
Abstract
N6-methyladenosine is a prevalent and abundant transcriptome modification, and its methylation regulates the various aspects of RNAs, including transcription, translation, processing and metabolism. The methylation of N6-methyladenosine is highly associated with numerous cellular processes, which plays important roles in the development of physiological process and diseases. The high prevalence of metabolic diseases poses a serious threat to human health, but its pathological mechanisms remain poorly understood. Recent studies have reported that the progression of metabolic diseases is closely related to the expression of RNA N6-methyladenosine modification. In this review, we aim to summarize the biological and clinical significance of RNA N6-methyladenosine modification in metabolic diseases, including obesity, type 2 diabetes, non-alcoholic fatty liver disease, hypertension, cardiovascular diseases, osteoporosis and immune-related metabolic diseases.
Similar content being viewed by others
Introduction
A total of 160 RNA modifications have been reported to participate in life activities and diseases progress, especially methylation [1]. In eukaryotic mRNA, there are several identified methylation modifications, such as N(7)-methylguanosine, N(6)-methyl-2′-O-methyladenosine, 2′-O-methylation, N(6)-methyladenosine (m6A) and 5-methylcytosine (m5C) [2]. Among them, m6A has been considered as the most abundant internal modification, since it was discovered from methylated nucleosides in mRNA of Novikoff hepatoma cells in the early 1970s [3]. m6A is enriched in stop codon and 3′ untranslated terminal region (UTR) and translates near 5′ UTR in a cap-independent manner [4,5,6], thereby regulating RNA transcription, translation, processing and metabolism [5, 6]. The process of m6A modification is reversible and can be regulated by three homologous factors jargonized as ‘writers’, ‘erasers’ and ‘readers’ [7, 8]. For example, ‘Writers’ are categorized as the components of that catalyze the formation of m6A methylation [7, 9]; ‘Erasers’ play an important role in m6A modification for their demethylated functions [10, 11]; ‘Readers’ are a group of molecules which can decode m6A methylation and generate functional signals [12, 13]. So far, m6A has been found not only in mRNAs, but also in a variety of non-coding RNAs including rRNA, tRNA, snRNA, miRNA, and lncRNA [14, 15]. For example, m6A methyltransferase-like 3 (METTL3) interacts with the microprocessor protein DGCR8 and modulates miR-873-5p mature process positively [16]. The expression of m6A demethylase fat mass and obesity-associated protein (FTO) can influence the steady state level of various miRNAs, including increased expression of hsa-miR-6505-5p, hsa-miR-651-5p and hsa-miR-493-5p, and decreased expression of hsa-miR-7-5p, hsa-miR-92a-1-5p and hsa-miR-6769a-3p [15]. In addition, m6A modification of lncRNAs can induce the proliferation, metastasis and apoptosis of cancer cells [17]. For example, alkB homolog 5 (ALKBH5) inhibits pancreatic cancer motility by demethylating lncRNA KCNK15-AS1 [18], and METTL16 can methylate diverse cellular RNAs in human embryonic kidney 293 cells, consisting of 8 pre-mRNAs, 355mRNAs, 68 lncRNAs and other type of RNAs [19].
m6A RNA modification is a widespread and reversible process, which is highly associated with multiple diseases such as metabolic diseases (MDs), infertility, virus infection and cancers [20,21,22,23]. In this review, we aim to summarize the biological features and therapeutic potentials of m6A modifications in MDs.
Metabolic diseases
MDs refer to the pathological results of metabolic disorders of proteins, fats, carbohydrates and other substances in the human body [24], including obesity, type 2 diabetes (T2D), non-alcoholic fatty liver disease (NAFLD), hypertension, osteoporosis, chronic kidney disease, cardiovascular disease and other related metabolic disorders [25]. Currently, there are over 1.9 billion adults and 340 million children and adolescents with overweight or obese [26], more than 415 million people with diabetes [27], and 6–35% (median 20%) of population with NAFLD [28] around the world. In the past decades, the various treatments were used to prevent and treat the aforementioned MDs but they are still limited [29]. For diabetes, the long term treatment is insufficient for controlling blood glucose by daily medicines take like metformin or subcutaneous injection of insulin, as blood glucose is easy fluctuated by the intake of food and physical activity [30]. For the treatment of NAFLD, although lifestyle modification, vitamin E, and clinical surgery as main methods are commonly used, there is no effective medicine to prevent the pathological development of it [31]. Recently, m6A RNA modification has been found to be involved in the development of MDs [32,33,34] (Table 1). Therefore, m6A modification might be potential targets for the therapy and early diagnosis of MDs.
m6A writers, erasers, readers
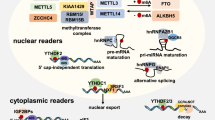
The regulators in m6A modification are categorized as ‘writers’ and ‘erasers’ (methylation and de-methylation, respectively) and ‘readers’ (recognition) [35,36,37] which were presented in Fig. 1. The m6A methylation begins to be installed by a large multiprotein writer complex, which includes the core METTL3 and METTL14 methyltransferase subunits and many other associated regulatory subunits [38]. METTL3 is a significant catalytic component [38, 39], and METTL14 as a homolog of METTL3 shares 43% identity with METTL3, which can help their RNA substrates recognize each other [39, 40]. These two proteins can form a stable heterodimer core METTL3–METTL14 complex that acts on the cellular m6A deposition of nuclear RNAs and increases the methyltransferase activities in mammals [38]. Meanwhile, Wilms’ tumor 1-associated protein (WTAP), Virilizer like m6A methyltransferase associated protein (VIRMA/KIAA1429), an E3 ubiquitin ligase for the E-cadherin complex (HAKAI), and zinc finger CCCH-type containing 13 (ZC3H13/KIAA0853) are adaptor proteins which may guide the METTL3–METTL14 heterodimer to its target mRNAs. Besides, RNA-binding protein 15 (RBM15) and RBM15B may participate in determining which sites can be methylated [9, 41,42,43,44,45,46,47,48,49,50,51].

The dynamic and reversible processes of m6A methylation and its biological functions. m6A RNA modification is a widespread and reversible process which is catalyzed by “writers”, consisting of METTL3, METTL14, WTAP, HAKAI, ZC3H13/KIAA0853, VIRMA/KIAA1429, RBM15B and RBM15. Meanwhile, the m6A methylation can be removed by m6A “erasers”, including FTO and ALKBH5. Besides, it is recognized by “readers”-YTHDF1, YTHDF2, YTHDF3, YTHDC1, YTHDC2, IGF2BP1, IGF2BP2, IGF2BP3, FMRP and PRRC2A. The biological functions of m6A methylation on stability, translation, splicing or nuclear export are highly involved in m6A methylation associated diseases
The demethylated process of m6A ‘erasers’ are dominated by two members of the a-ketoglutarate-dependent dioxygenase protein family, including FTO and ALKBH5 [10, 11]. ALKBH5 and FTO as powerful m6A demethylases can effectively demethylate m6Am and m6A, but the demethylation capacity of FTO is stronger than ALKBH5 [37, 52]. FTO is a significant fat mass and obesity associated gene with a full length of 400 kp, including nine exons, which mainly locates in the 16q12-q24 of the human chromosome [53]. It is currently recognized as the most robust predictor of polygenic obesity [53, 54] as its capability of encoding for several important energy regulating proteins [55,56,57,58].
