
Abstract
The gut microbiota of mammals is a complex ecosystem, which is essential for maintaining gut homeostasis and the host’s health. The high throughput sequencing allowed us to gain a deeper insight into the bacterial structure and diversity. In order to improve the health status of the endangered golden takins, we first characterized the fecal microbiota of healthy golden takins using high throughput sequencing of the 16S rRNA genes V3–V4 hypervariable regions. Our results showed that, Firstly, the gut microbiota community comprised 21 phyla, 40 classes, 62 orders, 96 families, and 216 genera. Firmicutes (67.59%) was the most abundant phylum, followed by Bacteroidetes (23.57%) and Proteobacteria (2.37%). Secondly, the golden takin maintained higher richness in spring than in the winter while community diversity and evenness was not significantly different. Thirdly, four female golden takins demonstrated highly similar microbiota and the five golden takin males had relatively highly similar microbiota. All of our results might indicate that the fecal microbiota of golden takins were influenced by the season and the animal’s sex. The findings provided theoretical basis regarding the gut microbiota of golden takins and may offer new insights to protect this endangered species.
Similar content being viewed by others
Introduction
The takin (Budorcas taxicolor) is a large Himalayan ungulate in the family Bovidae, which is classified into four subspecies based on distinct differences in physical characteristics and geographic location, including the golden takin (B. t.bedfordi), the Mishmi takin (B. t.taxicolor), the Sichuan takin (B. t. Tibetana), and the Bhutan takin (B. t.whitei). The golden takin and the Sichuan takin are endemic subspecies of China. Furthermore, relative to the other subspecies, the golden takin is listed as vulnerable by the International Union for Conservation of Nature (IUCN 2008) and is under state protection (category I) in China. The golden takin is a type of social bovid herbivore. As a ruminant, this animal primarily forages on the browse of trees and shrubs (Schaller et al. 1986). In the spring and summer, golden takins undertake seasonal migration to the higher elevations of the uppermost tree line. During the autumn and winter, they move to lower elevations with smaller groups (Ming et al. 2003).
The gut microbiota offers substantial benefits to the host, such as aiding in digestion, promoting immune system development and competing for niches with pathogens (Cadwell 2014). To date, there have been many studies investigating the gut microbiota in herbivores (Bian et al. 2013; Costa et al. 2012; Liu et al. 2014). For instance, Firmicutes and Bacteroidetes, represented by the Ruminococcaceae, Lachnospiraceae, Rikenellaceae and Prevotellaceae families (Bian et al. 2013), dominate the microbiota in the healthy rhinoceros. Costa et al. (2012) described the predominance of Firmicutes (68%) in the feces of healthy horses, followed by Bacteroidetes (14%) and Proteobacteria (10%). The core microbiota of donkeys is dominated by Firmicutes (64% males and 64% females) and Bacteroidetes (23% males and 21% females), followed by Verrucomicrobia, Euryarchaeota, Spirochaetes and Proteobacteria (Liu et al. 2014). Considerable evidence supports the complexity of the gut microbiota in many mammals and the necessity of gut bacterial communities to maintain gut homeostasis. However, surprisingly, little research was on the gut microbiota of the golden takin. Therefore, in present research, we characterized the fecal microbiota of nine golden takins by performing high-throughput sequencing. On this basis, we also compared gut microbiota variation from winter to spring, which is the season of animals likely to get sick. Our result indicated that season was a factor that might impact the composition of the gut microbiota.
Materials and methods
Animal selection
Nine healthy golden takins were enrolled in this study (Table 1). Four fresh fecal samples (Group A) were collected at Jinan wild animal park during the late morning in December 2015. Five fresh fecal samples (Group B) were collected at Jinan Animal Park during the late morning in April 2016. Group A and group B respectively represented the winter and spring feces samples. All of the animals were about 8 years old and the two environments were roughly same. The experiment was approved by the Qufu Normal University Animal Care and Use Committee. None of the animals had received anti-inflammatory drugs or antimicrobials within the prior 4 months, and none had a gastrointestinal-related disease. Samples were collected off the ground immediately after defecation, and each sample was instantly transferred into plastic tubes. The samples were stored at 4 °C with long-term storage at −80 °C for further analysis.
