
Abstract
The pathophysiology of atopic dermatitis (AD) is highly complex and understanding of disease endotypes may improve disease management. Immunoglobulins E (IgE) against human skin epitopes (IgE autoantibodies) are thought to play a role in disease progression and prolongation. These antibodies have been described in patients with severe and chronic AD, suggesting a progression from allergic inflammation to severe autoimmune processes against the skin. This review provides a summary of the current knowledge and gaps on IgE autoreactivity and self-reactive T cells in children and adults with AD based on a systematic search. Currently, the clinical relevance and the pathomechanism of IgE autoantibodies in AD needs to be further investigated. Additionally, it is unknown whether the presence of IgE autoantibodies in patients with AD is an epiphenomenon or a disease endotype. However, increased knowledge on the clinical relevance and the pathophysiologic role of IgE autoantibodies and self-reactive T cells in AD can have consequences for diagnosis and treatment. Responses to the current available treatments can be used for better understanding of the pathways and may shed new lights on the treatment options for patients with AD and autoreactivity against skin epitopes. To conclude, IgE autoantibodies and self-reactive T cells can contribute to the pathophysiology of AD based on the body of evidence in literature. However, many questions remain open. Future studies on autoreactivity in AD should especially focus on the clinical relevance, the contribution to the disease progression and chronicity on cellular level, the onset and therapeutic strategies.
Similar content being viewed by others


Background
Atopic dermatitis (AD) is an itchy, chronic relapsing skin condition affecting up to 25% of children and 2–8% of adults [1, 2]. First manifestations of AD usually appear in early childhood [3]. Allergic sensitizations in early childhood can act as eliciting or aggravating factors (atopic march) [4], which may continue in adulthood [5, 6] and exert a major disease burden, impaired quality of life and individual suffering. AD is an environmentally induced and IgE-mediated disease in which the allergic inflammation is causing a disturbed skin barrier, and can be aggravated by colonization with Staphylococcus aureus [7], Malassezia species [8,9,10] and pollutants [11].
The association between atopy and autoimmune diseases has gained interest in the last decades, likely because the incidences of both allergic- (AD, asthma, and rhinoconjunctivitis, allergic rhinitis) and autoimmune diseases (e.g. psoriasis, multiple sclerosis) are rising worldwide [12]. Patients with AD can be at higher risk for the development of co-morbid autoimmune diseases [13,14,15]. In addition, a combined allergic-autoimmune-driven response has been described in patients with moderate/severe AD [15,16,17,18,19,20,21,22]. However, it is still unclear whether IgE autoreactivity could be an endotype of AD or an epiphenomenon [21, 23]. Although several studies associate the presence of IgE autoreactivity with AD, the clinical relevance needs yet to be further investigated. Currently, the prevalence of autoreactive antibodies has mainly been investigated in adult patients with different disease backgrounds and age-matched healthy controls, while the pediatric profile is not well characterized. Development of autoreactive antibodies may already start in early childhood [20], likely due to the lack of immune stimulation. However, increased understanding of autoallergy in children may be of great importance with direct consequences for diagnosis and therapy.
The aim of this review is to summarize evidence on IgE autoreactivity in AD and the possible cellular pathways contributing to disease chronicity and severity. Additionally, we aim at comparing autoreactive profiles in children, adolescents and adults with AD to provide an overview of current knowledge and gaps. A systematic search was performed in PubMed using the following search strategy: Atopic dermatitis, atopic eczema, autoreactive, auto-IgE, autoantigen, autoallergy, autoimmunity, autoantibodies, autoreactive T cells (Additional file 1: Table S1). All available original studies on the association of immunoglobulin E (IgE) autoantibodies and T lymphocyte (T cell) autoreactivity in patients with AD were included. Studies in languages other than English, French, Dutch and German were excluded. The last update was on October 3, 2019. Eligible studies were screened by two independent reviewers (FB, KC) on title and abstract. Screening of full-text and data-extraction was performed (FB and SDV) and disagreements were resolved by discussion with a third reviewer (IKK). The PRISMA flow diagram [24] was used to depict the flow of the selection process (Fig. 1). In total, 27 original articles were included of which 18 on IgE autoantibodies, 7 on autoreactive T cells, 1 on IgG autoantibodies and 1 on sweat antigen (Fig. 1). An overview of the original articles on IgE autoreactivity in patients with AD can be found in Additional file 1: Table S2.

Flow diagram of the systematic search
The association of atopic dermatitis and autoimmunity
Atopy has been associated with classical autoimmune disorders and AD may co-occur with autoimmune diseases (psoriasis, rheumatoid arthritis, multiple sclerosis and type I diabetes mellitus). In a matched case–control study, including 8589 children with diagnosed AD before the age of 5 and 85,890 controls, the association between parental autoimmune disease and the development of AD in their offspring were studied [25]. The presence of maternal (not paternal), dermatologic or digestive autoimmune disease was associated with an increased risk for development of AD in their offspring. A follow-up study on a previous cohort consisting of 241 children with AD based on a questionnaire with a response of 145 adolescents (16–23 years), found an increased incidence of autoimmune disorders compared to non-atopic controls (9% vs. 1%) [26]. This suggested that infantile AD may increase the risk of autoimmune disorders in adulthood. A study on 8112 adult patients with AD and age and gender matched controls demonstrated an association of AD with 11 of 22 selected autoimmune diseases. Additionally, AD was linked with having multiple autoimmune comorbidities, and smoking increased the risk of autoimmune comorbidities compared to nonsmokers [15]. However, a study including 14,849 subjects from five cohorts evaluated the relation between specific-IgE positivity against airborne allergens and autoimmunity, but no associations between atopic predisposition and development of autoimmune disorders were found [27]. Contrarily, IgE-mediated diseases may reduce the risk of developing autoimmunity. Active T helper 1 (Th1)-inflammation may suppress development of atopy, and atopy may suppress the severity of autoimmunity [28, 29]. As a result of contradictory findings and varying study protocols, the underlying mechanism of AD and autoimmune disease comorbidities is still unclear.
IgE autoreactivity in atopic dermatitis
Patients with AD often display elevated levels of total serum IgE without clinically relevant allergy for the most common allergens. Autoreactive IgE antibodies may elicit an allergic-autoimmune process and contribute to perpetuation of inflammation. This can be a reason for the chronic relapsing course of AD. Studies on the presence of a combined allergy-autoimmune process in patients with AD are summarized here.
Prevalence of IgE autoreactivity in AD
The first evidence of the presence of IgE autoreactivity to human proteins in AD was described 25 years ago [16,17,18,19]. IgE autoantibodies were found in sera from patients with AD or have been determined via a positive response to autoantigens in vitro. The prevalence of autoreactivity in patients with AD was previously summarized and ranges from 23 to 91% based on 14 studies involving 2644 individuals [21]. Comparable results were obtained in the present review (Table 1, Additional file 1: Table S2). Most likely the variation in the prevalence of autoreactivity can be explained by different detection methods, cohort composition and the origin of the tested autoantigens. Autoreactive antibodies of IgG [30, 31] and IgM [32] have also been detected in patients with AD. In this review we only focus on IgE autoreactive antibodies.
The prevalence of IgE autoreactivity has mainly been studied in adults with AD [17, 33,34,35,36,37]. Only a few studies included children with AD [19, 20] or adolescents [19, 38], while AD usually starts in childhood and the percentage of children affected by AD is around 7 times higher than in adults. Due to insufficient numbers of studies on IgE autoreactivity both in children with AD and in healthy age-matched controls, the cause and thus, the start of the development of IgE autoantibodies is unknown.
