in stem cell biology. Nat Methods 3(10):801–806
Liu YJ et al (2017) Combination therapy with micellarized cyclopamine and temozolomide attenuate glioblastoma growth through Gli1 down-regulation. Oncotarget 8(26):42495–42509
Iorio AL et al (2017) Tumor response of temozolomide in combination with morphine in a xenograft model of human glioblastoma. Oncotarget 8(52):89595–89606
Wang H et al (2017) Combination therapy in a xenograft model of glioblastoma: enhancement of the antitumor activity of temozolomide by an MDM2 antagonist. J Neurosurg 126(2):446–459
Kim SS et al (2015) A tumor-targeting p53 nanodelivery system limits chemoresistance to temozolomide prolonging survival in a mouse model of glioblastoma multiforme. Nanomedicine 11(2):301–311
Lan F et al (2016) Sulforaphane reverses chemo-resistance to temozolomide in glioblastoma cells by NF-kappaB-dependent pathway downregulating MGMT expression. Int J Oncol 48(2):559–568
Lan F et al (2015) Sulforaphane enhances temozolomide-induced apoptosis because of down-regulation of miR-21 via Wnt/beta-catenin signaling in glioblastoma. J Neurochem 134(5):811–818
Sinnberg T et al (2009) Inhibition of PI3 K-AKT-mTOR signaling sensitizes melanoma cells to cisplatin and temozolomide. J Invest Dermatol 129(6):1500–1515
Presneau N et al (2009) Potential therapeutic targets for chordoma: PI3K/AKT/TSC1/TSC2/mTOR pathway. Br J Cancer 100(9):1406–1414
Prasad G et al (2011) Inhibition of PI3 K/mTOR pathways in glioblastoma and implications for combination therapy with temozolomide. Neuro Oncol 13(4):384–392
Lam FC et al (2018) Enhanced efficacy of combined temozolomide and bromodomain inhibitor therapy for gliomas using targeted nanoparticles. Nat Commun 9:1991
Nitta Y et al (2016) Nimotuzumab enhances temozolomide-induced growth suppression of glioma cells expressing mutant EGFR in vivo. Cancer Med 5(3):486–499
Nonnenmacher L et al (2015) RIST: a potent new combination therapy for glioblastoma. Int J Cancer 136(4):E173–E187
Lesueur P et al (2017) Poly-(ADP-ribose)-polymerase inhibitors as radiosensitizers: a systematic review of pre-clinical and clinical human studies. Oncotarget 8(40):69105–69124
Lesueur P et al (2018) Radiosensitization effect of talazoparib, a parp inhibitor, on glioblastoma stem cells exposed to low and high linear energy transfer radiation. Sci Rep 8:3664
Knudsen ES, Wang JY (2010) Targeting the RB-pathway in cancer therapy. Clin Cancer Res 16(4):1094–1099
Parrish KE et al (2015) Efflux transporters at the blood–brain barrier limit delivery and efficacy of cyclin-dependent kinase 4/6 inhibitor palbociclib (PD-0332991) in an orthotopic brain tumor model. J Pharmacol Exp Ther 355(2):264–271
Whittaker S et al (2017) Combination of palbociclib and radiotherapy for glioblastoma. Cell Death Discov 3:17033
Park J et al (2017) Expression of immune checkpoint molecules on tumor infiltrating lymphocytes in glioblastoma multiforme. J Immunol 198(1):196
Fecci PE et al (2007) Systemic CTLA-4 blockade ameliorates glioma-induced changes to the CD4(+) T cell compartment without affecting regulatory T-cell function. Clin Cancer Res 13(7):2158–2167
Pardoll DM (2012) The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 12(4):252–264
Zeng J et al (2013) Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int J Radiat Oncol Biol Phys 86(2):343–349
Raychaudhuri B et al (2015) Myeloid derived suppressor cell infiltration of murine and human gliomas is associated with reduction of tumor infiltrating lymphocytes. J Neurooncol 122(2):293–301
Raychaudhuri B et al (2011) Myeloid-derived suppressor cell accumulation and function in patients with newly diagnosed glioblastoma. Neuro Oncol 13(6):591–599
Kamran N et al (2017) Immunosuppressive myeloid cells’ blockade in the glioma microenvironment enhances the efficacy of immune-stimulatory gene therapy. Mol Ther 25(1):232–248
Jachimczak P et al (1996) Transforming growth factor-beta-mediated autocrine growth regulation of gliomas as detected with phosphorothioate antisense oligonucleotides. Int J Cancer 65(3):332–337
Schneider T et al (2008) Brain tumor therapy by combined vaccination and antisense oligonucleotide delivery with nanoparticles. J Neuroimmunol 195(1–2):21–27
Mathews Griner LA et al (2014) High-throughput combinatorial screening identifies drugs that cooperate with ibrutinib to kill activated B-cell-like diffuse large B-cell lymphoma cells. Proc Natl Acad Sci USA 111(6):2349–2354
Mott BT et al (2015) High-throughput matrix screening identifies synergistic and antagonistic antimalarial drug combinations. Sci Rep 5:13891
Berenbaum MC (1989) What is synergy? Pharmacol Rev 41(2):93–141
Loewe S (1953) The problem of synergism and antagonism of combined drugs. Arzneimittelforschung 3(6):285–290
Bliss CI (1939) The toxicity of poisons applied jointly 1. Ann Appl Biol 26(3):585–615
Yadav B et al (2015) Searching for drug synergy in complex dose–response landscapes using an interaction potency model. Comput Struct Biotechnol J 13:504–513
Authors’ contributions
All authors were involved in the conception, design and drafting of the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We apologize to those colleagues whose work has not been cited due to space limitation.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
Not applicable.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Funding
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Ghosh, D., Nandi, S. & Bhattacharjee, S. Combination therapy to checkmate Glioblastoma: clinical challenges and advances. Clin Trans Med 7, 33 (2018). https://doi.org/10.1186/s40169-018-0211-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40169-018-0211-8