‘Readers’, YT521-B homology (YTH) family proteins, contain a YTH domain that can specifically recognize m6A methylation. YTHDF1, YTHDF2, YTHDF3, and YTHDC2 are predominantly located in the cytoplasm, while YTHDC1 is mainly found in the nucleus [12, 35, 59,60,61,62]. Among them, YTHDF1, YTHDC2 can recognize and bind to the methyl tag on the RNA and influence the translation of the target RNA [60]. YTHDF2 can alter the distribution of various m6A-containing mRNAs in the cytoplasm and affect the stability of the target RNA [60]. A newly identified m6A reader family including insulin like growth factor 2 mRNA binding protein 1 (IGF2BP1), IGF2BP2 and IGF2BP3 can regulate gene expression by enhancing the stability of its target RNA [63]. In addition, fragile X mental retardation protein (FMRP) has showed to promote nuclear export of methylated mRNA targets during neural differentiation by reading m6A [64]. Another novel m6A reader, proline rich coiled-coil 2A (PRRC2A), controls myelination and oligodendrocyte specification by stabilizing target mRNA [65].
m6A methylation and T2D
The global prevalence of diabetes in adults is about 8% and it may increase to 10% by 2040 [66]. More than 90% of diabetes is T2D, which is characterized by hyperglycemia and dyslipidemia. Recent released studies have suggested that the m6A modification may play a critical role in the regulation of T2D [32, 67, 68]. For example, m6A highly stimulates glucose oxidation in rat adipocytes, which indicates that the proper level of m6A may be required to maintain certain concentration of blood glucose [67]. Many studies demonstrate that the content of m6A is negatively associated with the risk of T2D, as a significant reduction of m6A contents can been found in T2D patients [32], while, the increased mRNA expression of demethylase FTO is responsible for the reduction of m6A content, which may induce the complications of T2D, including obesity, cardiovascular diseases [68]. Meanwhile, high glucose stimulation contributes to the increase of FTO expression [32], and then further promotes the mRNA expression of forkhead box O1 (FOXO1), glucose-6-phosphatase catalytic subunit (G6PC), and diacylglycerol O-acyltransferase 2 (DGAT2) to participate in glucose and lipid metabolism [32]. Intriguingly, the levels of m6A methyltransferases (METTL3, METTL14, WTAP) mRNA expression are also significantly elevated in patients with T2D, but the expression of METTL3, METTL14, and KIAA1429 are negatively correlated with m6A content [32]. In addition, METTL3 inhibits hepatic insulin sensitivity via N6-methylation of FASN (fatty acid synthetase) mRNA and promoting fatty acid metabolism, which eventually results in the development of T2D [69]. In addition, METTL14 is essential for β-survival, differentiation and insulin secretion, the deficiency of METTL14 in β-cells increases cell death, changes cell differentiation and decreases β-cell mass and insulin secretion, leading to glucose intolerance and T2D [70]. Furthermore, the increased expression of m6A methylation upregulates the insulin/insulin-like growth factor 1 (IGF1)–AKT-pancreatic and duodenal homeobox 1 (PDX1) pathway by targeting METTL14 or METTL3 in human β-cells, which ultimately inhibits cell-cycle arrest and protects insulin secretion [71]. Besides, single nucleotide polymorphisms (SNPs) in FTO are also strongly associated with T2D, such as variant rs9939609 and rs17817449 of FTO gene [72], which are important for the development of insulin resistance and occurrence of T2D [73]. Together, m6A modulators might be potential therapeutic targets for maintaining glucose metabolism and preserving β-cell survival and insulin functions in T2D.
m6A methylation and obesity
Obesity is an increasing risk for its related chronic diseases like NAFLD, cardiovascular diseases, diabetes and cancers [74, 75]. Obesity or adipogenesis is usually characterized by increased cell size (hypertrophy) and fat cell numbers (hyperplasia) at the cellular level. Studies have suggested that FTO-mediated m6A demethylation is closely related with the upregulated ghrelin production, adipogenesis, fat mass and body weight [33, 76,77,78,79]. People with a high body mass index may commonly carry FTO risk alleles [80,81,82] and there are some SNPs of FTO positively associated with obesity. For instance, FTO (rs17817449) is positively correlated with obesity and plasma insulin, insulin resistance, percentage body fat and fat mass in a north Indian population [83]. FTO (rs3751812) can promote obesity by altering fat deposition and disturbing serum lipid profile [84]. FTO (rs9939609 T/A) is related to increased FTO expression, reduced m6A ghrelin mRNA methylation, and finally results in increased energy intake and obesity by upregulating the ghrelin expression [85]. m6A demethylase FTO can promote adipogenesis by inhibiting the Wnt/β-catenin signaling pathway in porcine intramuscular pre-adipocytes [86]. The knockdown of FTO decreases the expression of ATG5 (autophagy-related 5) and ATG7, leading to attenuation of autophagosome formation, thereby inhibiting autophagy and adipogenesis. Meanwhile, YTHDF2 decreases protein expression of ATG5 and ATG7 by shortening the lifespan of their m6A-modified mRNAs [87]. Furthermore, the effect of FTO on adipogenesis also appears to be regulated via enhanced expression of the pro-adipogenic short isoform of Runt-related transcription factor 1 (RUNX1), which can promote adipocyte proliferation [77]. In the contrast, WTAP, METTL3, METTL14 are negatively related with adipogenesis by promoting cell cycle transition in mitotic clonal expansion [88, 89]. Moreover, m6A-YTHDF2-FTO signaling way might be crucial for the development of obesity, m6A—binding protein YTHDF2 can methylate mRNAs of cyclin A2 (CCNA2) and cyclin dependent kinase 2 (CDK2), and then reduce their protein expression to prolong cell cycle progression and suppress adipogenesis [90]. The methylation effect of FTO on CCNA2 and CDK2 can be reversed by epigallocatechin gallate induced YTHDF2 expression [91]. The expression of METTL3 increases via the depletion of ZFP217 (zinc finger protein 217), reversely, METTL3 knockdown rescues the siZFP217-inhibited mitotic clonal expansion and promotes CCND1 (cyclin D1). Meanwhile, YTHDF2 recognizes and degrades the methylated CCND1 mRNA, leading to the downregulation of CCND1. Consequently, cell cycle progression is blocked, and adipogenesis is inhibited [92]. Taken together, m6A modification may be a novel potential biomarker of obesity.