It has been established that AD occurs more frequently in individuals with an Asian, African or Afro-American genetic background [39]. However, the association between ethnicity and the presence of IgE autoreactivity in AD has not been described so far.
Autoreactivity may develop already in early childhood
Autoreactivity may develop in early infancy [20] and atopy in early childhood may increase the development of autoimmune diseases later in life [20, 26, 28]. In a cross-sectional study including 346 children with active AD and 117 controls antinuclear antibodies (ANA) have been found in both groups, which increased with age [40]. Although not significantly different, in children with AD the ANA antibodies had a tendency to appear earlier, suggesting that active AD in early infancy can lead to earlier development of systemic autoreactivity [40].
Only few studies investigated IgE autoantibodies in children, adolescents and adults with AD. One study found that 22/51 patients with AD were positive for autoreactive antibodies [19]. To evaluate the IgE autoreactivity based on age, we subdivided the study population (Table 1). Additionally, a retrospective analysis of serological parameters from both children and adults with AD gave insights in the onset of IgE autoreactivity [20]. Twenty-three percent of adults with AD showed IgE reactivity to human epithelial antigens compared to the control groups. In addition, 15% of children younger than 1 year of age showed IgE autoreactivity. Children at the age of 2–13 years with moderate to severe AD, 80% were positive for IgE autoantibodies. Repeated serum determinations within 1 year demonstrated increased IgE autoreactivity in children at 2–6 years with AD and multiple sensitizations concluding that IgE autoreactivity can develop in early infancy [20]. The prevalence of IgE autoantibodies in newborns, children, adolescents and adults with AD based on their age is presented in Table 1.
Prevalence of IgE autoreactivity in other diseases
Often, AD is the starting point of the atopic march, in which patients with AD develop food allergy, allergic rhinitis, allergic rhinoconjunctivitis and allergic asthma later in life [41]. In case IgE autoantibodies start to develop in early childhood, it seems possible that these antibodies also play a role in co-morbid diseases of AD (and enhance the atopic march), as well as other inflammatory diseases. On the other hand, IgE autoantibodies have been identified in comparable frequency in children with or without AD (unpublished data Gutermuth and Schmidt-Weber). Thus, the pathophysiologic relevance of autoreactive IgE has not been investigated in-depth.
Other skin-related diseases have been associated with the presence of IgE autoreactive antibodies, such as chronic urticaria [42], IgE-mediated bullous pemphigoid [43]. Additionally, 65% of the patients with systemic lupus erythematosus produce IgE autoantibodies [44] and 83% in patients with active disease [45] as well as 50–60% of patients with rheumatoid arthritis [46]. Regarding other isotypes, in patients with rheumatoid arthritis, autoantibodies of subclasses IgG, IgM and IgA can be found in serum years before the disease onset and can predict disease development [47,48,49,50]. It was previously reported that IgE autoantibodies were neither present in healthy subjects nor in patients with allergic rhinoconjunctivitis, psoriasis or other inflammatory diseases based on results from 8 studies of 816 participants [21]. The prevalence of autoreactivity was not associated with age, gender, or disease duration, but rather linked to disease severity [21].
The presence of IgE autoreactivity in atopic dermatitis has been linked to disease severity
Since the discovery of autoreactivity in AD, the presence of IgE autoantibodies has been associated with disease severity in adult patients [17, 19, 34, 35, 51,52,53] and in children with AD [20]. IgE autoantibodies were mainly found in patients with moderate to severe AD, while they were absent in healthy controls (Table 2). Besides, IgE autoantibody levels decreased after successful treatment [20]. Therefore, the presence of IgE autoantibodies is described as a pathogenic factor in AD and as an indicator for chronic tissue damage. However, some studies also tested healthy individuals positive for autoreactivity. Skin tests with intradermal autologous sweat injections were positive in 56/66 patients with AD (84.4%), and in 3/27 healthy subjects (11.1%) [54]. Another study showed sweat antigen-induced histamine release from basophils in 47/61 (74.6%) patients with AD and 4/46 (8.7%) healthy subjects [55]. In a study on the autoreactivity to eight different cytotoxic T-lymphocyte (CTL)-directed peptides, no differences in IgE autoreactivity to 7 out of 8 CTL-directed epitopes were observed in patients with AD compared to healthy controls, while IgG autoreactivity was missing in a significant fraction of healthy donors. However, the study lacks functional appraisal of this finding [30]. Eukaryotic translation initiation factor 6 (eIF6)-specific serum IgG could be detected both in AD and healthy subjects. In addition, patients with AD and IgE autoantibodies showed no differences in SCORAD index compared to patients with AD without IgE autoantibodies (P < .7625), but had higher levels of total serum IgE [36]. These contradictory findings in healthy subjects may depend on the age of the patients (unpublished data Gutermuth and Schmidt-Weber) or the targets that were tested (Table 2).
When IgE autoantibodies are present in patients with severe disease, the level of autoantibodies may already increase before appearance of symptoms, as has been shown for IgG in patients with rheumatoid arthritis [47,48,49,50]. If so, the expansion of IgE autoantibodies may be useful as a marker or predictor for increased disease activity. However, prospective observational studies in children, adolescents and adults or birth cohorts are needed to investigate this. Alternatively, IgE autoreactivity might be an epiphenomenon which is elicited by a Th2-driven inflammation.
Targets of IgE autoantibodies
The first targets of IgE autoantibodies (autoantigens) that have been identified are epitopes of human keratinocyte proteins with partly unknown function (Hom s 1–5) [17,18,19, 37]. In more recent years, a wide range of autoantigens have been described. The most relevant targets are summarized in Table 2 and were previously listed by others [21, 22, 56]. Patients with the highest total serum IgE values, showed the highest frequency of anti-keratinocyte IgE [33]. Total protein purifications from epidermal carcinoma cell line A431 (17–91.7%) and epidermal keratinocyte suspensions (37%) were used to evaluate the presence of IgE autoreactivity in adult patients with AD. As specific targets, human manganese superoxide dismutase (MnSOD, 37.2%), RP1 (29%), eukaryotic translation initiation factor 6 (eIF6, 25.4%), dense fine speckles (DFS, 24.7%), Hom s2 (27.3%), Hom s4 (16.7%) and human thioredoxins (hTrx, 14.4%) have shown the highest percentages of autoreactivity, while no autoreactivity in healthy individuals (0%) was found for these targets (Table 2). This suggests that these autoantigens may be the most important ones in the pathophysiology in AD that are currently known. However, it is important to note that these data are based on only a few studies with relatively small sample sizes.
One study could not confirm a relationship between IgE and IgG subclass reactivity [51], which means that IgE autoantibodies can react to other antigens as IgG autoantibodies, while others found a positive association between IgG and IgE autoantibody reactivity’s [57]. Therefore, the role of this isotype relationship remains unclear. In addition, IgE responses to autoantigens can be boosted by seasonal exposure to pollen allergens in sensitized patients [51]. The presence of both exogenous allergens and autoallergens may therefore accelerate the immune response. It remains to be investigated what comes first; the exogenous allergens that facilitate the development of autoimmunity, or vice versa?
