
Abstract
Aspergillus sp. contain ppo genes coding for Ppo enzymes that produce oxylipins from polyunsaturated fatty acids. These oxylipins function as signal molecules in sporulation and influence the asexual to sexual ratio of Aspergillus sp. Fungi like Aspergillus nidulans and Aspergillus niger contain just ppo genes where the human pathogenic Aspergillus flavus and Aspergillus fumigatus contain ppo genes as well as lipoxygenases. Lipoxygenases catalyze the synthesis of oxylipins and are hypothesized to be involved in quorum-sensing abilities and invading plant tissue. In this study we used A. nidulans WG505 as an expression host to heterologously express Gaeumannomyces graminis lipoxygenase. The presence of the recombinant LOX induced phenotypic changes in A. nidulans transformants. Also, a proteomic analysis of an A. nidulans LOX producing strain indicated that the heterologous protein was degraded before its glycosylation in the secretory pathway. We observed that the presence of LOX induced the specific production of aminopeptidase Y that possibly degrades the G. graminis lipoxygenase intercellularly. Also the presence of the protein thioredoxin reductase suggests that the G. graminis lipoxygenase is actively repressed in A. nidulans.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Aspergillus sp. contains ppo genes coding for dioxygenases that belong to the linoleate diol synthase (LDS) protein family. These dioxygenases produce oxylipins called precocious sexual inducer (psi) factors, which is a collective term for C18:1, C18:2 and C18:3 derived oxylipins (Gao et al. [2007]). Oxylipins are signal molecules that are used by fungi to control sporulation (Brodhun and Feussner [2011]). These oxylipins are categorized in three groups depending on the position of hydroxyl groups on the polyunsaturated fatty acid (PUFA): psiB (8′-hydroxy-PUFA), psiC (5′,8′-dihydroxy-PUFA), and psiA, which has a δ-lactone ring at the 5′ position of the psiC oxylipin (Tsitsigiannis et al. [2004]). Depending on the PUFA it is further categorized as α (18:2, linoleic acid), β (18:1, oleic acid), and γ (18:3, linolenic acid) (Tsitsigiannis and Keller [2007]). Aspergillus nidulans contains three ppo genes: ppoA, ppoB, and ppoC coding for enzymes that synthesize the oxylipins psiBα, psiBβ, and psiBβ respectively (Brodhun and Feussner [2011]). Aspergillus niger also contains three ppo genes where it lacks ppoB but instead contains ppoD (Wadman et al. [2009]) In A. nidulans PpoA and PpoC have antagonistic roles and PpoB upregulates ppoA and represses ppoC (Tsitsigiannis et al. [2005]; Brodhun and Feussner [2011]). The deletion of the ppoA and ppoB genes increase the ratio of asexual to sexual sporulation, while deletion of the ppoC gene decreases the ratio of asexual to sexual sporulation. However, it is speculated that these sporulation phenotypes cannot be explained by the psiB oxylipin levels alone (Tsitsigiannis et al. [2005]). Therefore it is suggested that other oxylipins, produced by either Ppo’s or other enzymes, are involved in these phenotypic differences. Lipoxygenase (LOX) is a non-heme iron- or managese-containing dioxygenase present in a wide variety of organisms, including fungi (Heshof et al. [2013]). They catalyze the synthesis of oxylipins, but are absent in A. nidulans and A. niger (Wadman et al. [2009]). However, the human pathogenic Aspergillus flavus and Aspergillus fumigatus contain both ppo genes and lox genes (Brown et al. [2008]; Affeldt et al. [2012]). The distribution of ppo and lox genes is schematically given in Figure 1. Studies revealed that LOX in A. flavus is involved in quorum-sensing and phenotypic differences are seen when the gene is disrupted (Brown et al. [2008]; Affeldt et al. [2012]). The Gaeumannomyces graminis LOX is a secreted enzyme that is capable of producing 11S-HPODE and 13R-HPODE oxylipins and is hypothesized to be involved in invading plant tissue (Oliw [2002]). Plant oxylipins 9S- HPODE and 13S-HPODE induce sporogenic effects in A. nidulans similar to the ones produced by the psi factor (Calvo et al. [1999]). Both oxylipins caused decreased mycelial growth where 13S-HPODE in a 10–100 μM concentration also reduces mycotoxin production of aflatoxin and sterigmatocystin (Burow et al. [1997]). In this study we introduced the G. graminis LOX in the A. nidulans WG505 production host. The goal of the study was to verify whether A. nidulans is a suitable host for the heterologous expression of G. graminis LOX.