DNA extraction, 16S rRNA gene PCR and sequencing
Total genomic DNA was isolated from fecal samples using a QIAamp DNA Stool Mini Kit (Qiagen, Germany) according to the manufacturer’s recommended protocol. We used a UV–Vis spectrophotometer (NanoDrop 2000c, USA) to determine DNA quantification and quality. DNA concentrations were measured to ensure final concentrations greater than 20 ng/μl for PCR amplification of the V3–V4 hypervariable regions of the 16SrRNA gene using the primers 341F (CCTAYGGGRBGCASCAG) and 806R (GGACTACNNGGGTATCTAAT), as previously described (Hugerth et al. 2014). Amplification was performed in a 30-μl volume, and the amplified fragment size was approximately 410 bp. Specifically, PCR reactions contained 15 μl of Phusion Master Mix (2er), 1.5 µl of each primer, 10 µl of microbial genomic DNA, and 2 µl ddH2O to constitute a final volume of 30 µl. The following PCR protocol was used: 1 min at 98 °C for initial denaturation, 35 cycles of 10 s at 98 °C for denaturation, 30 s at 55 °C for annealing, and 30 s at 72 °C for elongation, and a final extension at 72 °C for 5 min. PCR products were mixed with same volume of 1× loading buffer (containing SYBR green) and separated by electrophoresis on a 2% agarose gel. Samples containing a bright band between 400 and 450 bp were chosen and mixed in equidensity ratios. Then, this mixture of PCR products was purified with a Qiagen Gel Extraction Kit (Qiagen, Germany). Sequencing libraries were generated using a TruSeq® DNA PCR-Free Sample Preparation Kit (Illumina, USA) following the manufacturer’s recommendations, and index codes were added. Library quality was assessed with a Qubit@ 2.0 Fluorometer (Thermo Scientific) and an Agilent Bioanalyzer 2100 system. Finally, the DNA library was sequenced on an IlluminaHiSeq 2500 platform, and 250-bp paired-end reads were generated.
Bioinformatics analysis
Paired-end reads were assigned to samples based on their unique barcodes and truncated by cutting off the barcodes and primer sequences. Paired-end reads were then merged using FLASH (V1.2.7) (Magoč and Salzberg 2011). The raw tags were subjected to specific filtering conditions to obtain high-quality clean tags using the QIIME (V1.7.0) (Caporaso et al. 2010) quality control process. The tags were compared with the reference database (Gold database) using the UCHIME algorithm (UCHIME Algorithm) (Edgar et al. 2011) to detect chimeric sequences, which were then removed. Finally, effective tags were obtained. Sequence analysis was performed using Uparse software (Uparse v7.0.1001) (Edgar 2013). Sequences demonstrating greater than or equal to 97% similarity were assigned to the same operational taxonomic unit (OTU). For each representative sequence, the GreenGene Database (Desantis et al. 2006) was used and the RDP classifier algorithm (Version 2.2) (Wang et al. 2007) was employed to annotate taxonomic information. To study the phylogenetic relationships between different OTUs and identify dominant species in different samples (groups), multiple sequence alignment was carried out using MUSCLE software (Version 3.8.31) (Edgar 2004). Ultimately, we obtained 54,748 sequences for diversity analysis. Alpha diversity was evaluated by calculating six indices, specifically the Observed-species, Chao1, Shannon, Simpson, ACE and Good-coverage indices. All indices were calculated with QIIME (V1.7.0) and displayed with R software (Version 2.15.3). Beta diversity based on both Weighted and Unweighted Unifrac distances was calculated using QIIME software (Version 1.7.0). Cluster analysis was preceded by principal component analysis (PCA) using the FactoMineR package and ggplot2 package in R software (Version 2.15.3). The data set supporting the results of this article is available in the Sequencing Read Archive (SRA) database, accession numbers SRP090712.
Results
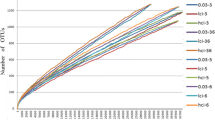
During quality control, a total of 670,356 high-quality sequences with an average length of 410 bp per sample were obtained from the nine fecal samples. The statistical estimates of species richness for the sequences from the nine samples that demonstrated a genetic distance of 3%, the total number of sequences, the coverage, and the number of OTUs were shown in Table 2. The rarefaction curves and rank abundance curves were generated using R software (Version 2.15.3). The rarefaction curves tended to approach the saturation plateau, while the rank abundance curves revealed a few dominant taxa and many low-abundance taxa comprising the bacterial community of the golden takin (Fig. 1). Based on ANOSIM analysis, significant differences were observed in bacterial community structure between group A and group B (P < 0.05). When analyzing differences between the two groups, the Alpha diversity index and box-plots intuitively reflected the median, degree of dispersion, maximum, minimum and outlier of species diversity within groups. The number of species in group A was lower than that in group B (Fig. 2), and this difference was significant (P < 0.05). However the difference of the community diversity and the species distribution uniformity between two groups were not significant (P > 0.05).