Pathomechanisms of autoimmunity
Autoreactive T cells
One of the most critical functions of the immune system is to avoid an effector response against self-antigens. During development in the thymus, T cells undergo positive selection for self-major histocompatibility complex (MHC) reactivity and a negative selection to destroy T cells with a high T cell receptor (TCR) affinity for self-peptides. T cells with lower affinity to self-peptides escape this selection and enter the periphery. The peripheral T cell population contains therefore approximately 4% of self-reactive T cells in healthy individuals [58]. Normally, peripheral tolerance mechanisms, such as suppression by T regulatory cells, anergy and ignorance, prevent these self-reactive T cells to become fully activated and cause pathological damage [59,60,61]. Like T regulatory cells, self-reactive T cells may even exert beneficial functions and contribute to tissue repair, to maintain tissue homeostasis, and stimulate the immune response against pathogens [58]. When the immune system is unable to distinguish between self and non-self-antigens, autoimmunity can develop as a consequence of loss of self-tolerance [62, 63]. Several pathways may underlie development of autoimmunity: (1) cross-reactivity due to molecular mimicry of exogenous and self-antigens, (2) epitope spreading; an autoimmune response secondary to the release of self-antigens due to tissue damage or chronic inflammation [64], (3) bystander activation; unspecific activation of T and B cells independent of TCR or B cell receptor signaling, such as cytokines, co-receptor expression and gap junctions [63], and (4) downregulation of regulatory T cells. Additional file 1: Table S3 provides an overview of studies on T-cell autoreactivity in AD.
Molecular mimicry
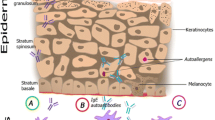
Molecular mimicry can occur when exogenous antigens activate autoreactive T or B cells due to structural homologies between the epitopes of foreign-antigens and self-antigens leading to autoimmunity [65]. Recognition of a range of MHC complexes is also called polyreactivity of TCRs or cross-reactivity. Evolutionary well conserved peptides in humans can share similar protein structures with pathogens which may lead to molecular mimicry after exposure to the pathogens. Serum IgE from subjects sensitized to thioredoxins (Trx) of Malassezia sympodialis (Mala s 13), Malassezia furfur or Aspergillus fumigatus were shown to cross-react with human Trx proteins leading to immediate allergic responses [22, 33, 66]. Likewise, the stress-inducible enzyme manganese superoxide dismutase (MnSOD) of fungal origin can also induce a humoral response against human MnSOD based on molecular mimicry. It was shown that specific-IgE to human MnSOD correlated with disease activity in 29/67 patients with AD [34]. Thus, molecular mimicry leads to antigen-specific T cells and antibodies that cross-react to both the foreign-peptide and the human-peptide. The latter can be demonstrated in an animal model using human peptides for sensitization subsequently followed by re-exposure to the human peptide leading to a Type-I hypersensitivity reaction. Antigen challenge of mice that were sensitized with human α-NAC showed an immediate allergen response with a mixed Th1 and Th2 reactivity and the presence of IgE and IgG antibodies against murine and human α-NAC [67]. Sequence similarities to human proteins can lead to binding of self-peptides in the skin and T cell activation, which may contribute to the pathophysiology of AD (Fig. 2). However, structural homology is unlikely to be a single pathway to elicit an autoimmune response and other factors such as loss of central tolerance, bystander activation and environmental or genetic factors also play a role in the development of autoimmunity [65].

Molecular mimicry can lead to IgE antibody production against self-antigens. An impaired barrier function facilitates access to environmental allergens and pathogens resulting in allergic sensitization and specific IgE antibody production. In the case of structural homologies of the epitopes (molecular mimicry), this can result in cross reactivity between exogenous antigens and self-antigens. DC dendritic cell, Th2 T helper 2 lymphocytes, IgE immunoglobulin E, IL-4 interleukin-4
Epitope spreading
Tissue damage or an impaired barrier function is crucial in the development of allergic sensitization. Beside genetic mutations, which can facilitate barrier defects, skin damage can be caused by mechanistic stress, such as scratching sequel of itch. In addition, exogenous factors [68,69,70] or mediators that are released during an ongoing chronic inflammation (IL-4, histamine, mast cell chymase) [71, 72] can facilitate an impaired barrier function. Endogenous proteins that are released following tissue damage can prime self-reactive T and B cells and become specific targets for autoantibodies. The role of epitope spreading is difficult to prove in humans, but is likely to contribute to AD. Tissue debris can be processed and presented on MHC-class II by peripheral antigen presenting cells (APCs) in lymphoid tissues. Activated and differentiated Th1 cells may migrate to the skin where they are re-exposed to the autoantigens by the resident APCs, such as dendritic cells and Langerhans cells. Autoreactive Th1 cells will then release cytokines and chemokines, such as tumour necrosis factor (TNF)-α, interferon (IFN)-γ and lymphotoxin, which directly accelerate further tissue damage. Macrophage inflammatory protein 1α and IFN-γ activate monocytes to differentiate into macrophages and facilitate bystander tissue destruction [64]. Cutaneous lymphocyte-associated antigen (CLA) is a skin-homing receptor expressed by more than 90% of skin-infiltrating T cells and 10–25% of the circulating T cells [73]. CLA+CD45RO+ T cells recognize allergens related to the skin and release higher levels of IL-13 and some IL-4 or IFN-γ, while CLA− CD45RO+ T cells produce higher amounts of IFN-γ [73, 74]. Higher amounts of IL-13 and IL-4 were released from CLA+ T cells of patients with AD compared to those of healthy controls [73]. IL-13 and IL-4 are needed for the isotype switch of B cells, while IFN-γ is a key factor that drives the Th1 response. Evidence was found that Hom s4 can induce the production and release of IFN-γ both in non-atopic and atopic individuals [37]. IL-17 was significantly upregulated by the α-chain of the nascent polypeptide-associated complex (α-NAC or Hom s2) [75] (Additional file 1: Table S3). In addition, the CLA+CD45RO+ T cells of patients with AD produce higher amounts of IL-31 than in healthy individuals [76]. IL-31R is expressed by keratinocytes and infiltrating macrophages [76], but also on sensory neurons [77] resulting in pruritus and inflammation, the hallmarks of AD, which drive the vicious cycle of skin damage.
It still remains speculative what the exact contribution of autoreactivity is in the pathophysiology of AD. A summary of the possible pathways and targets that may be involved in the process of allergy-autoimmunity in AD is presented in Fig. 3.

Hypothetic cellular pathways and targets of autoreactivity in atopic dermatitis. Skin damage can lead to release of self-peptides resulting in allergic sensitization to autoantigens via antigen presentation by dendritic cells (DC) to naïve T cells and class switch of B cells. Mast cells sensitized with IgE autoantibodies can directly interfere with the self-peptides that are released following skin damage resulting in histamine release. T cells with low binding affinity to self-peptides may escape selection and depletion in the thymus. Possibly, this population of cells may also be attracted to the skin where they are exposed to the self-peptides of the damaged skin. Cytokines (IL-4 and IFN-γ) produced by T-cells, and histamine by MC can directly exacerbate the skin lesion. Additionally, neurons in the skin can bind histamine and IL-31 specifically to the histamine 1 receptor (H1R) and IL-31 receptor (IL-31R) which results in itch and may lead to the chronicity of the impaired barrier function. Th2 T helper lymphocyte, EO eosinophils, ILC2 innate lymphoid cell, IgE immunoglobulin E, IL interleukin, IFN-γ Interferon gamma, TSLP thymic stromal lymphopoietin
New treatments as a model for better understanding of atopic dermatitis pathophysiology
Given the multiple pathomechanisms that contribute to AD, it is not always possible to find the underlying factor that drives the disease. Therefore, treatment responses of patients vary greatly. Patients with mild AD are relatively well-controlled with conventional therapies. Besides, in recent years, significant progress has been made for the development of pathway-specific biologic therapies for patients with severe and uncontrolled AD. Variations in treatment responses to pathway-specific effects can help to identify clinically relevant disease endotypes.