m6A methylation and NAFLD
NAFLD is the most common cause of chronic liver disease among children and adults all over the world [93,94,95], which is characterized by steatosis, ballooning degeneration, and fatty retention of liver parenchyma cells with no history of excessive alcohol intake or other known liver disease [96]. The pathological character of NAFLD is caused by metabolic dysregulation of de novo lipogenesis, fatty acid uptake, fatty acid oxidation, and triglycerides export [97, 98]. Previous studies have found that m6A alteration is highly related to the development of NAFLD [34, 99, 100]. The level of FTO is elevated in hepatic tissue at NAFLD patients with hyperglycemic and hyper-insulinemic [34], which can down-regulate mitochondrial content and up-regulate triglyceride (TG) deposition, while FTO (R316A) mutant lacking demethylation activity and could not regulate mitochondria and TG content. These indicate that FTO can affect mitochondrial content and fat metabolism by modulating m6A levels in hepatocytes [101]. In addition, the activation of phosphatidylinositol 3-kinase (PI3K)/AKT signaling pathway may improve the development of NAFLD by suppressing FTO mediated hepatocyte regeneration [102]. Enhanced FTO expression can increase expression of lipogenic genes, containing fatty acid synthase (FASN), stearoyl-CoA desaturase (SCD) and monoacylglycerol O-acyltransferase 1 (MOGAT1), and intracellular TG level in HepG2 cells [101], which finally promotes hepatic fat accumulation. Meanwhile, these effects can be effectively reversed by betaine (a methyl donor) [101, 103]. Increased FTO levels are also highly involved in hepatic oxidative stress and lipid deposition which participate in the process of NAFLD [99]. Currently, dietary curcumin can affect the expression of METTL3, METTL14, ALKBH5, FTO, and YTHDF2 mRNAs, and finally improve lipopolysaccharide-induced liver injury and hepatic lipid metabolism disruption by increasing m6A methylation level in the liver of piglets [104]. In addition, the knockdown of METTL3 or YTHDF2 can increase the lifetime and expression of peroxisome proliferator activated receptor alpha (PPARα) mRNA, resulting in a reduction of lipid accumulation [105]. In summary, m6A modulators have potentials in the therapeutic function of NAFLD.
m6A methylation in hypertension and cardiovascular diseases
Recent studies show that m6A modification is closely related to blood pressure (BP) and cardiovascular diseases [106]. For example, the m6A-SNP (Lys67Arg, rs197922) in golgi SNAP receptor complex member 2 gene is positively associated with hypertension in white individuals [107]. In addition, the m6A-SNPs (Arg389Gly, rs1801253; Ser49Gly, rs1801253) can develop hypertension as they can encode β1-adrenoreceptor, a hypertension-susceptibility candidate gene [108, 109]. rs9847953 and rs197922 have regulatory potentials to alter BP related gene expression, mRNA stability and homeostasis [110]. The m6A RNA modifications also involve in various mechanisms of cardiovascular diseases. For example, FTO overexpression in mouse models of myocardial infarction decreases fibrosis and enhanced angiogenesis [111]. In addition, cardiac growth is controlled by METTL3, which drives cardiomyocyte hypertrophy by catalyzing methylation of m6A on certain subsets of mRNAs. Whereas, diminished METTL3 promotes eccentric cardiomyocyte remodeling and dysfunction [112]. Moreover, METTL3 upregulation inhibits cellular autophagic flux and promotes apoptosis in hypoxia/reoxygenation-treated cardiomyocytes [113]. In summary, targeting m6A through its relative enzymes may be used as a potential diagnostic or a novel therapeutic strategy for hypertension and cardiovascular diseases in the future.
m6A methylation and osteoporosis
Osteoporosis is one of the most significant bone metabolic diseases, especially aged-related osteoporosis. The low bone mass and excessive accumulation of adipose tissue in bone marrow milieu can result in architectural deterioration of the skeleton, the decrease of bone strength and an increased risk of fragility fractures [114, 115]. Recent released studies has suggested that m6A modification and its regulatory enzymes such as FTO, METTL3 are the key factors for osteoporosis [116,117,118]. The deletion of METTL3 in porcine bone marrow stem cells could promote adipogenesis and adipogenic differentiation via janus kinase 1 (JAK1)/signal transducer and activator of transcription 5 (STAT5)/CCAAT/enhancer binding protein β (C/EBPβ) pathway [119]. Also, the deletion of METTL3 in bone marrow mesenchymal stem cells disrupts cell fate and promotes osteoporosis pathological phenotypes (decreasing bone mass with incompetent osteogenic potential and increasing marrow adiposity with enhanced adipogenic potential) by reducing m6A methylation level in mice via parathyroid hormone (PTH)/parathyroid hormone 1 receptor (PTH1R) signaling axis [118]. In addition, the abundance of FTO can promote the differentiation of adipocyte and osteoblast from bone marrow mesenchymal stem cells by growth differentiation factor 11 (GDF11) and peroxisome proliferator-activated receptor gamma (PPARγ) in a C/EBPα-dependent manner [116]. Interestingly, FTO expression in the bone is up-regulated during aging and osteoporosis, while the expression of METTL3 is not affected by age [116]. In the contrast, FTO in osteoblasts can enhance the stability of mRNAs which protect osteoblasts from genotoxic damage through Hspa1a–NF-κB signaling way [120]. Besides, bone mineral density-associated m6A-SNPs may also play significant roles in the pathology of osteoporosis, including m6A-SNP rs17787930. rs1110720 and rs11614913 [117]. All in all, the levels of m6A methylation or regulators are strongly associated with osteoporosis.
m6A methylation and immune-related MDs
The interactions between immune and metabolic responses play an important role in pathological development and chronic inflammation [121], including insulin resistance, insulin unresponsiveness, hepatic fat deposition and excessive adipose tissue development [122]. m6A methylation emerges as an significant role in immune-related MDs, for example, ALKBH5 is highly up-expressed in organs enriched in immune cells with frequent immune reactions, including thymus, spleen and thyroid [10, 123]. Also, METTL3-mediated m6A of CD40, CD80 and toll-like receptors 4 (TLR4) signaling adaptor TIR domain containing adaptor protein (TIRAP) transcripts enhance their translation in dendritic cells for stimulating T cell activation and the development of T lymphocytes in the thymus [124, 125]. Furthermore, the deletion of METTL3 in mouse T cells disrupts T cell homeostasis and differentiation by targeting the interleukin 7 (IL-7)/STAT5/cytokine inducible SH2 containing protein (SOCS) pathways [126]. In addition, m6A modification prevents TLRs activation upon binding of native mRNAs such as mRNAs with m5C, 5-methyluridine, 2-thiouridine substrate, m6A, which cannot active TLR3, TLR7 or TLR8, while unmodified RNA could activate all these human TLRs [127]. Thus, the study of m6A methylation on immune response may provide a new insight for the treatment of immune-related MDs, and more related mechanisms need to be clarified.
Conclusions and perspectives
m6A modification is highly involved in RNA stability, localization, turnover and translation efficiency, which is crucial for the biological functions [128]. The mRNA m6A methylation has a wide range of effects on MDs. The researches can be conducted by many experimental methods such as m6A-seq (m6A-specific methylated RNA immunoprecipitation with next-generation sequencing), PA-m6A-seq (photo-crosslinking-assisted m6A-sequencing), and LC–MS/MS (liquid chromatography linked to tandem mass spectrometry) [4, 129, 130]. Apart from the expensive experimental screening of m6A sites in RNAs, some bioinformatics tools have been developed for large-scale identification of m6A modification sites, including SCARLET (site-specific cleavage and radioactive-labeling followed by ligation-assisted extraction and thin-layer chromatography), TargetM6A, RNA-methylPred, iRNA-Methyl and pRNAm-PC [131,132,133,134,135]. This m6A related regulatory system will promote targeted therapy for MDs.