Rarefaction curve analysis of nine golden takins. Repeated samples of OTU subsets were performed to evaluate whether further sampling would likely yield additional taxa, as indicated by whether the curve reached a plateau value. The y-axis indicates the number of OTUs detected, and the x-axis indicates the number of taxa in the sequence subset analyzed. Rank abundance curves were used to estimate the richness and evenness of taxa present in the samples. The y-axis indicates the relative abundance of OTUs, and the x-axis indicates the number of OTUs according to the relative abundance from large to small. The larger the span curve on the x-axis, the higher the species richness. The smoother the curve on the y-axis, the more even the species distribution

A comparison of community alpha diversities between winter and spring samples in golden takins. Diversity was measured by calculating the Shannon index, and richness was determined by calculating the Observed-species index
Taxonomic composition
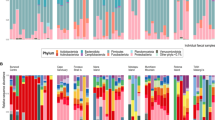
A total of 20 prokaryotic phyla were identified from the nine golden takins (Fig. 3a). The majority of their gut flora belonged to two phyla: Firmicutes (66.37–69.23%) and Bacteroidetes (21.26–26.55%). These two phyla represented more than 90% of the total sequences in all nine samples. However, Euryarchaeota belonged to the Archaeal domain, and the remainder of the identified bacteria belonged to the bacterial domain. Furthermore, variations were noted in the microbiota among samples, e.g. Synergistetes was only found in samples J1, J6, J7, and J8; Fusobacteria was only identified in samples J6 and J7; Gemmatimonadetes was only observed in sample J7; Deferribacteres were identified in sample J6; Chloroflexi was only present in J1, J5, and J8.

Gut bacterial composition at the phylum level per sample (a) and the relative abundance of the top ten biological species at the family level (b) and the genus level (c)
At the family level, we detected 96 families, and the abundance of unclassified bacteria in the samples was 17.04%. The distribution proportion was greater than 0.5% for 10 families (Fig. 3b). Among these 10 families, Ruminococcaceae was predominant, with an abundance of 49.61%, followed by Rikenellaceae (10.93%), Lachnospiraceae (6.41%), Christensenellaceae (3.68%), Clostridiales_vadinBB60_group (3.15%), Prevotellaceae (2.92%), Bacteroidaceae (2.64%), Bacteroidales_RF16_group (2.32%), Verrucomicrobiaceae (0.79%), and Bacillaceae (0.50%). The most abundant families were Ruminococcaceae, Rikenellaceae and Lachnospiraceae, which comprised approximately 66.95% of total sequences.
At the genus level, the percentage of unclassified bacteria averaged 42.52% (ranging from 36.7 to 50.27%), and the remainder were classified into 216 genera. The distribution proportion was greater than 0.5% in 10 genera (Fig. 3c). Among these 10 genera, Ruminococcaceae_UCG-010 was predominant, with an abundance of 21.44%, followed by Ruminococcaceae_UCG-005 (13.93%), Rikenellaceae_RC9_gut_group (7.30%), Christensenellaceae_R-7_group (3.56%), Alistipes (2.84%), Bacteroides (2.64%), [Eubacterium]_coprostanoligenes_group (2.36%), Ruminococcaceae_UCG-013 (2.11%), Akkermansia (0.79%), and Bacillus (0.50%). Of the 10 genera, Ruminococcaceae_UCG-010, Ruminococcaceae_UCG-005, Christensenellaceae_R-7_group, Ruminococcaceae_UCG-013, Bacillus, and [Eubacterium]_coprostanoligenes_group belong to Firmicutes. Rikenellaceae_RC9_gut_group, Alistipes and Bacteroides belong to Bacteroidetes, and Akkermansia belongs to Verrucomicrobia.
To know the bacterial community profiles at the genus level, we plotted a clustered heatmap. The result indicated that the sample J1, J2, J3 and J4 (group A) grouped together, while the sample J5, J6, J7 and J8 (group B) clustered together (Fig. 4). To compare the microbial communities across the different samples, we plotted another heatmap based on Weighted Unifrac and Unweighted Unifrac distances (Fig. 5). The similarity of the samples was consistent with the clustering results. In addition, the PCA score plot revealed the separation of group A from group B (Fig. 6), which was consistent with the heatmap results.