Patients with AD often show high serum IgE levels. Treatment with the recombinant humanized monoclonal antibody omalizumab, targeting the Fc of IgE, was thought to be a promising approach to control AD. However, a low response rate to omalizumab was found [78]. A systematic review of omalizumab treatment in severe AD showed that 62.5% had some benefit of the treatment, while no effect was found in 37.5% of the patients [79]. Other skin related diseases, such as bullous pemphigoid [80] and chronic spontaneous urticaria [81] can be successfully treated with omalizumab, highlighting the importance of IgE in the disease pathophysiology. As both bullous pemphigoid and chronic spontaneous urticaria are associated with the presence of IgE autoreactive antibodies, omalizumab could possibly be effective in patients with AD and IgE autoantibodies.
The monoclonal antibody rituximab induces B cell depletion and impairs the interaction between autoreactive Th cells [82] and antigen-presenting B cells. Rituximab also reduces the expression of CD40 and CD80 on B cells as well as the expression of CD40L, CD69, ICOS and HLA-DR on CD4+ Th cells, reducing the interaction between immune cells [83]. Rituximab directly induces CD4+CD25+ regulatory T cells [84]. Rituximab is effective to treat patients with autoimmune diseases, such as SLE, pemphigus vulgaris and rheumatoid arthritis. This highlights the role of B cells and autoreactive T cells in these diseases. Only limited trials have been published on rituximab treatment in AD with some conflicting results. Rituximab treatment can improve AD [85] or can be combined with omalizumab [86]. However, treatment of two patients with AD could not confirm these data [87]. It might be interesting to study the effect of rituximab on clinical improvement on selected patients with AD and autoreactivity.
IL-5 is a key player in the pathogenesis of eosinophilic diseases. Anti‐IL‐5 antibody (mepolizumab) therapy is effective to treat a subgroup of patients with persistent airway eosinophilia and moderate to severe asthma [88] as well as eosinophilic esophagitis [89]. The effect of mepolizumab on clinical improvement has also been studied in patients with AD [90]. A clinical trial including 18 actively and 22 placebo treated subjects demonstrated reduced numbers of blood eosinophils after mepolizumab and significant but only moderate improvement [90]. More evidence is needed to state that IL-5 may not be the crucial cytokine the pathomechanism of AD.
Systemic treatment with cyclosporine was shown to improve skin symptoms in patients with AD, and to reduce IgE autoreactivity [53, 91]. After ceasing treatment with cyclosporine, autoreactivity reappeared. Cyclosporine treatment suppresses the pathways involved in the autoimmune process, such as suppression of T cells as well as the restoration of the skin barrier which reduces the contact to the (auto)antigens [53].
Dysregulation of the Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway can contribute to autoinflammatory diseases including AD, psoriasis, rheumatoid arthritis, cutaneous lupus erythematosus, lichen planus [92]. Inhibitors of the JAK/STAT pathway are small molecules with immunosuppressive effects targeting JAK/STAT-mediated pathways by preventing the phosphorylation of JAK kinases, and thereby activation of STAT transcription factors. Most pathways in the AD pathogenesis are regulated by JAK/STAT kinases, such as the IL-4 receptor α (binding of IL-4 and IL-13). Therefore, treatment with JAK/STAT inhibitors can result in reduced cytokine production and effectively reduce the immune response. Interestingly, treatment with tofacitinib, a JAK1/JAK3 inhibitor, can also directly target keratinocytes and fibroblasts in 3D human skin equivalents in absence of immune cells [93]. IFN-γ signaling is mediated by the JAK/STAT pathway and is downregulated by JAK2 inhibitors, such as ruxolitinib (JAK1/JAK2 inhibitor). Oral or topical tofacitinib has been shown to be effective to treat autoimmune inflammatory diseases in phase 2 and 3 studies and is a promising treatment in AD [94,95,96,97,98,99,100]. It is unknown whether JAK/STAT inhibition may contribute to reduced levels of IgE autoantibodies. If so, it may affect IgE autoreactivity in an indirect way.
Dupilumab is a human monoclonal antibody against the IL-4 receptor α. Phase 3, clinical trials have proven the clinical effectiveness of dupilumab treatment in patients with moderate/severe AD [101,102,103]. A recent study of 138 adult patients with severe AD also confirmed the clinical effectiveness with improved symptoms of AD and self-reported clinical scores for pain/discomfort, itch, anxiety and depression, and quality of life [104]. The responses to both dupilumab treatment and JAK inhibitors in patients with severe AD confirm the importance of the cytokines IL-4 and IL-13 in the disease pathomechanism. Future studies are needed to evaluate the effects on the production of IgE autoantibodies in patients with AD.
Conclusion
Increased understanding of the contribution of IgE autoantibodies to the pathophysiology of AD may lead to improved diagnosis, treatment and prognosis. In addition, early detection may increase successful disease management. Therefore, moving from phenotypes to endotypes will help to understand the pathomechanism of AD, prediction of the course of disease and development of personalized therapy. Progress has been made to find evidence for the presence of IgE autoantibodies in AD and many targets have been identified (Table 2). Based on the body of evidence in the literature, it can be accepted that IgE autoantibodies and T cells against epitopes in the human skin can contribute to the pathophysiology of AD. However, the clinical relevance of IgE autoantibodies and self-reactive T cells in AD is still unclear, although, there are many studies showing an association between IgE autoreactivity and severity of clinical symptoms.
Currently, many questions on the role of autoantibodies in the pathomechanism of AD are still open:
-
Is there an autoreactive endotype of AD or is this an epiphenomenon?
-
Do the IgE autoantibodies start to develop in early childhood?
-
Can IgE autoantibodies have a beneficial role in healthy individuals especially in children?
-
Can this role show a switch to a pathogenic response?
-
Do they expand prior to disease progression and can they be used as predictive marker?
-
What is the subtype of self-reactive T cells and what is their exact role in AD?
-
What are the therapeutic implications of the presence of IgE autoreactivity in patients with severe AD?
Recently, four distinct clusters of patients with AD have been identified based on serum biomarker- and cytokine profiles regardless their clinical presentation [105]. Identification of biomarkers can identify downstream pathways that are involved in AD and clustering based on serum biomarker profiles may therefore be useful to explain the biological mechanisms of AD. It is clear that more research is warranted to find answers to the questions. Future perspectives include the clinical characterization of the individuals with AD and autoreactivity, the onset of IgE autoantibodies development, investigation of the contribution of the IgE autoantibodies and self-reactive T cells to the severity and chronicity of AD as well as the response to immune regulatory treatments in this group of patients.
Availability of data and materials
We performed a systematic search on Pubmed.
Abbreviations
- AD:
-
Atopic dermatitis
- ANA:
-
Antinuclear antibodies
- APC:
-
Antigen presenting cell
- CLA:
-
Cutaneous lymphocyte-associated antigen
- DFS:
-
Dense fine speckles
- hTrx:
-
Human thioredoxins
- hMnSOD:
-
Human manganese superoxide dismutase
- IFN-γ:
-
Interferon gamma
- Ig:
-
Immunoglobulin
- IL:
-
Interleukin
- JAK:
-
Janus kinase
- MHC :
-
Major histocompatibility complex
- STAT:
-
Signal transducer and activator of transcription
- TCR:
-
T cell receptor
- TNF-α:
-
Tumor necrosis factor alpha
- Th1:
-
T helper 1 lymphocytes
- Th2:
-
T helper 2 lymphocytes
References
Wollenberg A, Barbarot S, Bieber T, Christen-Zaech S, Deleuran M, Fink-Wagner A, et al. Consensus-based European guidelines for treatment of atopic eczema (atopic dermatitis) in adults and children: part I. J Eur Acad Dermatol Venereol. 2018;32(5):657–82.