Strategies for m6A-targeted drugs design are on the following: Firstly, virtual screening can be used to discover the potential compounds for experimental validation by using the drug-like SPECS database which contains about 100,000 compounds [136]; Secondly, the mechanistic study and kinetics analysis can be used to select the best m6A inhibitor or methyl donor [136]; In addition, differential scanning fluorometry- and liquid chromatography-based assays are applied to screen related compounds [55]; Furthermore, we can also synthetize m6A related compounds by utilizing a modular approach [137].
Currently, several promising agents may have potentials to treat MDs by targeting m6A, such as m6A inhibitors. It is known that FTO negatively regulated m6A levels and positively regulated adipogenesis, thus we can use FTO inhibitors (rhein, radicicol, epigallocatechin gallate, entacapone and meclofenamic acid) [91, 136, 138,139,140] to remove the potential effect of FTO. In addition, ALKBH5 is positively related to the frequent immune reactions [123], if we rule out the effects of ALKBH5 on immune cells via using ALKBH5 inhibitor (IOX3) [141], the immune-related MDs will be improved. Also, cycloleucine (a methylation inhibitor), S-adenosylhomocysteine (a competitive inhibitor for some adenosylmethionine-dependent methyltransferases) can be applied to downregulate m6A methylation directly [88, 101, 142]. In the contrast, many m6A regulators are useful for the improvement of MDs, for instance, METTL3, METTL14, YTHDF2 are negatively correlated with adipogenesis [87, 89]. Therefore, betaine, a methyl donor [88, 101], could be employed to upregulate m6A methylation directly. All in all, it’s still a long journey for the special m6A-targeted drugs for MDs, but the development and application of more m6A inhibitors or methyl donors will provide important clues to the development of m6A special drugs for MDs.
So far, the studies on mRNA m6A methylation remain poorly understood. For example, almost all the known demethylases belong to the AlkB family, and whether other proteins in or out the AlkB family are also involved in mRNA demethylation needs to be further studied. Variations in methylated and demethylated genes need to be further explored. The functions of m6A modification on non-coding RNAs, such as miRNA, circRNA, piRNA and lncRNA need to be unveiled in the metabolic processing. Accordingly, m6A—as one of the abundant basic modifications of circRNAs, lncRNA and miRNA [143,144,145], may have a promising future in early diagnosis on MDs through identifying downregulated or upregulated m6A methylation levels or mediators levels. The RNA m6A methyltransferases and demethylases can selectively methylate or demethylate the MDs-related genes [146, 147]. The immune cell responses play an important role in the pathological development of MDs, however, the roles of m6A modifications in immune-related MDs are poorly understood. Based on the functions of m6A modifications in immune responses, thus we speculated that m6A modifications in immune-related MDs might be important.
There are many problems in the m6A dominated diagnosis and therapies of MDs. Firstly, the biological functions of m6A modification in MDs needs to be further clarified. Secondly, the functions of m6A modification on risk factors of MDs such as aging, infection and cancers are still a tip of the iceberg. Finally, the m6A related treatment of MDs merely focus on FTO inhibitors, so the novel therapeutics targeting m6A related potents and specific small-molecule m6A modification inhibitors need to be further identified or developed through small-molecule compound library screening or chemical synthesis.
Availability of data and materials
All data reviewed and described is either included in this manuscript or available online in the relevant publications.
Abbreviations
- m6A:
-
N(6)-Methyladenosine
- m5C:
-
5-Methylcytosine
- UTR:
-
Untranslated terminal region
- METTL3:
-
m6A methyltransferase-like 3
- FTO:
-
Fat mass and obesity-associated protein
- ALKBH5:
-
alkB homolog 5
- MDs:
-
Metabolic diseases
- T2D:
-
Type 2 diabetes
- NAFLD:
-
Non-alcoholic fatty liver disease
- WTAP:
-
Wilms’ tumor 1-associated protein
- VIRMA/KIAA1429:
-
Virilizer like m6A methyltransferase associated protein
- HAKAI:
-
An E3 ubiquitin ligase for the E-cadherin complex
- ZC3H13/KIAA0853:
-
Zinc finger CCCH-type containing 13
- RBM15:
-
RNA-binding protein 15
- YTH:
-
YT521-B homology
- IGF2BP1:
-
Insulin like growth factor 2 mRNA binding protein 1
- FMRP:
-
Fragile X mental retardation protein
- PRRC2A:
-
Proline rich coiled-coil 2A
- FOXO1:
-
Forkhead box O1
- G6PC:
-
Glucose-6-phosphatase catalytic subunit
- DGAT2:
-
Diacylglycerol O-acyltransferase 2
- FASN:
-
Fatty acid synthetase
- IGF1:
-
Insulin-like growth factor 1
- PDX1:
-
Pancreatic and duodenal homeobox 1
- SNPs:
-
Single nucleotide polymorphisms
- ATG5:
-
Autophagy-related 5
- RUNX1:
-
Runt-related transcription factor 1
- CCNA2:
-
Cyclin A2
- CDK2:
-
Cyclin dependent kinase 2
- ZFP217:
-
Zinc finger protein 217
- CCND1:
-
Cyclin D1
- TG:
-
Triglyceride
- PI3K:
-
Phosphatidylinositol 3-kinase
- FASN:
-
Fatty acid synthase
- SCD:
-
Stearoyl-CoA desaturase
- MOGAT1:
-
Monoacylglycerol O-acyltransferase 1
- PPARA:
-
Peroxisome proliferator activated receptor alpha
- BP:
-
Blood pressure
- JAK1:
-
Janus kinase 1
- STAT5:
-
Signal transducer and activator of transcription 5
- C/EBPβ:
-
CCAAT/enhancer binding protein β
- PTH:
-
Parathyroid hormone
- PTH1R:
-
Parathyroid hormone 1 receptor
- GDF11:
-
Growth differentiation factor 11
- PPARγ:
-
Peroxisome proliferator-activated receptor gamma
- TLR4:
-
Toll-like receptors 4
- TIRAP:
-
TIR domain containing adaptor protein
- IL-7:
-
Interleukin 7
- SOCS:
-
Cytokine inducible SH2 containing protein
- m6A-seq:
-
m6A-specific methylated RNA immunoprecipitation with next-generation sequencing
- PA-m6A-seq:
-
Photo-crosslinking-assisted m6A-sequencing
- LC–MS/MS:
-
Liquid chromatography linked to tandem mass spectrometry
- SCARLET:
-
Site-specific cleavage and radioactive-labeling followed by ligation-assisted extraction and thin-layer chromatography
References
Boccaletto P, Machnicka MA, Purta E, et al. MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic Acids Res. 2018;46(D1):D303–7.
Liu J, Jia G. Methylation modifications in eukaryotic messenger RNA. J Genet Genomics Yi chuan xue bao. 2014;41(1):21–33.
Desrosiers R, Friderici K, Rottman F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc Natl Acad Sci USA. 1974;71(10):3971–5.
Dominissini D, Moshitch-Moshkovitz S, Schwartz S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485(7397):201–6.
Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell. 2012;149(7):1635–46.
Meyer KD, Patil DP, Zhou J, et al. 5′ UTR m(6)A promotes cap-independent translation. Cell. 2015;163(4):999–1010.
Li A, Chen YS, Ping XL, et al. Cytoplasmic m(6)A reader YTHDF3 promotes mRNA translation. Cell Res. 2017;27(3):444–7.
Chen XY, Zhang J, Zhu JS. The role of m(6)A RNA methylation in human cancer. Mol Cancer. 2019;18(1):103.
Schwartz S, Mumbach MR, Jovanovic M, et al. Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5′ sites. Cell Rep. 2014;8(1):284–96.
Zheng G, Dahl JA, Niu Y, et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. 2013;49(1):18–29.
Jia G, Fu Y, Zhao X, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7(12):885–7.
Wang X, Zhao BS, Roundtree IA, et al. N(6)-methyladenosine modulates messenger rna translation efficiency. Cell. 2015;161(6):1388–99.
Haussmann IU, Bodi Z, Sanchez-Moran E, et al. m(6)A potentiates Sxl alternative pre-mRNA splicing for robust Drosophila sex determination. Nature. 2016;540(7632):301–4.
Pan T. N6-methyl-adenosine modification in messenger and long non-coding RNA. Trends Biochem Sci. 2013;38(4):204–9.
Berulava T, Rahmann S, Rademacher K, Klein-Hitpass L, Horsthemke B. N6-adenosine methylation in MiRNAs. PLoS ONE. 2015;10(2):e0118438.
Wang J, Ishfaq M, Xu L, Xia C, Chen C, Li J. METTL3/m(6)A/miRNA-873-5p attenuated oxidative stress and apoptosis in colistin-induced kidney injury by modulating Keap1/Nrf2 pathway. Front Pharmacol. 2019;10:517.
Jacob R, Zander S, Gutschner T. The dark side of the epitranscriptome: chemical modifications in long non-coding RNAs. Int J Mol Sci. 2017;18(11):2387.
He Y, Hu H, Wang Y, et al. ALKBH5 inhibits pancreatic cancer motility by decreasing long non-coding RNA KCNK15-AS1 methylation. Cell Physiol Biochem. 2018;48(2):838–46.
Warda AS, Kretschmer J, Hackert P, et al. Human METTL16 is a N(6)-methyladenosine (m(6)A) methyltransferase that targets pre-mRNAs and various non-coding RNAs. EMBO Rep. 2017;18(11):2004–14.
Wei W, Ji X, Guo X, Ji S. Regulatory role of N(6) -methyladenosine (m(6) A) methylation in RNA processing and human diseases. J Cell Biochem. 2017;118(9):2534–43.
Lichinchi G, Gao S, Saletore Y, et al. Dynamics of the human and viral m(6)A RNA methylomes during HIV-1 infection of T cells. Nat Microbiol. 2016;1:16011.
Lin S, Choe J, Du P, Triboulet R, Gregory RI. The m(6)A methyltransferase METTL3 promotes translation in human cancer cells. Mol Cell. 2016;62(3):335–45.
Yang Y, Huang W, Huang JT, et al. Increased N6-methyladenosine in human sperm RNA as a risk factor for asthenozoospermia. Sci Rep. 2016;6:24345.
Shi S, Kong N, Feng C, et al. Drug delivery strategies for the treatment of metabolic diseases. Adv Healthc Mater. 2019;8:e1801655.
Hotamisligil GS, Erbay E. Nutrient sensing and inflammation in metabolic diseases. Nat Rev Immunol. 2008;8(12):923–34.
WHO. WHO obesity and overweight: key facts; 2018. https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight. Accessed 16 Feb 2018.
Ogurtsova K, da Rocha Fernandes JD, Huang Y, et al. IDF Diabetes Atlas: global estimates for the prevalence of diabetes for 2015 and 2040. Diabetes Res Clin Pract. 2017;128:40–50.
Bellentani S. The epidemiology of non-alcoholic fatty liver disease. Liver Int. 2017;37(Suppl 1):81–4.
Dohrmann CE. Target discovery in metabolic disease. Drug Discov Today. 2004;9(18):785–94.
Polimeni G, Trifiro G, Ingrasciotta Y, Caputi AP. The advent of biosimilars for the treatment of diabetes: current status and future directions. Acta Diabetol. 2015;52(3):423–31.
Neuschwander-Tetri BA. Non-alcoholic fatty liver disease. BMC Med. 2017;15(1):45.
Yang Y, Shen F, Huang W, et al. Glucose is involved in the dynamic regulation of m6A in patients with type 2 diabetes. J Clin Endocrinol Metab. 2019;104(3):665–73.
Dina C, Meyre D, Gallina S, et al. Variation in FTO contributes to childhood obesity and severe adult obesity. Nat Genet. 2007;39(6):724–6.
Guo J, Ren W, Li X, et al. Altering of FTO in the serum and livers of NAFLD patients: a correlation analysis. Int J Clin Exp Med. 2018;11:6046–53.
Shi H, Wang X, Lu Z, et al. YTHDF3 facilitates translation and decay of N(6)-methyladenosine-modified RNA. Cell Res. 2017;27(3):315–28.
Wang X, Feng J, Xue Y, et al. Corrigendum: structural basis of N(6)-adenosine methylation by the METTL3–METTL14 complex. Nature. 2017;542(7640):260.
Wei J, Liu F, Lu Z, et al. Differential m(6)A, m(6)Am, and m(1)A demethylation mediated by FTO in the cell nucleus and cytoplasm. Mol Cell. 2018;71(6):973–985.e975.
Liu J, Yue Y, Han D, et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. 2014;10(2):93–5.
Wang X, Feng J, Xue Y, et al. Structural basis of N(6)-adenosine methylation by the METTL3–METTL14 complex. Nature. 2016;534(7608):575–8.
Sledz P, Jinek M. Structural insights into the molecular mechanism of the m(6)A writer complex. eLife. 2016;5:e18434.
Knuckles P, Lence T, Haussmann IU, et al. Zc3h13/Flacc is required for adenosine methylation by bridging the mRNA-binding factor Rbm15/Spenito to the m(6)A machinery component Wtap/Fl(2)d. Genes Dev. 2018;32(5–6):415–29.
Wen J, Lv R, Ma H, et al. Zc3h13 regulates nuclear RNA m(6)A methylation and mouse embryonic stem cell self-renewal. Mol Cell. 2018;69(6):1028–1038.e1026.
Yue Y, Liu J, Cui X, et al. VIRMA mediates preferential m(6)A mRNA methylation in 3′UTR and near stop codon and associates with alternative polyadenylation. Cell Discov. 2018;4:10.
Geula S, Moshitch-Moshkovitz S, Dominissini D, et al. Stem cells m6A mRNA methylation facilitates resolution of naive pluripotency toward differentiation. Science (New York, NY). 2015;347(6225):1002–6.
Roundtree IA, Evans ME, Pan T, He C. Dynamic RNA modifications in gene expression regulation. Cell. 2017;169(7):1187–200.