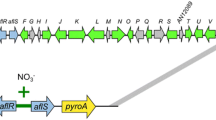
Distribution of ppo and lox genes amongst four different Aspergillus species. The black box indicates the presence of the ppo gene. The blue box indicates the presence of a lox gene coding for an intracellular LOX, and the yellow box represents a lox gene coding for an extracellular LOX. A. nidulans and A. niger have three different ppo genes. A. flavus has four different ppo genes and a lox gene. A. fumigatus has three different ppo genes and two lox genes.
Materials and methods
Expression of G. graminis LOX in A. nidulans
Heterologous expression of G. graminis LOX in A. nidulans was performed according to previous studies (Nyyssölä et al. [2012]). The gene encoding the G. graminis LOX AAK81882.1 was codon-optimized for expression in A. niger and synthesized by DNA 2.0 (Menlo Park, USA) [GenBank KM248327]. For expression the promoter and secretion signal of the xlnD gene from A. niger [GI:74626559] replaced the native secretion signal of G. graminis (Van Peij et al. [1997], Van der Straat et al. [2014]). With help of the Xba I and Bam HI restriction sites the synthesized gene was incorporated into a pUC19 vector and was used to transform A. nidulans WG505, that is a pyrA derivative of A. nidulans WG 096 (ATTC 48756) (Nyyssölä et al. [2012]). The transformants were plated on MMS plates and incubated for 4 days at 37°C (Kusters-van Someren et al. [1991]). Transformants were analyzed by PCR for verification of integration of the lox gene into the genome. A. nidulans was grown for 48 h at 37°C at 250 rpm in 100 ml MM + 50 mM D-xylose using 500 ml Erlenmeyer flasks. The culture broth was separated from the mycelium by funnel filtration and both were submerged into liquid nitrogen to freeze and preserve the materials. The culture broth and the mycelium were stored at −80°C until further research.
Growth and induction of A. nidulans wild type and transformant
A. nidulans WG505 and an isogenic transformant expressing the G. graminis LOX (A. nidulans GG-LOX) were cultivated on agar plates containing complete medium (6.0 g/l NaNO3, 1.5 g/l KH2PO4, 0.5 g/l KCl, 0.5 MgSO4 · 7 H2O, 2 g/l peptone, 1 g/l yeast extract, 1 g/l casamino acids, 0.3 g/l yeast ribonucleic acids, 2 ml/l vitamin solution, 1 ml/l Vishniac solution, 50 mM D-(+)xylose, 12% agar) (Pontecorvo et al. [1953]). Also, A. nidulans was grown at 37°C in 500 ml Erlenmeyers containing 100 ml complete medium without agar and inoculated with 106 spores/ml. Both media contained 50 mM D-xylose to induce the xlnD promotor and stimulate production of G. graminis LOX.
