
Abstract
Compounds designed to display polypharmacology may have utility in treating complex diseases, where activity at multiple targets is required to produce a clinical effect. In particular, suitable compounds may be useful in treating neurodegenerative diseases by promoting neuronal survival in a synergistic manner via their multi-target activity at the adenosine A1 and A2A receptors (A1R and A2AR) and phosphodiesterase 10A (PDE10A), which modulate intracellular cAMP levels. Hence, in this work we describe a computational method for the design of synthetically feasible ligands that bind to A1 and A2A receptors and inhibit phosphodiesterase 10A (PDE10A), involving a retrosynthetic approach employing in silico target prediction and docking, which may be generally applicable to multi-target compound design at several target classes. This approach has identified 2-aminopyridine-3-carbonitriles as the first multi-target ligands at A1R, A2AR and PDE10A, by showing agreement between the ligand and structure based predictions at these targets. The series were synthesized via an efficient one-pot scheme and validated pharmacologically as A1R/A2AR–PDE10A ligands, with IC50 values of 2.4–10.0 μM at PDE10A and Ki values of 34–294 nM at A1R and/or A2AR. Furthermore, selectivity profiling of the synthesized 2-amino-pyridin-3-carbonitriles against other subtypes of both protein families showed that the multi-target ligand 8 exhibited a minimum of twofold selectivity over all tested off-targets. In addition, both compounds 8 and 16 exhibited the desired multi-target profile, which could be considered for further functional efficacy assessment, analog modification for the improvement of selectivity towards A1R, A2AR and PDE10A collectively, and evaluation of their potential synergy in modulating cAMP levels.

Similar content being viewed by others

Background
Neurodegeneration involves the progressive loss of the structure and function of neurons, which is common in Parkinson’s, Huntington’s disease and schizophrenia [1]. Recently, there has been substantial interest in the search for alternative non-dopamine (non-DA) based approaches for the treatment of neurodegenerative diseases, as the classical DA-based approaches have long been associated with many undesirable side effects such as dyskinesia, hallucinations, and on/off effects [2]. Given that the adenosine neuromodulation system (via the adenosine A1 and A2A receptors) has been identified as a key target for the management of neurodegenerative diseases, this qualifies its targeting as a potential promising non-DA based treatment approach [3, 4]. Indeed, modulation of cAMP levels has proven to have benefits in neuronal survival in an adenosine receptor-dependent manner [5]. In addition, recent findings suggest that phosphodiesterase 10A (PDE10A) also plays a role in neurodegenerative diseases such as Parkinson’s, Huntington’s disease, and schizophrenia [6,7,8]. Inhibition of PDE10A resulting in maintenance of elevated intracellular cAMP concentrations, has been suggested to be effective in the treatment of these diseases. Thus multi-target ligands that bind to different adenosine receptors subtypes (A1 and A2A receptors) while simultaneously inhibit PDE10A might be synergistic in modulating cAMP levels, which is of therapeutic potential for neurodegenerative diseases [9,10,11].
Conceptually, multi-target drugs work by creating a combination effect on multiple targets in the biological network simultaneously, which may (through e.g. synergistic effects) decrease the therapeutic dose required, thus increasing therapeutic efficacy, preventing drug resistance, and reducing target-related adverse effects [12,13,14]. Also, another advantage of multi-target drugs over other types of treatments such as combination therapies, is a reduced likelihood of drug–drug interactions [15, 16].
However, it remains a challenging task for medicinal chemists to design drugs with a specific multi-target profile and to achieve selectivity for specific targets over off-target effects with suitable pharmacokinetic properties [17, 18]. In fact, the field of multi-target drug design has recently become an active field of research in the pharmaceutical industry, where around 20 designed multi-target drugs have either reached advanced development stages or are already approved [14, 19, 20].
In particular, for Central Nervous System (CNS) diseases, there has been growing interest in exploiting the multi-target profiles of existing compounds to investigate their potential applicability as drugs. For example, multi-target profiles of drugs and drug candidates affecting the dopaminergic system have been investigated. Examples include Aripiprazole, Amitriptyline, Chlorpromazine, and Clozapine [21]. In addition, various multi-target based virtual screening protocols for multi-target drug design have been developed [13, 22,23,24]. Examples of ligand-based protocols include in silico target prediction and Chemogenomic and pharmacophore-based approaches, which resulted in the discovery of CNS drugs with multi-target combinations such as MAO-A/MAO-B/AChE/BuChE, AChE/BuChE, and H3-R/HMT/AChE/BuChE [21,22,23,24]. Structure-based approaches such as docking and molecular dynamics calculations have also been employed for the discovery of new multi-target ligands such as BuChE inhibitors/hCB2R and MAO-A/MAO-B/AChE/BuChE ligands to treat neurodegenerative diseases [25].
In this work, we offer a computational strategy for designing synthetically feasible ligands that bind to A1R and A2AR, and inhibit PDE10A—a novel multi-target combination of G protein-coupled receptors (GPCRs) and an enzyme, which has not, to our knowledge, been previously exploited. The designed ligands with this multi-target combination are intended as starting points for future development of multi-target drugs treating neurodegenerative diseases. It should be noted here that in the current study we only consider affinity of ligands to the above receptors, which we also experimentally validate as outlined below. However, for therapeutically relevant purposes also functional effects and optimization of selectivity towards A1R, A2AR and PDE10A need to be considered, which will be the area of a future study.
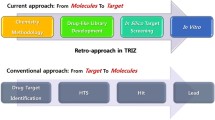
The workflow of the current study is shown in Fig. 1. Starting with a focused chemical space consisting of known actives against A1R, A2AR and PDE10A, new synthetically feasible compounds were established via RECAP (Retrosynthetic Combinatorial Analysis Procedure) [26, 27], which fragments molecules at pre-defined bonds and recombines them in a combinatorial manner, and were then evaluated in silico, using target prediction and ligand/protein docking. Compounds with favorable assessments in both steps were carried forward for substructural analysis. This analysis identified compound series with the highest frequency of prediction as multi-target ligands against the desired set of targets, which is of advantage from the practical side, given their synthetic accessibility via a common synthetic route.