Patil DP, Chen CK, Pickering BF, et al. m(6)A RNA methylation promotes XIST-mediated transcriptional repression. Nature. 2016;537(7620):369–73.
Meyer KD, Jaffrey SR. Rethinking m(6)A readers, writers, and erasers. Annu Rev Cell Dev Biol. 2017;33:319–42.
Ma H, Wang X, Cai J, et al. N(6-)Methyladenosine methyltransferase ZCCHC4 mediates ribosomal RNA methylation. Nat Chem Biol. 2019;15(1):88–94.
Lence T, Akhtar J, Bayer M, et al. m(6)A modulates neuronal functions and sex determination in Drosophila. Nature. 2016;540(7632):242–7.
Ping XL, Sun BF, Wang L, et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014;24(2):177–89.
He C. Grand challenge commentary: RNA epigenetics? Nat Chem Biol. 2010;6(12):863–5.
Mauer J, Luo X, Blanjoie A, et al. Reversible methylation of m(6)Am in the 5′ cap controls mRNA stability. Nature. 2017;541(7637):371–5.
Sanchez-Pulido L, Andrade-Navarro MA. The FTO (fat mass and obesity associated) gene codes for a novel member of the non-heme dioxygenase superfamily. BMC Biochem. 2007;8:23.
Hinney A, Nguyen TT, Scherag A, et al. Genome wide association (GWA) study for early onset extreme obesity supports the role of fat mass and obesity associated gene (FTO) variants. PLoS ONE. 2007;2(12):e1361.
Aik W, Demetriades M, Hamdan MK, et al. Structural basis for inhibition of the fat mass and obesity associated protein (FTO). J Med Chem. 2013;56(9):3680–8.
Caruso V, Bahari H, Morris MJ. The beneficial effects of early short-term exercise in the offspring of obese mothers are accompanied by alterations in the hypothalamic gene expression of appetite regulators and FTO (fat mass and obesity associated) gene. J Neuroendocrinol. 2013;25(8):742–52.
Gao X, Shin YH, Li M, Wang F, Tong Q, Zhang P. The fat mass and obesity associated gene FTO functions in the brain to regulate postnatal growth in mice. PLoS ONE. 2010;5(11):e14005.
Guo Y, Liu H, Yang TL, et al. The fat mass and obesity associated gene, FTO, is also associated with osteoporosis phenotypes. PLoS ONE. 2011;6(11):e27312.
Roundtree IA, Luo GZ, Zhang Z, et al. YTHDC1 mediates nuclear export of N(6)-methyladenosine methylated mRNAs. Life. 2017;6:e31311.
Wang X, Lu Z, Gomez A, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505(7481):117–20.
Jain D, Puno MR, Meydan C, et al. ketu mutant mice uncover an essential meiotic function for the ancient RNA helicase YTHDC2. eLife. 2018;7:e30919.
Hsu PJ, Zhu Y, Ma H, et al. Ythdc2 is an N(6)-methyladenosine binding protein that regulates mammalian spermatogenesis. Cell Res. 2017;27(9):1115–27.
Huang H, Weng H, Sun W, et al. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol. 2018;20(3):285–95.
Edens BM, Vissers C, Su J, et al. FMRP modulates neural differentiation through m(6)A-dependent mRNA nuclear export. Cell Rep. 2019;28(4):845–854.e845.
Wu R, Li A, Sun B, et al. A novel m(6)A reader Prrc2a controls oligodendroglial specification and myelination. Cell Res. 2019;29(1):23–41.
Lascar N, Brown J, Pattison H, Barnett AH, Bailey CJ, Bellary S. Type 2 diabetes in adolescents and young adults. Lancet Diabetes Endocrinol. 2018;6(1):69–80.
Souness JE, Stouffer JE, Chagoya de Sanchez V. Effect of N6-methyladenosine on fat-cell glucose metabolism. Evidence for two modes of action. Biochem Pharmacol. 1982;31(24):3961–71.
Shen F, Huang W, Huang JT, et al. Decreased N(6)-methyladenosine in peripheral blood RNA from diabetic patients is associated with FTO expression rather than ALKBH5. J Clin Endocrinol Metab. 2015;100(1):E148–54.
Xie W, Ma LL, Xu YQ, Wang BH, Li SM. METTL3 inhibits hepatic insulin sensitivity via N6-methyladenosine modification of Fasn mRNA and promoting fatty acid metabolism. Biochem Biophys Res Commun. 2019;518(1):120–6.
Liu J, Luo G, Sun J, et al. METTL14 is essential for beta-cell survival and insulin secretion. Biochim Biophys Acta. 2019;1865(9):2138–48.
De Jesus DF, Zhang Z, Kahraman S, et al. m6A mRNA methylation regulates human β-cell biology in physiological states and in type 2 diabetes. Nat Metab. 2019;1:765–74.
Sabarneh A, Ereqat S, Cauchi S, et al. Common FTO rs9939609 variant and risk of type 2 diabetes in Palestine. BMC Med Genet. 2018;19(1):156.
Younus LA, Algenabi AHA, Abdul-Zhara MS, Hussein MK. FTO gene polymorphisms (rs9939609 and rs17817449) as predictors of type 2 diabetes mellitus in obese Iraqi population. Gene. 2017;627:79–84.
Wang YC, McPherson K, Marsh T, Gortmaker SL, Brown M. Health and economic burden of the projected obesity trends in the USA and the UK. Lancet (London, England). 2011;378(9793):815–25.
Hinnouho GM, Czernichow S, Dugravot A, et al. Metabolically healthy obesity and the risk of cardiovascular disease and type 2 diabetes: the Whitehall II cohort study. Eur Heart J. 2015;36(9):551–9.
Fischer J, Koch L, Emmerling C, et al. Inactivation of the Fto gene protects from obesity. Nature. 2009;458(7240):894–8.
Merkestein M, Laber S, McMurray F, et al. FTO influences adipogenesis by regulating mitotic clonal expansion. Nat Commun. 2015;6:6792.
Zhao X, Yang Y, Sun BF, et al. FTO-dependent demethylation of N6-methyladenosine regulates mRNA splicing and is required for adipogenesis. Cell Res. 2014;24(12):1403–19.
Hess ME, Bruning JC. The fat mass and obesity-associated (FTO) gene: obesity and beyond? Biochem Biophys Acta. 2014;1842(10):2039–47.
Haupt A, Thamer C, Staiger H, et al. Variation in the FTO gene influences food intake but not energy expenditure. Exp Clin Endocrinol Diabetes. 2009;117(4):194–7.
Cecil JE, Tavendale R, Watt P, Hetherington MM, Palmer CN. An obesity-associated FTO gene variant and increased energy intake in children. N Engl J Med. 2008;359(24):2558–66.
Ronkainen J, Mondini E, Cinti F, et al. Fto-deficiency affects the gene and MicroRNA expression involved in brown adipogenesis and browning of white adipose tissue in mice. Int J Mol Sci. 2016;17(11):1851.
Prakash J, Srivastava N, Awasthi S, et al. Association of FTO rs17817449 SNP with obesity and associated physiological parameters in a north Indian population. Ann Hum Biol. 2011;38(6):760–3.