Double dendrogram showing the bacterial distribution among the fecal samples of nine golden takins. The abundances of the top 35 genera were sorted for the analysis. The heatmap plot depicts the relative percentage of each bacterial genus (variables clustering on the y-axis) within each sample (x-axis clustering). The relative values for the bacterial genus are depicted by color intensity in the legend indicated at the top right of the figure. Clusters based on the distance of the nine samples along the x-axis and the bacterial genera along the y-axis are indicated in the upper part and left of the figure, respectively. Different colors on the x-axis represent different groups, and different colors on the y-axis represent which phylum they belong to. The meaning of the different colors is indicated at the top right of the figure

Heatmap of Weighted Unifrac and Unweighted Unifrac distances. In the same square, a fluctuation in two values indicates the Weighted Unifrac and Unweighted Unifrac distances. The number in the diagram pane is the discrepancy coefficient between any two samples. The smaller the discrepancy coefficient between two samples, the smaller the difference in species diversity

PCA of bacterial community structures of the gut microbiota in the two sample groups. Each symbol represents an individual gut microbiota. Black and red symbols represent the winter group and the spring group, respectively
Analysis of species abundance
We performed t test and Mann–Whitney U test to evaluate the significant differences (P < 0.05) among species. At the phylum level, Actinobacteria was significantly more abundant in group B than in group A. At the class level, Coriobacteriia and Deltaproteobacteria were significantly less abundant in group A than in group B. At the order level, Coriobacteriales was also significantly higher in group B. At the family level, Coriobacteriaceae was significantly higher in group B, while Ruminococcaceae and Bacteroidales_S24-7_group were higher in group A (Table 3). Eighteen of the 216 genera were significantly different (P < 0.05) between group A and group B (Table 4). Among these 18 genera, Parabacteroides, Sphaerochaeta, Ruminiclostridium_1 and Phascolarctobacterium were significantly higher in group A, while the others were significantly higher in group B. Additionally, 13 of the 18 genera belonged to Firmicutes.
Discussion
In present study, we first performed high-throughput sequencing to characterize the microbial community in the golden takin. Similar to previous researches (Costa et al. 2012; Oikonomou et al. 2013), the golden takin gut microbiota was also dominated by Firmicutes (67.59%) and Bacteroidetes (23.57%), followed by Proteobacteria (2.37%). Firstly, Firmicutes was an advantaged group of bacteria in the gastrointestinal tract and fecal flora of various animals (Ley et al. 2008). The advantage exhibited by Firmicutes might be related to the anatomical physiology and diet of specific species that feed primarily upon insoluble fiber and in which the cecum and colon were the major sites of fermentation (Costa et al. 2012). Within Firmicutes, Clostridiales (65.42%) dominated in the feces of golden takins. According to previous studies, Clostridiales accounted for 24–59% of human fecal samples and was also the dominant order in the canine gut microbiota; thus, this family was an important index of intestinal bacterial ecosystem function and metabolic differences (Wang et al. 2005). Additionally, Ruminococcaceae dominated in the nine golden takins, and Lachnospiraceae was present at elevated levels; these bacteria were regarded as fiber-degraders in the herbivore rumen and hindgut (Bian et al. 2013; Jami and Mizrahi 2012). Trace amounts of another major fiber-degrader in the rumen, Fibrobacter, were detected in the feces of the nine golden takins. Secondly, Bacteroidetes were the second-most predominant components of the gut flora in the nine golden takins, which were consistent with previous researches (Bian et al. 2013; Costa et al. 2012; Oikonomou et al. 2013). Conversely, Bacteroidetes constitutes the dominant group in the rumens of other ruminants (Bhatt et al. 2013; Jami and Mizrahi 2012). Bacteroidetes degraded high-molecular-weight organic matter, such as protein and carbohydrates, and complemented the host genome to degrade resistant foods such as plant cell wall compounds (including cellulose, pectin, and xylan) (Thoetkiattikul et al. 2013). Bacteroides also degraded host sources of carbohydrates, primarily derived from gastrointestinal tract secretions, such as mucins and chondroitin sulfate polysaccharides (Salyers et al. 1977). Additionally, Bacteroidetes produces butyrate, which was the product of colonic fermentation. Bacteroidetes possessed antitumor properties, which played important roles in maintaining intestinal health (Kim and Milner 2007), and participated in toxic or mutagenic bile acid metabolism and transformation (Smith et al. 2005). In our study, the families Rikenellaceae, Prevotellaceae and Bacteroidaceae dominated within this phylum. Thus, Bacteroidetes played an equally important role as Firmicutes in the digestive system of herbivorous animals. Thirdly, Proteobacteria constituted a large group of bacteria comprising various bacterial taxa that are capable of catabolizing a wide range of feedstuff components (Evans et al. 2010), which advantages these bacteria in the panda (Wei et al. 2012). These differences may be caused by different community structures in the host diet and bowel. In our study, at the genus level, the highest species distribution proportions were observed for Ruminococcaceae_UCG-010 and Ruminococcaceae_UCG-00, which belonged to Ruminococcaceae and were involved in fiber digestion. Fourthly, unclassified bacteria in the nine samples accounted for 18.82–22.48% of sequences. According to previous studies, many animal gut microbiomes contained certain proportions of unclassified bacteria (Bian et al. 2013; Costa et al. 2012; Wu et al. 2016). The reason of this might due to the limited database of 16S RNA gene sequences and little research on classification of fecal microbes. Unclassified bacteria and their functions in different hosts required further research.