Eichenfield LF, Tom WL, Chamlin SL, Feldman SR, Hanifin JM, Simpson EL, et al. Guidelines of care for the management of atopic dermatitis: section 1. Diagnosis and assessment of atopic dermatitis. J Am Acad Dermatol. 2014;70(2):338–51.
Illi S, von Mutius E, Lau S, Nickel R, Grüber C, Niggemann B, et al. The natural course of atopic dermatitis from birth to age 7 years and the association with asthma. J Allergy Clin Immunol. 2004;113(5):925–31.
Spergel JM. Epidemiology of atopic dermatitis and atopic march in children. Immunol Allergy Clin N Am. 2010;30(3):269–80.
Mortz CG, Andersen KE, Bindslev-Jensen C. Recall bias in childhood atopic diseases among adults in the Odense Adolescence Cohort Study. Acta Derm Venereol. 2015;95(8):968–72.
Garmhausen D, Hagemann T, Bieber T, Dimitriou I, Fimmers R, Diepgen T, et al. Characterization of different courses of atopic dermatitis in adolescent and adult patients. Allergy. 2013;68(4):498–506.
Nakamura Y, Oscherwitz J, Cease KB, Chan SM, Muñoz-Planillo R, Hasegawa M, et al. Staphylococcus δ-toxin promotes mouse allergic skin disease by inducing mast cell degranulation. Nature. 2013;503(7476):397–401.
Schmid-Grendelmeier P, Scheynius A, Crameri R. The role of sensitization to Malassezia sympodialis in atopic eczema. Chem Immunol Allergy. 2006;91:98–109.
Fischer Casagrande B, Flückiger S, Linder MT, Johansson C, Scheynius A, Crameri R, et al. Sensitization to the yeast Malassezia sympodialis is specific for extrinsic and intrinsic atopic eczema. J Investig Dermatol. 2006;126(11):2414–21.
Selander C, Zargari A, Möllby R, Rasool O, Scheynius A. Higher pH level, corresponding to that on the skin of patients with atopic eczema, stimulates the release of Malassezia sympodialis allergens. Allergy. 2006;61(8):1002–8.
Krämer U, Behrendt H. Luftverschmutzung und atopisches Ekzem. Hautarzt. 2019;70(3):169–84.
Bach J-F. The effect of infections on susceptibility to autoimmune and allergic diseases. N Engl J Med. 2002;347(12):911–20.
Silverberg JI, Gelfand JM, Margolis DJ, Boguniewicz M, Fonacier L, Grayson MH, et al. Association of atopic dermatitis with allergic, autoimmune, and cardiovascular comorbidities in US adults. Ann Allergy Asthma Immunol. 2018;121(5):604–12.
Schmitt J, Schwarz K, Baurecht H, Hotze M, Fölster-Holst R, Rodríguez E, et al. Atopic dermatitis is associated with an increased risk for rheumatoid arthritis and inflammatory bowel disease, and a decreased risk for type 1 diabetes. J Allergy Clin Immunol. 2016;137(1):130–6.
Andersen YMF, Egeberg A, Gislason GH, Skov L, Thyssen JP. Autoimmune diseases in adults with atopic dermatitis. J Am Acad Dermatol. 2017;76(2):274–80.
Valenta R, Duchene M, Pettenburger K, Sillaber C, Valent P, Bettelheim P, et al. Identification of profilin as a novel pollen allergen; IgE autoreactivity in sensitized individuals. Science. 1991;253(5019):557–60.
Valenta R, Steiner R, Seiberler S, Maurer D, Sperr WR, Valent P, et al. Immunoglobulin E response to human proteins in atopic patients. J Investig Dermatol. 1996;107(2):203–8.
Valenta R, Natter S, Seiberler S, Wichlas S, Maurer D, Hess M, et al. Molecular characterization of an autoallergen, Hom s 1, identified by serum IgE from atopic dermatitis patients 11 part of this manuscript was previously published in the proceedings of the 21st Symposium of the Collegium Internationale Allergologicum “Allergy—A Disease of Modern Society”, Int Arch Allergy Immunol 113:209–212, 1998. J Investig Dermatol. 1998;111(6):1178–83.
Natter S, Seiberler S, Hufnagl P, Binder BR, Hirschl AM, Ring J, et al. Isolation of cDNA clones coding for IgE autoantigens with serum IgE from atopic dermatitis patients. FASEB J. 1998;12(14):1559–69.
Mothes N, Niggemann B, Jenneck C, Hagemann T, Weidinger S, Bieber T, et al. The cradle of IgE autoreactivity in atopic eczema lies in early infancy. J Allergy Clin Immunol. 2005;116(3):706–9.
Tang TS, Bieber T, Williams HC. Does “autoreactivity” play a role in atopic dermatitis? J Allergy Clin Immunol. 2012;129(5):1209–15.
Roesner LM, Werfel T. Autoimmunity (or not) in atopic dermatitis. Front Immunol. 2019;10:2128.
Cipriani F, Ricci G, Leoni MC, Capra L, Baviera G, Longo G, et al. Autoimmunity in atopic dermatitis: biomarker or simply epiphenomenon? J Dermatol. 2014;41(7):569–76.
Moher D, Liberati A, Tetzlaff J, Altman DG. Preferred reporting items for systematic reviews and meta-analyses: The PRISMA Statement. PLoS Med. 2009;6(7):e1000097.
Hamann CR, Egeberg A, Silverberg JI, Gislason G, Skov L, Thyssen JP. Association between parental autoimmune disease and atopic dermatitis in their offspring: a matched case–control study. J Eur Acad Dermatol Venereol. 2019;33(6):1143–51.
Kokkonen J, Niinimäki A. Increased incidence of autoimmune disorders as a late complication in children with early onset dermatitis and/or milk allergy. J Autoimmun. 2004;22(4):341–4.
Skaaby T, Husemoen LLN, Thuesen BH, Fenger RV, Linneberg A. Specific IgE positivity against inhalant allergens and development of autoimmune disease. Autoimmunity. 2015;48(5):282–8.
Rabin RL, Levinson AI. The nexus between atopic disease and autoimmunity: a review of the epidemiological and mechanistic literature. Clin Exp Immunol. 2008;153(1):19–30.
Eyerich S, Onken AT, Weidinger S, Franke A, Nasorri F, Pennino D, et al. Mutual antagonism of T cells causing psoriasis and atopic eczema. N Engl J Med. 2011;365(3):231–8.
Kawamoto N, Yamada A, Ohkouchi S, Maeda T, Tanaka S, Hashimoto T, et al. IgG reactive to CTL-directed epitopes of self-antigens is either lacking or unbalanced in atopic dermatitis patients. Tissue Antigens. 2003;61(5):352–61.
Watanabe K, Muro Y, Sugiura K, Tomita Y. IgE and IgG4 autoantibodies against DFS70/LEDGF in atopic dermatitis. Autoimmunity. 2011;44(6):511–9.
Szakos E, Lakos G, Aleksza M, Gyimesi E, Páll G, Fodor B, et al. Association between the occurrence of the anticardiolipin IgM and mite allergen-specific IgE antibodies in children with extrinsic type of atopic eczema/dermatitis syndrome. Allergy. 2004;59(2):164–7.
Kortekangas-Savolainen O, Peltonen S, Pummi K, Kalimo K, Savolainen J. IgE-binding components of cultured human keratinocytes in atopic eczema/dermatitis syndrome and their crossreactivity with Malassezia furfur. Allergy. 2004;59(2):168–73.
Schmid-Grendelmeier P, Flückiger S, Disch R, Trautmann A, Wüthrich B, Blaser K, et al. IgE-mediated and T cell–mediated autoimmunity against manganese superoxide dismutase in atopic dermatitis. J Allergy Clin Immunol. 2005;115(5):1068–75.