Qureshi SA, Mumtaz A, Shahid SU, Shabana NA. rs3751812, a common variant in fat mass and obesity-associated (FTO) gene, is associated with serum high- and low-density lipoprotein cholesterol in Pakistani individuals. Nutrition (Burbank, Los Angeles County, Calif). 2017;39–40:92–5.
Karra E, O’Daly OG, Choudhury AI, et al. A link between FTO, ghrelin, and impaired brain food-cue responsivity. J Clin Investig. 2013;123(8):3539–51.
Chen X, Luo Y, Jia G, Liu G, Zhao H, Huang Z. FTO promotes adipogenesis through inhibition of the Wnt/beta-catenin signaling pathway in porcine intramuscular preadipocytes. Anim Biotechnol. 2017;28(4):268–74.
Wang X, Wu R, Liu Y, et al. m(6)A mRNA methylation controls autophagy and adipogenesis by targeting Atg5 and Atg7. Autophagy. 2019; 1–15. https://doi.org/10.1080/15548627.2019.1659617.
Wang X, Zhu L, Chen J, Wang Y. mRNA m(6)A methylation downregulates adipogenesis in porcine adipocytes. Biochem Biophys Res Commun. 2015;459(2):201–7.
Kobayashi M, Ohsugi M, Sasako T, et al. The RNA methyltransferase complex of WTAP, METTL3, and METTL14 regulates mitotic clonal expansion in adipogenesis. Mol Cel Biol. 2018;38(16):e00116-18.
Wu R, Liu Y, Yao Y, et al. FTO regulates adipogenesis by controlling cell cycle progression via m(6)A-YTHDF2 dependent mechanism. Biochim Biophys Acta. 2018;1863(10):1323–30.
Wu R, Yao Y, Jiang Q, et al. Epigallocatechin gallate targets FTO and inhibits adipogenesis in an mRNA m(6)A-YTHDF2-dependent manner. Int J Obes (2005). 2018;42(7):1378–88.
Liu Q, Zhao Y, Wu R, et al. ZFP217 regulates adipogenesis by controlling mitotic clonal expansion in a METTL3-m(6)A dependent manner. RNA Biol. 2019;16:1–9.
Anderson EL, Howe LD, Fraser A, et al. Weight trajectories through infancy and childhood and risk of non-alcoholic fatty liver disease in adolescence: the ALSPAC study. J Hepatol. 2014;61(3):626–32.
Lawlor DA, Callaway M, Macdonald-Wallis C, et al. Nonalcoholic fatty liver disease, liver fibrosis, and cardiometabolic risk factors in adolescence: a cross-sectional study of 1874 general population adolescents. J Clin Endocrinol Metab. 2014;99(3):E410–7.
Swiderska-Syn M, Suzuki A, Guy CD, et al. Hedgehog pathway and pediatric nonalcoholic fatty liver disease. Hepatology (Baltimore, MD). 2013;57(5):1814–25.
Kleiner DE, Brunt EM, Van Natta M, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology (Baltimore, MD). 2005;41(6):1313–21.
Cobbina E, Akhlaghi F. Non-alcoholic fatty liver disease (NAFLD)—pathogenesis, classification, and effect on drug metabolizing enzymes and transporters. Drug Metab Rev. 2017;49(2):197–211.
Liu W, Cao H, Yan J, Huang R, Ying H. ‘Micro-managers’ of hepatic lipid metabolism and NAFLD. Wiley Interdiscip Rev RNA. 2015;6(5):581–93.
Guo J, Ren W, Li A, et al. Fat mass and obesity-associated gene enhances oxidative stress and lipogenesis in nonalcoholic fatty liver disease. Dig Dis Sci. 2013;58(4):1004–9.
Luo Z, Zhang Z, Tai L, Zhang L, Sun Z, Zhou L. Comprehensive analysis of differences of N(6)-methyladenosine RNA methylomes between high-fat-fed and normal mouse livers. Epigenomics. 2019;11(11):1267–82.
Kang H, Zhang Z, Yu L, Li Y, Liang M, Zhou L. FTO reduces mitochondria and promotes hepatic fat accumulation through RNA demethylation. J Cell Biochem. 2018;119(7):5676–85.
Li S, Wang X, Zhang J, et al. Exenatide ameliorates hepatic steatosis and attenuates fat mass and FTO gene expression through PI3K signaling pathway in nonalcoholic fatty liver disease. Braz J Med Biol Res Revista brasileira de pesquisas medicas e biologicas. 2018;51(8):e7299.
Zhang L, Qi Y, Aluo Z, Liu S, Zhang Z, Zhou L. Betaine increases mitochondrial content and improves hepatic lipid metabolism. Food Funct. 2019;10(1):216–23.
Lu N, Li X, Yu J, et al. Curcumin attenuates lipopolysaccharide-induced hepatic lipid metabolism disorder by modification of m(6)A RNA methylation in piglets. Lipids. 2018;53(1):53–63.
Zhong X, Yu J, Frazier K, et al. Circadian clock regulation of hepatic lipid metabolism by modulation of m(6)A mRNA methylation. Cell Rep. 2018;25(7):1816–1828.e1814.
Paramasivam A, Vijayashree Priyadharsini J, Raghunandhakumar S. N6-adenosine methylation (m6A): a promising new molecular target in hypertension and cardiovascular diseases. Hypertens Res. 2019;43:153–4.
Meyer TE, Shiffman D, Morrison AC, et al. GOSR2 Lys67Arg is associated with hypertension in whites. Am J Hypertens. 2009;22(2):163–8.
Kong H, Li X, Zhang S, Guo S, Niu W. The beta1-adrenoreceptor gene Arg389Gly and Ser49Gly polymorphisms and hypertension: a meta-analysis. Mol Biol Rep. 2013;40(6):4047–53.
Wang H, Liu J, Liu K, et al. beta1-adrenoceptor gene Arg389Gly polymorphism and essential hypertension risk in general population: a meta-analysis. Mol Biol Rep. 2013;40(6):4055–63.
Mo XB, Lei SF, Zhang YH, Zhang H. Examination of the associations between m(6)A-associated single-nucleotide polymorphisms and blood pressure. Hypertens Res. 2019;42:1582–9.
Mathiyalagan P, Adamiak M, Mayourian J, et al. FTO-dependent N(6)-methyladenosine regulates cardiac function during remodeling and repair. Circulation. 2019;139(4):518–32.
Dorn LE, Lasman L, Chen J, et al. The N(6)-methyladenosine mRNA methylase METTL3 controls cardiac homeostasis and hypertrophy. Circulation. 2019;139(4):533–45.
Song H, Feng X, Zhang H, et al. METTL3 and ALKBH5 oppositely regulate m(6)A modification of TFEB mRNA, which dictates the fate of hypoxia/reoxygenation-treated cardiomyocytes. Autophagy. 2019;15(8):1419–37.
Canalis E, Giustina A, Bilezikian JP. Mechanisms of anabolic therapies for osteoporosis. N Engl J Med. 2007;357(9):905–16.
Devlin MJ, Rosen CJ. The bone-fat interface: basic and clinical implications of marrow adiposity. Lancet Diabetes Endocrinol. 2015;3(2):141–7.