The PCA and heatmap analyses indicated that the separation was observed between the microbiota of group A and group B. However, within groups, there was a high percent identity, suggesting the fecal microbiota of golden takins may be influenced by different seasons. Moreover, rarefaction curves revealed lower phylogenetic diversity and numbers of observed species (OTUs) in the winter microbiota than in the spring microbiota. At the same time, the spring microbiota exhibited higher numbers of OTUs with significant differences (Fig. 2). Sun et al. (2016) also found that the dietary shift from winter to spring affected the Tibetan macaque gut microbial composition and diversity. For our research, the diets of the nine golden takins in winter and spring were roughly the same. The significant differences (P < 0.05) among species showed that the relative abundances of Firmicutes and Bacteroidetes in spring were slightly decreased compared with those in winter, Acidobacteria differed significantly between the two seasons and was significantly higher in the spring at the phylum level. According to previous studies, Acidobacteria was present in many wetland soils and contained a number of genes encoding cellulases and hemicellulases. Thus, these bacteria might play an important roles in plant residue degradation (Kanokratana et al. 2011; Pankratov et al. 2011). At the class level, Coriobacteriia and Deltaproteobacteria were significantly more abundant in the spring than in the winter. Coriobacteriia belonged to Actinobacteria, and Deltaproteobacteria belonged to Proteobacteria. Actinobacteria was an important secondary metabolite (enzyme and antibiotics) producer that also played this role in animal intestines, and the secondary metabolites of Actinobacteria are potent antibiotics (Bull et al. 2005; Cundliffe 2006). We hypothesized that the rise in Coriobacteriia facilitated better resistance to harmful microorganisms external to the intestinal environment, and the rise in Deltaproteobacteria was attributable to decreased food quality. Instestingly, 18 genera were significantly different (P < 0.05) between spring and winter, and 13 of these 18 genera belonged to Firmicutes; among these 13 genera, two genera exhibited higher abundances in the winter than in the spring. In contrast, the other 11 genera were less abundant in the winter than in the spring. Thus, Firmicutes might play different roles in different seasons (Carey et al. 2012; Ley et al. 2008; Taglialatela et al. 2009). The weather in spring was highly variable, which was suitable for the proliferation of a variety of pathogenic microorganisms; thus, food might contain greater numbers of pathogenic microorganisms. We suspected that the seasonal differences might be due to the feeding quality, and the intestinal flora of the golden takin might change in the spring to enhance resistance and facilitate the ability to withstand changes in the outside world.
Previous researches also showed that the fecal microbiota of many mammals might be influenced by sex (Bian et al. 2013; Liu et al. 2014; Wu et al. 2016). In present study, the four female golden takins demonstrated highly similar microbiota and the five golden takin males had relatively highly similar microbiota, although J6 and J7 were relatively far from the other three samples, which might indicate that the fecal microbiota of golden takins might also be influenced by the animal’s sex. Sex-related differences might be induced by sex hormones and their effects on gene expression as well as the immune system but also might be attributable to innate physiological differences between males and females (Mcclelland and Smith 2011; Zhao et al. 2013). However, the physiological mechanisms underlying sexual influence were uncertain.