Altrichter S, Kriehuber E, Moser J, Valenta R, Kopp T, Stingl G. Serum IgE autoantibodies target keratinocytes in patients with atopic dermatitis. J Investig Dermatol. 2008;128(9):2232–9.
Zeller S, Rhyner C, Meyer N, Schmid-Grendelmeier P, Akdis CA, Crameri R. Exploring the repertoire of IgE-binding self-antigens associated with atopic eczema. J Allergy Clin Immunol. 2009;124(2):278–85.
Aichberger KJ, Mittermann I, Reininger R, Seiberler S, Swoboda I, Spitzauer S, et al. Hom s 4, an IgE-reactive autoantigen belonging to a new subfamily of calcium-binding proteins, can induce Th cell type 1-mediated autoreactivity. J Immunol. 2005;175(2):1286–94.
Mittermann I, Reininger R, Zimmermann M, Gangl K, Reisinger J, Aichberger KJ, et al. The IgE-reactive autoantigen Hom s 2 induces damage of respiratory epithelial cells and keratinocytes via induction of IFN-γ. J Investig Dermatol. 2008;128(6):1451–9.
Brunner PM, Guttman-Yassky E. Racial differences in atopic dermatitis. Ann Allergy Asthma Immunol. 2019;122(5):449–55.
Ress K, Metsküla K, Annus T, Putnik U, Lepik K, Luts K, et al. Antinuclear antibodies in atopic dermatitis: a cross-sectional study on 346 children. Int J Dermatol. 2015;54(1):24–8.
Schneider L, Hanifin J, Boguniewicz M, Eichenfield LF, Spergel JM, Dakovic R, et al. Study of the atopic march: development of atopic comorbidities. Pediatr Dermatol. 2016;33(4):388–98.
Maurer M, Altrichter S, Schmetzer O, Scheffel J, Church MK, Metz M. Immunoglobulin E-mediated autoimmunity. Front Immunol. 2018;9:689.
Iwata Y, Komura K, Kodera M, Usuda T, Yokoyama Y, Hara T, et al. Correlation of IgE autoantibody to BP180 With a severe form of bullous pemphigoid. Arch Dermatol. 2008;144(1):41–8.
Dema B, Pellefigues C, Hasni S, Gault N, Jiang C, Ricks TK, et al. Autoreactive IgE is prevalent in systemic lupus erythematosus and is associated with increased disease activity and nephritis. PLoS ONE. 2014;9(2):e90424.
Sanjuan MA, Sagar D, Kolbeck R. Role of IgE in autoimmunity. J Allergy Clin Immunol. 2016;137(6):1651–61.
Scott DL, Wolfe F, Huizinga TW. Rheumatoid arthritis. Lancet. 2010;376(9746):1094–108.
Rantapää-Dahlqvist S, de Jong BAW, Berglin E, Hallmans G, Wadell G, Stenlund H, et al. Antibodies against cyclic citrullinated peptide and IgA rheumatoid factor predict the development of rheumatoid arthritis. Arthritis Rheumat. 2003;48(10):2741–9.
Nielen MMJ, van Schaardenburg D, Reesink HW, van de Stadt RJ, van der Horst-Bruinsma IE, de Koning MHMT, et al. Specific autoantibodies precede the symptoms of rheumatoid arthritis: a study of serial measurements in blood donors. Arthritis Rheumat. 2004;50(2):380–6.
Shi J, van de Stadt LA, Levarht EWN, Huizinga TWJ, Hamann D, van Schaardenburg D, et al. Anti-carbamylated protein (anti-CarP) antibodies precede the onset of rheumatoid arthritis. Ann Rheumat Dis. 2014;73(4):780–3.
van de Stadt LA, de Koning MHMT, van de Stadt RJ, Wolbink G, Dijkmans BAC, Hamann D, et al. Development of the anti-citrullinated protein antibody repertoire prior to the onset of rheumatoid arthritis. Arthritis Rheumat. 2011;63(11):3226–33.
Seiberler S, Bugajska-Schretter A, Hufnagl P, Binder BR, Stöckl J, Spitzauer S, et al. Characterization of IgE-reactive autoantigens in atopic dermatitis 1. Subcellular distribution and tissue-specific expression. IAA. 1999;120(2):108–16.
Valenta R, Seiberler S, Natter S, Mahler V, Mossabeb R, Ring J, et al. Autoallergy: a pathogenetic factor in atopic dermatitis? J Allergy Clin Immunol. 2000;105(3):432–7.
Kinaciyan T, Natter S, Kraft D, Stingl G, Valenta R. IgE autoantibodies monitored in a patient with atopic dermatitis under cyclosporin A treatment reflect tissue damage. J Allergy Clin Immunol. 2002;109(4):717–9.
Hide M, Tanaka T, Yamamura Y, Koro O, Yamamoto S. IgE-mediated hypersensitivity against human sweat antigen in patients with atopic dermatitis. Acta Derm Venereol. 2002;82(5):335–40.
Tanaka A, Tanaka T, Suzuki H, Ishii K, Kameyoshi Y, Hide M. Semi-purification of the immunoglobulin E-sweat antigen acting on mast cells and basophils in atopic dermatitis. Exp Dermatol. 2006;15(4):283–90.
Holmes J, Fairclough LC, Todd I. Atopic dermatitis and autoimmunity: the occurrence of autoantibodies and their association with disease severity. Arch Dermatol Res. 2019;311(3):141–62.
Ochs RL, Muro Y, Si Y, Ge H, Chan EK, Tan EM. Autoantibodies to DFS 70 kd/transcription coactivator p75 in atopic dermatitis and other conditions. J Allergy Clin Immunol. 2000;105(6 Pt 1):1211–20.
Richards DM, Kyewski B, Feuerer M. Re-examining the nature and function of self-reactive T cells. Trends Immunol. 2016;37(2):114–25.
Ribatti D. Sir Frank Macfarlane Burnet and the clonal selection theory of antibody formation. Clin Exp Med. 2009;9(4):253.
Mueller DL. Mechanisms maintaining peripheral tolerance. Nat Immunol. 2010;11(1):21–7.
Boehncke W-H, Brembilla NC. Autoreactive T-lymphocytes in inflammatory skin diseases. Front Immunol. 2019;10:1198.
Lleo A, Invernizzi P, Gao B, Podda M, Gershwin ME. Definition of human autoimmunity—autoantibodies versus autoimmune disease. Autoimmun Rev. 2010;9(5):A259–66.
Pacheco Y, Acosta-Ampudia Y, Monsalve DM, Chang C, Gershwin ME, Anaya J-M. Bystander activation and autoimmunity. J Autoimmun. 2019;1(103):102301.
Vanderlugt CL, Miller SD. Epitope spreading in immune-mediated diseases: implications for immunotherapy. Nat Rev Immunol. 2002;2(2):85–95.
Rojas M, Restrepo-Jiménez P, Monsalve DM, Pacheco Y, Acosta-Ampudia Y, Ramírez-Santana C, et al. Molecular mimicry and autoimmunity. J Autoimmun. 2018;1(95):100–23.
Limacher A, Glaser AG, Meier C, Schmid-Grendelmeier P, Zeller S, Scapozza L, et al. Cross-reactivity and 1.4-Å crystal structure of Malassezia sympodialis thioredoxin (Mala s 13), a Member of a New Pan-Allergen Family. J Immunol. 2007;178(1):389–96.
Bünder R, Mittermann I, Herz U, Focke M, Wegmann M, Valenta R, et al. Induction of autoallergy with an environmental allergen mimicking a self protein in a murine model of experimental allergic asthma. J Allergy Clin Immunol. 2004;114(2):422–8.