Shen GS, Zhou HB, Zhang H, et al. The GDF11-FTO-PPARgamma axis controls the shift of osteoporotic MSC fate to adipocyte and inhibits bone formation during osteoporosis. Biochim Biophys Acta. 2018;1864(12):3644–54.
Mo XB, Zhang YH, Lei SF. Genome-wide identification of m(6)A-associated SNPs as potential functional variants for bone mineral density. Osteoporos Int. 2018;29(9):2029–39.
Wu Y, Xie L, Wang M, et al. Mettl3-mediated m(6)A RNA methylation regulates the fate of bone marrow mesenchymal stem cells and osteoporosis. Nat Commun. 2018;9(1):4772.
Yao Y, Bi Z, Wu R, et al. METTL3 inhibits BMSC adipogenic differentiation by targeting the JAK1/STAT5/C/EBPbeta pathway via an m(6)A-YTHDF2-dependent manner. FASEB J. 2019;33(6):7529–44.
Zhang Q, Riddle RC, Yang Q, et al. The RNA demethylase FTO is required for maintenance of bone mass and functions to protect osteoblasts from genotoxic damage. Proc Natl Acad Sci USA. 2019;116(36):17980–9.
Hotamisligil GS. Foundations of immunometabolism and implications for metabolic health and disease. Immunity. 2017;47(3):406–20.
Burcelin R, Garidou L, Pomie C. Immuno-microbiota cross and talk: the new paradigm of metabolic diseases. Semin Immunol. 2012;24(1):67–74.
Tsujikawa K, Koike K, Kitae K, et al. Expression and sub-cellular localization of human ABH family molecules. J Cell Mol Med. 2007;11(5):1105–16.
Wang H, Hu X, Huang M, et al. Mettl3-mediated mRNA m(6)A methylation promotes dendritic cell activation. Nat Commun. 2019;10(1):1898.
Huang Y, Qiu AW, Peng YP, Liu Y, Huang HW, Qiu YH. Roles of dopamine receptor subtypes in mediating modulation of T lymphocyte function. Neuro Endocrinol Lett. 2010;31(6):782–91.
Li HB, Tong J, Zhu S, et al. m(6)A mRNA methylation controls T cell homeostasis by targeting the IL-7/STAT5/SOCS pathways. Nature. 2017;548(7667):338–42.
Kariko K, Buckstein M, Ni H, Weissman D. Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity. 2005;23(2):165–75.
Scholler E, Weichmann F, Treiber T, et al. Interactions, localization, and phosphorylation of the m(6)A generating METTL3-METTL14-WTAP complex. RNA (New York, NY). 2018;24(4):499–512.
Chen K, Lu Z, Wang X, et al. High-resolution N(6) -methyladenosine (m(6) A) map using photo-crosslinking-assisted m(6) A sequencing. Angew Chem Int Ed Engl. 2015;54(5):1587–90.
Berton T, Mayhoub F, Chardon K, et al. Development of an analytical strategy based on LC-MS/MS for the measurement of different classes of pesticides and theirs metabolites in meconium: application and characterisation of foetal exposure in France. Environ Res. 2014;132:311–20.
Zhou Y, Zeng P, Li YH, Zhang Z, Cui Q. SRAMP: prediction of mammalian N6-methyladenosine (m6A) sites based on sequence-derived features. Nucleic Acids Res. 2016;44(10):e91.
Li GQ, Liu Z, Shen HB, Yu DJ. Target M6A: identifying N(6)-methyladenosine sites from RNA sequences via position-specific nucleotide propensities and a support vector machine. IEEE Trans Nanobiosci. 2016;15(7):674–82.
Jia CZ, Zhang JJ, Gu WZ. RNA-MethylPred: a high-accuracy predictor to identify N6-methyladenosine in RNA. Anal Biochem. 2016;510:72–5.
Chen W, Feng P, Ding H, Lin H, Chou KC. iRNA-Methyl: identifying N(6)-methyladenosine sites using pseudo nucleotide composition. Anal Biochem. 2015;490:26–33.
Liu Z, Xiao X, Yu DJ, Jia J, Qiu WR, Chou KC. pRNAm-PC: predicting N(6)-methyladenosine sites in RNA sequences via physical-chemical properties. Anal Biochem. 2016;497:60–7.
Chen B, Ye F, Yu L, et al. Development of cell-active N6-methyladenosine RNA demethylase FTO inhibitor. J Am Chem Soc. 2012;134(43):17963–71.
Zheng G, Cox T, Tribbey L, et al. Synthesis of a FTO inhibitor with anticonvulsant activity. ACS Chem Neurosci. 2014;5(8):658–65.
Wang R, Han Z, Liu B, et al. Identification of natural compound radicicol as a potent FTO inhibitor. Mol Pharm. 2018;15(9):4092–8.
Peng S, Xiao W, Ju D, et al. Identification of entacapone as a chemical inhibitor of FTO mediating metabolic regulation through FOXO1. Sci Transl Med. 2019;11(488):eaau7116.
Huang Y, Yan J, Li Q, et al. Meclofenamic acid selectively inhibits FTO demethylation of m6A over ALKBH5. Nucleic Acids Res. 2015;43(1):373–84.
Aik W, Scotti JS, Choi H, et al. Structure of human RNA N(6)-methyladenine demethylase ALKBH5 provides insights into its mechanisms of nucleic acid recognition and demethylation. Nucleic Acids Res. 2014;42(7):4741–54.
Kloor D, Osswald H. S-Adenosylhomocysteine hydrolase as a target for intracellular adenosine action. Trends Pharmacol Sci. 2004;25(6):294–7.
Zhou C, Molinie B, Daneshvar K, et al. Genome-wide maps of m6A circRNAs identify widespread and cell-type-specific methylation patterns that are distinct from mRNAs. Cell Rep. 2017;20(9):2262–76.
Chhabra R. miRNA and methylation: a multifaceted liaison. Chembiochem. 2015;16(2):195–203.
Patil DP, Pickering BF, Jaffrey SR. Reading m(6)A in the transcriptome: m(6)A-binding proteins. Trends Cell Biol. 2018;28(2):113–27.
Berulava T, Buchholz E, Elerdashvili V, et al. Changes in m6A RNA methylation contribute to heart failure progression by modulating translation. Eur J Heart Fail. 2019;22:54–66.
Zhou J, Wan J, Shu XE, et al. N(6)-methyladenosine guides mRNA alternative translation during integrated stress response. Mol Cell. 2018;69(4):636–647.e637.
Acknowledgements
This work was financially supported by the Sichuan Science and Technology Department (Grant Number: 2016Jy0156).
Funding
This work was supported by Sichuan Science and Technology Department (Grant Number: 2016Jy0156).
Author information
Authors and Affiliations
Contributions
YL, KX and HZ contributed to drafting and editing the manuscript. WW, MS and CH contributed to analyzing the data and editing the table and figure. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not involved into ethics.
Consent for publication
All authors have contributed to this study and approve its submission.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, Y., Wang, J., Huang, C. et al. RNA N6-methyladenosine: a promising molecular target in metabolic diseases. Cell Biosci 10, 19 (2020). https://doi.org/10.1186/s13578-020-00385-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13578-020-00385-4