Fermentation of A. nidulans
A. nidulans WG505 and A. nidulans GG-LOX were fermented using a New Brunswick BioFlo® 310 (Eppendorf, Nijmegen, The Netherlands). The batch-phase was performed using 3 l of medium for 24 h (5 g/kg glucose, 0.5 g/kg KH2PO4, 0.5 g/kg MgSO4. 7 H2O, 4.0 g/kg (NH4)2SO4, 1 g/kg yeast extract, 0.1 g/kg Struktol J673). When the glucose was completely consumed the fermentation was fed 2 l using D-xylose medium at a speed of 0.35 g/min (75 g/kg D-xylose, 3.1 g/kg KH2PO4, 14.85 g/kg (NH4)2SO4, 24.4 g/kg yeast extract). Samples were taken every 2 h to test for the presence of the G. graminis LOX.
mRNA isolation and identification of the G. graminis LOX
Mycelium from A. nidulans WG505 and A. nidulans GG-LOX was submerged in peqGOLD TriFast (peqLAB, De Meern, The Netherlands) and disrupted using glass beads and a MP FastPrep-24 beadbeater (MP Biomedicals, Eindhoven, The Netherlands). The RNA isolated was treated with DNase I and transcribed to cDNA using Omniscript RT enzyme (Qiagen, Venlo, The Netherlands). The resulting cDNA was submitted to PCR using the forward primer 5′-TGAGTTGCAGAACTGGATCG-3′ and reverse primer 5′-GCAGAACGCCAGAAAACTTC-3′ for detection of the G. graminis lox mRNA. cDNA from positive reactions were sequenced (Baseclear, Leiden, The Netherlands). As a positive control the pyruvate kinase (pkiA) gene was amplified using the forward 5′-GCCAGTCTTGAACTGAACGC-3′ primer and the reverse 5′-GCCAGATCTTGACGTTGAAGTC-3′ primer (de Graaff et al. [1992]). Amplified gDNA results in a 304 bp fragment while amplified cDNA results in a 204 bp fragment due to the existence of an intron in the fragment.
Mutagenesis of the G. graminis LOX
To test whether the activity of the G. graminis LOX interferes with the correct synthesis and secretion of the protein a double site-directed H306Q-H310E mutagenesis was performed resulting in inactive A. nidulans GG-LOX mutants. These mutations do not affect protein expression levels but result in a catalytically inactive enzyme (Cristea et al. [2005]). For mutagenesis the QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent Technologies, Amstelveen, The Netherlands) was used. The forward 5′-GTTCTACTCCCAAATGTACCAGGTGCTGTTCGAGACCATCCCGGAG-3′ primer and the reverse 5′-CTCCGGGATGGTCTCGAACAGCACCTGGTACATTTGGGAGTAGAAC-3′ primer were designed as advised by the protocol of the mutagenesis kit. Positive transformants were identified by sequencing (Baseclear, Leiden, The Netherlands).
Western blot analysis
To identify the presence of G. graminis LOX a western blot analysis was performed. 20 μl of culture broth and disrupted mycelium was isolated and run on a 10% SDS-PAGE with a voltage of 100 V for 1 h using Tris-HEPES-SDS Running Buffer (Thermo Scientific PI28398). Afterwards the gel was rinsed with dH2O and pre-soaked with CAPS-blot buffer. A nitrocellulose membrane was used to absorb the proteins from the SDS-PAGE and was blotted overnight at 70 mA. The membrane was washed for 30 min in TBST and afterwards it was blocked using TBST + 1% BSA for 30 min. Rabbit polyclonal antibody of G. graminis (Eurogentec, Seraing, Belgium) was used in a 1/1000 dilution to detect LOX on the membrane. After 3x washes with TBST for 10 min the membrane was incubated for 30 min using a 1/1000 dilution of secondary anti-rabbit peroxidase antibody (Sigma-Aldrich Lot. A0545-1ML). After the final 3 × 10 min wash step with TBST the membrane was submitted to AP-detection. This was done by mixing two solvents: 60 mg of 4-chloro-1-naphtol to 20 ml methanol (A) and by adding 60 μl of 30% ice-cold H2O2 to 100 ml TBS (B). Prior to use solvents A and B were mixed and the membrane was added to this mixture. The reaction was stopped with dH2O after 30 min of incubation.