In conclusion, we describe the predominant fecal bacterial populations in golden takin and offered the taxonomic baseline for further research investigating the intestinal ecosystems in golden takins. We also found that the composition of gut microbiota was influenced by the season which might help to strengthen the management of feed quality in the spring to prevent disease in these animals. These observations also increased our understanding of the bacterial ecosystems in this vulnerable animal.
Abbreviations
- DNA:
-
deoxyribonucleic acid
- PCR:
-
polymerase chain reaction
- PCA:
-
principal component analysis
- OTUs:
-
operational taxonomic unit
References
Bhatt VD, Dande SS, Patil NV, Joshi CG (2013) Molecular analysis of the bacterial microbiome in the forestomach fluid from the dromedary camel (Camelus dromedarius). Mol Biol Rep 40(4):3363–3371
Bian G, Ma L, Su Y, Zhu W (2013) The microbial community in the feces of the white rhinoceros (Ceratotherium simum) as determined by barcoded pyrosequencing analysis. PLoS ONE 8(7):e70103–e70103
Bull AT, Stach JEM, Ward AC, Goodfellow M (2005) Marine actinobacteria: perspectives, challenges, future directions. Antonie Van Leeuwenhoek 87(1):65–79
Cadwell K (2014) Expanding the role of the virome: commensalism in the gut. J Virol 89(4):1951–1953
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7(5):335–336
Carey HV, Walters WA, Knight R (2012) Seasonal restructuring of the ground squirrel gut microbiota over the annual hibernation cycle. Am J Physiol Regul Integr Comp Physiol 304(1):R33–R42
Costa MC, Arroyo LG, Allenvercoe E, Stämpfli HR, Kim PT, Sturgeon A, Weese JS (2012) Comparison of the fecal microbiota of healthy horses and horses with colitis by high throughput sequencing of the V3-V5 region of the 16S rRNA gene. PLoS ONE 7(7):267–278
Cundliffe E (2006) Antibiotic production by actinomycetes: the Janus faces of regulation. J Ind Microbiol Biotechnol 33(7):500–506
Desantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL (2006) Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72(7):5069–5072
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32(5):1792–1797
Edgar RC (2013) UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods 10(10):996–998
Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R (2011) UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27(16):2194–2200
Evans NJ, Brown JM, Murray RD, Getty B, Birtles RJ, Hart CA, Carter SD (2010) Characterization of novel bovine gastrointestinal tract Treponema isolates and comparison with bovine digital dermatitis treponemes. Appl Environ Microbiol 77(1):138–147
Hugerth LW, Wefer HA, Lundin S, Jakobsson HE, Lindberg M, Rodin S, Engstrand L, Andersson AF (2014) DegePrime, a program for degenerate primer design for broad-taxonomic-range PCR in microbial ecology studies. Appl Environ Microbiol 80(16):5116–5123
Jami E, Mizrahi I (2012) Composition and similarity of bovine rumen microbiota across individual animals. PLoS ONE 7(3):e33306
Kanokratana P, Uengwetwanit T, Rattanachomsri U, Bunterngsook B, Nimchua T, Tangphatsornruang S, Plengvidhya V, Champreda V, Eurwilaichitr L (2011) Insights into the phylogeny and metabolic potential of a primary tropical peat swamp forest microbial community by metagenomic analysis. Microb Ecol 61(3):518–528
Kim YS, Milner JA (2007) Dietary modulation of colon cancer risk. J Nutr 137(11 Suppl):2576S–2579S
Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, Bircher JS, Schlegel ML, Tucker TA, Schrenzel MD, Knight R (2008) Evolution of mammals and their gut microbes. Science 320(5883):1647–1651
Liu X, Fan H, Ding X, Hong Z, Nei Y, Liu Z, Li G, Guo H (2014) Analysis of the gut microbiota by high-throughput sequencing of the V5-V6 regions of the 16S rRNA gene in donkey. Curr Microbiol 68(5):657–662
Magoč T, Salzberg SL (2011) FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27(21):2957–2963
Mcclelland EE, Smith JM (2011) Gender specific differences in the immune response to infection. Arch Immunol Et Ther Exp 59(3):203–213