Golebski K, Röschmann KIL, Toppila-Salmi S, Hammad H, Lambrecht BN, Renkonen R, et al. The multi-faceted role of allergen exposure to the local airway mucosa. Allergy. 2013;68(2):152–60.
Toppila-Salmi S, Renkonen J, Joenväärä S, Mattila P, Renkonen R. Allergen interactions with epithelium. Curr Opin Allergy Clin Immunol. 2011;11(1):29–32.
Kortekaas Krohn I, Seys SF, Lund G, Jonckheere A-C, de Dierckx Casterlé I, Ceuppens JL, et al. Nasal epithelial barrier dysfunction increases sensitization and mast cell degranulation in the absence of allergic inflammation. Allergy. 2019;75:1155–64.
Zhou X, Wei T, Cox CW, Jiang Y, Roche WR, Walls AF. Mast cell chymase impairs bronchial epithelium integrity by degrading cell junction molecules of epithelial cells. Allergy. 2019;74(7):1266–76.
Steelant B, Seys SF, Van Gerven L, Van Woensel M, Farré R, Wawrzyniak P, et al. Histamine and T helper cytokine-driven epithelial barrier dysfunction in allergic rhinitis. J Allergy Clin Immunol. 2018;141(3):951–63.
Akdis M, Akdis CA, Weigl L, Disch R, Blaser K. Skin-homing, CLA+ memory T cells are activated in atopic dermatitis and regulate IgE by an IL-13-dominated cytokine pattern: IgG4 counter-regulation by CLA-memory T cells. J Immunol. 1997;159(9):4611–9.
Werfel T, Morita A, Grewe M, Renz H, Wahn U, Krutmann J, et al. Allergen specificity of skin-infiltrating t cells is not restricted to a type-2 cytokine pattern in chronic skin lesions of atopic dermatitis. J Investig Dermatol. 1996;107(6):871–6.
Hradetzky S, Roesner LM, Balaji H, Heratizadeh A, Mittermann I, Valenta R, et al. Cytokine effects induced by the human autoallergen α-NAC. J Invest Dermatol. 2014;134(6):1570–8.
Bilsborough J, Leung DYM, Maurer M, Howell M, Boguniewcz M, Yao L, et al. IL-31 is associated with cutaneous lymphocyte antigen–positive skin homing T cells in patients with atopic dermatitis. J Allergy Clin Immunol. 2006;117(2):418–25.
Cevikbas F, Wang X, Akiyama T, Kempkes C, Savinko T, Antal A, et al. A sensory neuron-expressed IL-31 receptor mediates T helper cell-dependent itch: involvement of TRPV1 and TRPA1. J Allergy Clin Immunol. 2014;133(2):448–60.
Belloni B, Andres C, Ollert M, Ring J, Mempel M. Novel immunological approaches in the treatment of atopic eczema. Curr Opin Allergy Clin Immunol. 2008;8(5):423–7.
Holm JG, Agner T, Sand C, Thomsen SF. Omalizumab for atopic dermatitis: case series and a systematic review of the literature. Int J Dermatol. 2017;56(1):18–26.
Fairley JA, Baum CL, Brandt DS, Messingham KAN. Pathogenicity of IgE in autoimmunity: successful treatment of bullous pemphigoid with omalizumab. J Allergy Clin Immunol. 2009;123(3):704–5.
Gericke J, Metz M, Ohanyan T, Weller K, Altrichter S, Skov PS, et al. Serum autoreactivity predicts time to response to omalizumab therapy in chronic spontaneous urticaria. J Allergy Clin Immunol. 2017;139(3):1059–61.
Eming R, Nagel A, Wolff-Franke S, Podstawa E, Debus D, Hertl M. Rituximab exerts a dual effect in pemphigus vulgaris. J Invest Dermatol. 2008;128(12):2850–8.
Nagel A, Hertl M, Eming R. B-cell-directed therapy for inflammatory skin diseases. J Invest Dermatol. 2009;129(2):289–301.
Sfikakis PP, Souliotis VL, Fragiadaki KG, Moutsopoulos HM, Boletis JN, Theofilopoulos AN. Increased expression of the FoxP3 functional marker of regulatory T cells following B cell depletion with rituximab in patients with lupus nephritis. Clin Immunol. 2007;123(1):66–73.
Simon D, Hösli S, Kostylina G, Yawalkar N, Simon H-U. Anti-CD20 (rituximab) treatment improves atopic eczema. J Allergy Clin Immunol. 2008;121(1):122–8.
Sánchez-Ramón S, Eguíluz-Gracia I, Rodríguez-Mazariego ME, Paravisini A, Zubeldia-Ortuño JM, Gil-Herrera J, et al. Sequential combined therapy with omalizumab and rituximab: a new approach to severe atopic dermatitis. J Investig Allergol Clin Immunol. 2013;23(3):190–6.
Šedivá A, Kayserová J, Vernerová E, Poloučková A, Čapková Š, Špíšek R, et al. Anti-CD20 (rituximab) treatment for atopic eczema. J Allergy Clin Immunol. 2008;121(6):1515–6.
Menzella F, Lusuardi M, Galeone C, Taddei S, Zucchi L. Profile of anti-IL-5 mAb mepolizumab in the treatment of severe refractory asthma and hypereosinophilic diseases. J Asthma Allergy. 2015;8:105–14.
Straumann A, Conus S, Grzonka P, Kita H, Kephart G, Bussmann C, et al. Anti-interleukin-5 antibody treatment (mepolizumab) in active eosinophilic oesophagitis: a randomised, placebo-controlled, double-blind trial. Gut. 2010;59(1):21–30.
Oldhoff JM, Darsow U, Werfel T, Katzer K, Wulf A, Laifaoui J, et al. Anti-IL-5 recombinant humanized monoclonal antibody (mepolizumab) for the treatment of atopic dermatitis. Allergy. 2005;60(5):693–6.
Lucae S, Schmid-Grendelmeier P, Wüthrich B, Kraft D, Valenta R, Linhart B. IgE responses to exogenous and endogenous allergens in atopic dermatitis patients under long-term systemic cyclosporine A treatment. Allergy. 2016;71(1):115–8.
Cotter DG, Schairer D, Eichenfield L. Emerging therapies for atopic dermatitis: JAK inhibitors. J Am Acad Dermatol. 2018;78(3 Suppl 1):S53–62.
Clarysse K, Pfaff CM, Marquardt Y, Huth L, Kortekaas Krohn I, Kluwig D, et al. JAK1/3 inhibition preserves epidermal morphology in full-thickness 3D skin models of atopic dermatitis and psoriasis. J Eur Acad Dermatol Venereol. 2019;33(2):367–75.
Vu M, Heyes C, Robertson SJ, Varigos GA, Ross G. Oral tofacitinib: a promising treatment in atopic dermatitis, alopecia areata and vitiligo. Clin Exp Dermatol. 2017;42(8):942–4.
Nakagawa H, Nemoto O, Igarashi A, Nagata T. Efficacy and safety of topical JTE-052, a Janus kinase inhibitor, in Japanese adult patients with moderate-to-severe atopic dermatitis: a phase II, multicentre randomized, vehicle-controlled clinical study. Br J Dermatol. 2018;178(2):e169.
Bissonnette R, Papp KA, Poulin Y, Gooderham M, Raman M, Mallbris L, et al. Topical tofacitinib for atopic dermatitis: a phase IIa randomized trial. Br J Dermatol. 2016;175(5):902–11.
Fukuyama T, Ehling S, Cook E, Bäumer W. Topically administered janus-kinase inhibitors tofacitinib and oclacitinib display impressive antipruritic and anti-inflammatory responses in a model of allergic dermatitis. J Pharmacol Exp Ther. 2015;354(3):394–405.