Immunoprecipitation using G. graminis LOX antibody and proteomics analysis
A. nidulans WG505 and A. nidulans GG-LOX were grown with 50 mM D-xylose for 48 h and the culture broth and mycelium were separated by funnel filtration The mycelium was disrupted three times by French Press with a pressure of 1,000 psi and the resulting cell free extract was prepared by centrifugation. Samples of 5 ml from both culture broth and cell free extract were taken and incubated with 5 ml of polyclonal G. graminis antibody (Eurogentec, Seraing, Belgium). The samples were incubated overnight with a stirring speed of 200 rpm at 4°C. Immunoprecipitation was performed using 100 μl Dynabeads Protein G Magnetic Beads (Life Technology, Bleiswijk, The Netherlands). After immunoprecipitation the proteins were separated on a 10% SDS-PAGE at 100 V. The proteins were cut from the SDS-PAGE and submitted to in-gel digestion with 100 ng trypsin/sample in 50 mM ammonium bicarbonate buffer. Afterwards the samples were diluted 1:1 using 2% trifluoroacetic acid to acidify the proteins. The samples were purified by binding the proteins to a reversed-phase C18 column and washing with 0.1% formic acid. Then the proteins were eluted with 80% acetonitrile + 0.1% formic acid. Finally the samples were analyzed by LC-MS/MS in the Radboud Proteomics Centre (Radboud University, Nijmegen, The Netherlands). The resulting data were analyzed using MaxQuant software (Cox and Mann [2008]).
Results
Phenotype differences in A. nidulans
After 48 h of growth in liquid cultures and on agar plates A. nidulans GG-LOX showed a different phenotype from the one of the wild type. As shown in Figure 2, the mycelium of A. nidulans GG-LOX showed a browner colour when compared to A. nidulans WG505. Also, the pellet size of the transformant was smaller than the one of the wild type strain. The amount of spores formed was also increased in the LOX expressing strain compared to the wild type, both in liquid culture and in agar plates, which could be caused by the produced oxylipins of the G. graminis LOX (data not shown).

A. nidulans wild type and transformant on plates and liquid media after 48 h of growth. All cultures were induced with 50 mM D-xylose. Phenotypic changes are found, the transformant expressing G. graminis LOX becomes brown while the wild type does not.
mRNA identification of the G. graminis lox gene
In order to confirm that the G. graminis LOX gene was successfully transcribed in the A. nidulans GG-LOX transformant mRNA was isolated from both the wild type and the A. nidulans GG-LOX. The analysis was also performed in the wild type strain that was used as a negative control. The mRNA sequence of the transformant resulted in 100% homology with the G. graminis lox gene. This implies the gene was correctly transcribed and the production of the G. graminis LOX was stopped at another stage during protein synthesis.
Western blot analysis
A western blot analysis was performed on three different transformants carrying the G. graminis lox gene (T1-T3) and three different G. graminis lox H306Q-H310E mutant transformants (M1-M3) for which the results are shown in Figure 3. Western blot analysis revealed A. nidulans has two different protein band fingerprints seen in samples WT, T2, T3, M2 and T1, M1, M3 (Figure 3a). This coincides with the single and double band found in the western blot analysis of the intracellular proteins of A. nidulans (Figure 3d). The presence of the bands found in the wild type samples showed non-selective binding of polyclonal antibodies. Production of the G. graminis LOX was neither detected in the supernatants nor in the cell free extract. Also the mutant version of the G. graminis LOX was not be detected in the M1-M3 samples. Since the expression of the G. graminis LOX was not be detected at protein level by SDS-PAGE nor by western blot, a proteomic analysis was performed of the proteins isolated by immunoprecipitation using the polyclonal antibodies against G. graminis LOX.