Ming L, Wei F, Groves P, Feng Z, Hu J (2003) Genetic structure and phylogeography of the takin (Budorcas taxicolor). Can J Zool 81(3):462–468(7)
Oikonomou G, Teixeira AG, Foditsch C, Bicalho ML, Machado VS, Bicalho RC (2013) Fecal microbial diversity in pre-weaned dairy calves as described by pyrosequencing of metagenomic 16S rDNA. Associations of Faecalibacterium species with health and growth. PLoS ONE 8(4):7377–7382
Pankratov TA, Ivanova AO, Dedysh SN, Liesack W (2011) Bacterial populations and environmental factors controlling cellulose degradation in an acidic Sphagnum peat. Environ Microbiol 13(7):1800–1814
Salyers AA, Vercellotti JR, West SE, Wilkins TD (1977) Fermentation of mucin and plant polysaccharides by strains of Bacteroides from the human colon. Appl Environ Microbiol 33(2):319–322
Schaller GB, Teng Q, Pan W, Qin Z, Wang X, Jinchu HU, Shen H (1986) Feeding behavior of Sichuan takin (Budorcas taxicolor). Mammalia 50(3):311–322
Smith CJ, Rocha ER, Paster BJ (2006) The medically important bacteroides spp. in health and disease. In: Dworkin M, Falkow S, Rosenberg E, Schleifer K-H, Stackebrandt E (eds) The prokaryotes: volume 7: proteobacteria: delta, epsilon subclass. Springer, New York, pp 381–427. doi:10.1007/0-387-30747-8_14
Sun B et al (2016) Marked variation between winter and spring gut microbiota in free-ranging Tibetan Macaques (Macaca thibetana). Sci Rep 6:26035. doi:10.1038/srep26035
Taglialatela JP, Russell JL, Pope SM, Morton T, Bogart S, Reamer LA, Schapiro SJ, Hopkins WD (2009) A core gut microbiome in obese and lean twins. Nature 457(7228):480–484
Thoetkiattikul H, Mhuantong W, Laothanachareon T, Tangphatsornruang S, Pattarajinda V, Eurwilaichitr L, Champreda V (2013) Comparative analysis of microbial profiles in cow rumen fed with different dietary fiber by tagged 16S rRNA gene pyrosequencing. Curr Microbiol 67(2):130–137
Wang M, Ahrné S, Jeppsson B, Molin G (2005) Comparison of bacterial diversity along the human intestinal tract by direct cloning and sequencing of 16S rRNA genes. FEMS Microbiol Ecol 54(2):219–231
Wang Q, Garrity GM, Tiedje JM, Cole JR (2007) Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73(16):5261–5267
Wei F, Fang Z, Xiao Y (2012) Evidence for lignin oxidation by the giant panda fecal microbiome. PLoS ONE 7(11):e50312
Wu X, Zhang H, Chen J, Shang S, Wei Q, Yan J, Tu X (2016) Comparison of the fecal microbiota of dholes high-throughput Illumina sequencing of the V3–V4 region of the 16S rRNA gene. Appl Microbiol Biotechnol 100(8):1–10
Zhao L, Wang G, Siegel P, He C, Wang H, Zhao W, Zhai Z, Tian F, Zhao J, Zhang H (2013) Quantitative genetic background of the host influences gut microbiomes in chickens. Sci Rep 3(5):1970
Authors’ contributions
Conceived and designed the experiments: HHZ, JC. Performed the experiments: JC, XYW. Analyzed the date: JC, HXZ. Contributed materials: JC, JKY, SS, YC. Wrote the paper: JC. Modify the manuscript: XXT. All authors read and approved the final manuscript.
Acknowledgements
We thank Ying Gao (Jinan Wild Animal Park), Xuchang Xie (Jinan Animal Park) for their assistance in the fecal samples collection.
Competing interests
The authors declare that they have no competing interests.
Availability of date and materials
The raw data obtained had been deposited in the NCBI Sequence Read Archive (SRA) with the Bio Project ID- PRJNA344965, Accession: SRP090712, ID: 090712.
Ethical approval and consent to participate
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
Funding
This work was supported in part by grants from the Special Fund for Forest Scientific Research in the Public Welfare (No. 201404420), the National Natural Science Fund of China (Nos. 31372220, 31672313), Science and technology research plan of Shandong Province (No. 2013GSF11707).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Chen, J., Zhang, H., Wu, X. et al. Characterization of the gut microbiota in the golden takin (Budorcas taxicolor bedfordi). AMB Expr 7, 81 (2017). https://doi.org/10.1186/s13568-017-0374-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13568-017-0374-5