Levy LL, Urban J, King BA. Treatment of recalcitrant atopic dermatitis with the oral Janus kinase inhibitor tofacitinib citrate. J Am Acad Dermatol. 2015;73(3):395–9.
O’Shea JJ, Laurence A, McInnes IB. Back to the future: oral targeted therapy for RA and other autoimmune diseases. Nat Rev Rheumatol. 2013;9(3):173–82.
Meyer DM, Jesson MI, Li X, Elrick MM, Funckes-Shippy CL, Warner JD, et al. Anti-inflammatory activity and neutrophil reductions mediated by the JAK1/JAK3 inhibitor, CP-690,550, in rat adjuvant-induced arthritis. J Inflamm. 2010;11(7):41.
Simpson EL, Bieber T, Guttman-Yassky E, Beck LA, Blauvelt A, Cork MJ, et al. Two phase 3 trials of dupilumab versus placebo in atopic dermatitis. N Engl J Med. 2016;375(24):2335–48.
Blauvelt A, de Bruin-Weller M, Gooderham M, Cather JC, Weisman J, Pariser D, et al. Long-term management of moderate-to-severe atopic dermatitis with dupilumab and concomitant topical corticosteroids (LIBERTY AD CHRONOS): a 1-year, randomised, double-blinded, placebo-controlled, phase 3 trial. Lancet. 2017;389(10086):2287–303.
de Bruin-Weller M, Thaçi D, Smith CH, Reich K, Cork MJ, Radin A, et al. Dupilumab with concomitant topical corticosteroid treatment in adults with atopic dermatitis with an inadequate response or intolerance to ciclosporin A or when this treatment is medically inadvisable: a placebo-controlled, randomized phase III clinical trial (LIBERTY AD CAFÉ). Br J Dermatol. 2018;178(5):1083–101.
Ariëns LFM, van der Schaft J, Bakker DS, Balak D, Romeijn MLE, Kouwenhoven T, et al. Dupilumab is very effective in a large cohort of difficult-to-treat adult atopic dermatitis patients: first clinical and biomarker results from the BioDay registry. Allergy. 2019.
Thijs JL, Strickland I, Bruijnzeel-Koomen CAFM, Nierkens S, Giovannone B, Csomor E, et al. Moving toward endotypes in atopic dermatitis: identification of patient clusters based on serum biomarker analysis. J Allergy Clin Immunol. 2017;140(3):730–7.
Roesner LM, Heratizadeh A, Wieschowski S, Mittermann I, Valenta R, Eiz-Vesper B, et al. Alpha-NAC-specific autoreactive CD8+ T cells in atopic dermatitis are of an effector memory type and secrete IL-4 and IFN-γ. J Immunol. 2016;196(8):3245–52.
Glatz M, Buchner M, von Bartenwerffer W, Schmid-Grendelmeier P, Worm M, Hedderich J, et al. Malassezia spp.-specific immunoglobulin E level is a marker for severity of atopic dermatitis in adults. Acta Derm Venereol. 2015;95(2):191–6.
Mossabeb R, Seiberler S, Mittermann I, Reininger, Spitzauer S, Natter S, et al. Characterization of a novel isoform of alpha-nascent polypeptide-associated complex as IgE-defined autoantigen. J Invest Dermatol. 2002;119(4):820–9.
Guarneri F, Costa C, Foti C, Hansel K, Guarneri C, Guarneri B, et al. Frequency of autoallergy to manganese superoxide dismutase in patients with atopic dermatitis: experience of three Italian dermatology centres. Br J Dermatol. 2015;173(2):559–62.
Vilhelmsson M, Glaser AG, Martinez DB, Schmidt M, Johansson C, Rhyner C, et al. Mutational analysis of amino acid residues involved in IgE-binding to the Malassezia sympodialis allergen Mala s 11. Mol Immunol. 2008;46(2):294–303.
Andersson A, Rasool O, Schmidt M, Kodzius R, Flückiger S, Zargari A, et al. Cloning, expression and characterization of two new IgE-binding proteins from the yeast Malassezia sympodialis with sequence similarities to heat shock proteins and manganese superoxide dismutase. Eur J Biochem. 2004;271(10):1885–94.
Czech W, Stadler BM, Schôpf E, Kapp A. IgE autoantibodies in atopic dermatitis-occurrence of different antibodies against the CH3 and the CH4 epitopes of IgE. Allergy. 1995;50(3):243–8.
Mayer C, Appenzeller U, Seelbach H, Achatz G, Oberkofler H, Breitenbach M, et al. Humoral and cell-mediated autoimmune reactions to human acidic ribosomal P2 protein in individuals sensitized to Aspergillus fumigatus P2 protein. J Exp Med. 1999;189(9):1507–12.
Balaji H, Heratizadeh A, Wichmann K, Niebuhr M, Crameri R, Scheynius A, et al. Malassezia sympodialis thioredoxin-specific T cells are highly cross-reactive to human thioredoxin in atopic dermatitis. J Allergy Clin Immunol. 2011;128(1):92–9.
Heratizadeh A, Mittermann I, Balaji H, Wichmann K, Niebuhr M, Valenta R, et al. The role of T-cell reactivity towards the autoantigen α-NAC in atopic dermatitis. Br J Dermatol. 2011;164(2):316–24.
Kapitein B, Aalberse JA, Klein MR, de Jager W, Hoekstra MO, Knol EF, et al. Recognition of self-heat shock protein 60 by T cells from patients with atopic dermatitis. Cell Sterss Chaperones. 2013;18(1):87–95.
Lind SM, Kuylenstierna C, Moll M, Jordö DE, Winqvist O, Lundeberg L, et al. IL-18 skews the invariant NKT-cell population via autoreactive activation in atopic eczema. Eur J Immunol. 2009;39(8):2293–301.
Acknowledgements
Not applicable.
Funding
IKK is holder of the Fonds Wetenschappelijk Onderzoek (FWO) Post-Doctoral mandate (12W2219N), and holder of a Sanofi Genzyme Type 2 Innovation Grant (0000000122).
Author information
Authors and Affiliations
Contributions
FB: discussed the design of the manuscript, screening of systematic search, reading of included articles, data extraction, tables, discussed the design of the manuscript, reviewed the manuscript. SDV: screening of systematic search, reading of included articles, data extraction, tables, reviewed the manuscript. KC: screening of systematic search, reviewed the manuscript. JR: discussed the design of the manuscript, tables and figures, reviewed the manuscript. CBSW: reviewed the manuscript. JG: reviewed the manuscript. IKK: discussed the design of the manuscript, third reviewer, reading of included articles, figures, tables, wrote the first draft of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1.
Systematic search strategy. Table S2. Systematic search on IgE autoantibodies in patients with atopic dermatitis. AD: atopic dermatitis, RC: rhino conjunctivitis, NA: nonatopic, HC: healthy controls without allergic symptoms, PS: psoriasis, CA: contact allergy, UA: urticaria. Table S3. Autoreactive T cells in patients with atopic dermatitis. AD: atopic dermatitis, RC: rhinoconjunctivitis, HC: healthy controls, NA: non-atopic, CD: chronic dermatoses, PS: psoriasis, ns: non-sensitized, ABPA: allergic bronchopulmonary aspergillosis. * median.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Badloe, F.M.S., De Vriese, S., Coolens, K. et al. IgE autoantibodies and autoreactive T cells and their role in children and adults with atopic dermatitis. Clin Transl Allergy 10, 34 (2020). https://doi.org/10.1186/s13601-020-00338-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13601-020-00338-7