SDS-PAGE and western blot analysis of G. graminis LOX expression A. nidulans . Samples T1-T3 are transformants of A. nidulans carrying the G. graminis lox gene, while M1-M3 are transformants of A. nidulans carrying the mutated G. graminis lox gene. As positive controls 10 ng, 100 ng, and 1000 ng of the G. graminis LOX were applied. The wild type (WT) sample is used as a negative control. a) SDS-PAGE of the proteins in the culture broth of A. nidulans; b) SDS-PAGE of intracellular proteins of A. nidulans; c) Western blot of the proteins in the culture broth reacting to the antibodies of the G. graminis LOX; d) Western blot of intracellular proteins reacting to the antibodies of G. graminis LOX.
Comparative proteomics analysis of LOX immuno-precipitated fractions from A. nidulans WG505 and A. nidulans GG-LOX strains
Immunoprecipitation was done on both cell free extract and culture broth for the identification of G. graminis LOX. Three different proteins were identified in the A. nidulans WG505 wild type and the A. nidulans carrying the G. graminis LOX as shown in Figure 4. Analysis of the A. nidulans transformant revealed intracellular production of G. graminis LOX (1) but the LOX was not found in the culture broth. Also, aminopeptidase Y (2) was identified in the A. nidulans transformant. A third difference is the transformant showed production of thioredoxin reductase (3). This protein functions as a defense agent against oxidative damage and could well be a reflection of the production and activity of G. graminis LOX (Missall and Lodge [2005]). Finally a difference is found in the production of the PpoC enzyme. In the A. nidulans WG505 wild type this protein is present in the cell free extract and the culture broth. However, only small traces of PpoC were found in the transformant.

Immunoprecipitation of G. graminis LOX in cell free extract (CFE) and culture broth (CB) of A. nidulans wild type (WT) and the transformant (T) using polyclonal antibodies against G. graminis LOX. In sample Tcfe a protein band is found at ~67 kDa, which corresponds to the size G. graminis LOX using proteomics (1), a protein band of ~54 kDa represents aminopeptidase Y (2), and a protein band ~41 kDa is thioredoxin reductase (3).
Fermentation of A. nidulans
To verify whether the uncontrolled pH, substrate consumption, and oxygen consumption in shake flasks were promoting the degradation of the G. graminis LOX a 3 to 5 l fed-batch fermentation was run. Biomass was generated using D-glucose as the carbon source before A. nidulans was induced using D-xylose. Samples were taken every 2 h to test for the presence of the G. graminis LOX in the medium. However, no G. graminis LOX could be detected.
Discussion
Phenotypic differences between the wild type and the transformant expressing the G. graminis LOX were found on both agar plate and in liquid cultures. After 48 h the culture broth of A. nidulans GG-LOX changed towards a brown colour. This indicates A. nidulans might be stressed due to the production of G. graminis LOX. Based on these phenotypic changes and the absence of G. graminis LOX activity, it was investigated to study at what stage the LOX production was stopped. By mRNA analysis G. graminis lox mRNA could be detected thus, it can be concluded the lox gene was successfully transcribed. Therefore the production failed at a different level in the protein synthesis process. Proteins from both strains were isolated and analyzed using G. graminis polyclonal antibodies using different methods. Proteomics data revealed production of three proteins in the A. nidulans transformant that were not found in the wild type. The presence of significant amounts of protease aminopeptidase Y and the intercellular location of the G. graminis LOX suggest the LOX is not secreted but is effectively intracellular degraded. Figure 4 shows G. graminis LOX is produced as the 67 kDa version instead of the highly glycosylated one as is found in G. graminis. This indicates the G. graminis LOX is degraded before it is glycosylated in the secretory pathway. Previous research showed expression of the G. graminis LOX in Pichia pastoris does not need to be glycosylated to be active and this non-glycosylated LOX could be formed by proteases from the expression host (Cristea et al. [2005]). Also, production of thioredoxin reductase suggests A. nidulans responds the oxidative stress caused by the G. graminis LOX. Another difference found is the presence of PpoC in the wild type while only traces were found in the transformant. This result suggests the presence and activity of G. graminis LOX interferes with the ppo oxylipin pathway in A. nidulans. The distribution of ppo and lox genes in Aspergillus sp. show A. nidulans and A. niger have three ppo genes and no lox genes. A. fumigatus contains three ppo genes and two lox genes, and A. flavus contains four ppo genes and one lox gene (Brown et al. [2008]; Affeldt et al. [2012]; Wadman et al. [2009]; Tsitsigiannis et al. [2005]). This implies the balance of LOX activity in Aspergillus is monitored by another oxylipin-producing coding gene. The presence of thioredoxin reductase suggests oxidative stress to the A. nidulans GG-LOX transformant and the G. graminis LOX is intercellular active. Previous studies showed a concentration of 10–100 μM 9S-HPODE and 13S-HPODE had effect on mycelial growth (Burow et al. [1997]). Our results show a phenotypic difference between A. nidulans wild type and transformant suggesting the oxylipin balance is disturbed by G. graminis LOX activity. One might speculate on the repressing activity of G. graminis LOX on ppoC, since PpoC was only slightly present in the A. nidulans transformant. The repressing and up-regulating function of PpoB on ppoA and ppoC are in line with this hypothesis (Tsitsigiannis et al. [2005]).
Heterologous production of G. graminis LOX was successfully performed in P. pastoris and Trichoderma reesei (Cristea et al. [2005]; Nyyssölä et al. [2012]). A difference between P. pastoris and T. reesei compared to Aspergillus sp. is the presence of these ppo genes. The double site-directed H306Q-H310E mutagenesis show that G. graminis LOX activity is not causing the low yield of the G. graminis LOX. However, the presence of the protease aminopeptidase Y suggests the LOX is degraded. We hypothesize the introduction of the G. graminis LOX disturbs the oxylipin balance in A. nidulans resulting in a different phenotype and thioredoxin reductase is induced to neutralize the oxidative stress that is caused by the G. graminis LOX. Based on the protein composition between A. nidulans WG505 and A. nidulans GG-LOX, we conclude heterologous production of G. graminis LOX using A. nidulans as an expression system is not effective for industrial purposes.
References
Affeldt KJ, Brodhagen M, Keller NP: Aspergillus oxylipin signaling and quorum sensing pathways depend on G protein-coupled receptors. Toxins 2012, 4(9):695–717. doi:10.3390/toxins4090695
Brodhun F, Feussner I: Oxylipins in fungi. FEBS J 2011, 278(7):1047–1063. doi:10.1111/j.1742–4658.2011.08027.x
Brown SH, Zarnowski R, Sharpee WC, Keller NP: Morphological transitions governed by density dependence and lipoxygenase activity in Aspergillus flavus . Appl Environ Microbiol 2008, 74(18):5674–5685. doi:10.1128/AEM.00565–08
Burow GB, Nesbitt TC, Dunlap J, Keller NP: Seed lipoxygenase products modulate Aspergillus mycotoxin biosynthesis. Mol Plant Microbe In 1997, 10(3):380–387.
Calvo AM, Hinze LL, Gardner HW, Keller NP: Sporogenic effect of polyunsaturated fatty acids on development of Aspergillus spp. Appl Environ Microbiol 1999, 65(8):3668–3673.
Cox J, Mann M: MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 2008, 26(12):1367–1372. doi:10.1038/nbt.1511
Cristea M, Engström K, Su C, Hörnsten L, Oliw EH: Expression of manganese lipoxygenase in Pichia pastoris and site-directed mutagenesis of putative metal ligands. Arch Biochem Biophys 2005, 434(1):201–211.
de Graaff LH, van den Broeck H, Visser J: Isolation and characterization of the Aspergillus niger pyruvate kinase gene. Curr Genet 1992, 22(1):21–27.
Gao X, Shim WB, Göbel C, Kunze S, Feussner I, Meeley R, Balint-Kurti P, Kolomiets M: Disruption of a maize 9-lipoxygenase results in increased resistance to fungal pathogens and reduced levels of contamination with mycotoxin fumonisin. Mol Plant Microbe In 2007, 20(8):922–933.
Heshof R, Jylhä S, Haarmann T, Jørgensen AL, Dalsgaard TK, de Graaff LH: A novel class of fungal lipoxygenases. Appl Microbiol Biotechnol 2013, 98(3):1261–1270. doi:10.1007/s00253–013–5392-x
Kusters-van Someren MA, Harmsen JAM, Kester HCM, Visser J: Structure of the Aspergillus niger pelA gene and its expression in Aspergillus niger and Aspergillus nidulans . Curr Genet 1991, 20(4):293–299.
Missall TA, Lodge JK: Thioredoxin reductase is essential for viability in the fungal pathogen Cryptococcus neoformans . Eukaryot Cell 2005, 4(2):487–489.
Nyyssölä A, Heshof R, Haarmann T, Eidner J, Westerholm-Parvinen A, Langfelder K, Kruus K, de Graaff LH, Buchert J (2012) Methods for identifying lipoxygenase producing microorganisms on agar plates. AMB Express 2(17): doi:10.1186/2191–0855–2-17
Oliw EH: Plant and fungal lipoxygenases. Prostag Oth Lipid M 2002, 68–69: 313–323.
Pontecorvo G, Roper JA, Hemmons LM, Macdonald KD, Bufton AW: The genetics of Aspergillus nidulans . Adv Genet 1953, 5: 141–238.
Tsitsigiannis DI, Keller NP: Oxylipins as developmental and host-fungal communication signals. Trends Microbiol 2007, 15(3):109–118.
Tsitsigiannis DI, Zarnowski R, Keller NP: The lipid body protein, PpoA, coordinates sexual and asexual sporulation in Aspergillus nidulans . J Biol Chem 2004, 279(12):11344–11353.
Tsitsigiannis DI, Kowieski TM, Zarnowski R, Keller NP: Three putative oxylipin biosynthetic genes integrate sexual and asexual development in Aspergillus nidulans . Microbiology 2005, 151(6):1809–1821. doi:10.1099/mic.0.27880–0
van der Straat L, Vernooij M, Lammers M, van den Berg W, Schonewille T, Cordewener J, van der Meer I, Koops A, de Graaff LH (2014) Expression of the Aspergillus terreus itaconic acid biosynthesis cluster in Aspergillus niger. Microb Cell Fact 13(11): doi:10.1186/1475–2859–13–11
van Peij NN, Brinkmann J, Vrsanská M, Visser J, de Graaff LH: beta-Xylosidase activity, encoded by xlnD , is essential for complete hydrolysis of xylan by Aspergillus niger but not for induction of the xylanolytic enzyme spectrum. Eur J Biochem 1997, 245(1):164–173.
Wadman MW, de Vries RP, Kalkhove SI, Velding GA, Vliegenthart JF (2009) Caracterization of oxylipins and dioxygenase genes in the asexual fungus Aspergillus niger. BMC Microbiol 9(59): doi:10.1186/1471–2180–9-59
Acknowledgements
The authors thank Jasper Sloothaak for his technical assistance in the proteomics analysis. The authors gratefully acknowledge the financial support provided by the European Research Project (Novel enzyme tools for production of functional oleochemicals from unsaturated lipids (ERA-NOEL), ERA-IB/BIO/0001/2008).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
RH carried out the molecular cloning of A. nidulans GG-LOX, mRNA analysis, the western blot experiments, the immunoprecipitation, the proteomics analysis, fermentation experiments, and drafted the manuscript. JPvS was responsible for the mutagenesis work and analysis presented in this paper. JATR conceived and performed the proteomics sample preparation and contributed to the manuscript. RH and LHdG designed the experiments participated to draft the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Heshof, R., van Schayck, J.P., Tamayo-Ramos, J.A. et al. Heterologous expression of Gaeumannomyces graminis lipoxygenase in Aspergillus nidulans. AMB Expr 4, 65 (2014). https://doi.org/10.1186/s13568-014-0065-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13568-014-0065-4