The computational strategy for rational design of A1R/A2AR–PDE10A multi-target ligands started with a focused chemical space consisting of known actives of A1R, A2AR and PDE10A, and formed new synthetically feasible compounds which were subjected to target prediction and docking for synthesis and pharmacological evaluation
A series of 2-aminopyridine-3-carbonitriles were selected for prospective validation of the pipeline, a series which was synthetically accessible via a one pot synthetic scheme i.e. providing products with the desired properties: cost-effective, synthetically efficient and available in a timely fashion [28, 29].
Subsequently the synthesized compounds were experimentally tested and confirmed as A1R/A2AR–PDE10A multi-target ligands. Selectivity against other subtypes of both protein families confirmed the pharmacological profile of the compound series, and structure activity relationships (SAR) were also deduced. Hence, in this work we report a successful computational strategy, which allowed the discovery of the first A1R/A2AR–PDE10A multi-target ligands. The novel A1R/A2AR–PDE10A ligands are sought to display a combination effect in modulating the A1R, A2AR, and PDE10A targets simultaneously similar to that of combination compounds of Adenosine receptors and PDEs, reported by Rickles et al., which were synergistic in modulating cAMP levels [10].
Results and discussion
Design of synthetically feasible A1R/A2AR–PDE10A multi-target ligands
Human enzyme and receptor data were extracted from ChEMBL 20 [30]. Substructure analysis of A1R, A2AR ligands and PDE10A inhibitors with Ki and IC50 values less than or equal to 1 µM revealed that the most frequently occurring common heterocycles among the actives against the three target classes were pyridine, pyrimidine, piperazine, and 1H-pyrazole (Additional file 1: Figure S1). Subsequently, A1R (2104), A2AR (2489) and PDE10A inhibitors (679) containing those frequent heterocycles were subjected to RECAP analysis/synthesis in MOE (see Methods for details) [26]. As a result, 458,839 (potentially) synthetically accessible ligands were formed in silico. This list of candidates was filtered to those retaining the common heterocycles (listed above), in order to create a focused chemical space characteristic of A1R, A2AR and PDE10A (with the simultaneous trade-off of reduced novelty), giving rise to 22,233 compounds.
Target prediction of the designed RECAP library
To assess the likelihood of active compounds against A1R, A2AR and PDE10A, PIDGIN 1.0 (Prediction including Inactivity), a tool which uses ECFP 4 circular Morgan fingerprints and trained on ChEMBL actives and PubChem inactives, was used to perform in silico target prediction for the focused RECAP library (22,233 compounds) [24]. Subsequent enrichment analysis of the predictions was done using an estimation score, average ratio as developed by Liggi et al. [31] and via Chi square test [32]. For targets to be considered as enriched according to these methods, the estimation score and the Chi square test p value should be less than or equal to 0.01 and 0.05, respectively. Hence, upon analyzing the enrichment parameters for the A1R, A2AR and PDE10A targets that were predicted for the focused RECAP library (Additional file 1: Figure S2), the three targets were predicted with an estimation score equal to 0 (enriched) as well as average ratios less than 0.1 (enriched) with Chi squared p values < 0.005. The percentage of RECAP compounds of the focused library that were predicted as actives against the A1R, A2AR and PDE10A targets were 51.1, 52.8, and 24.5% respectively. These numbers are relatively high, which however is understandable given that the input to the RECAP analysis consisted of experimentally established known ligands of the above protein targets.
Docking of the compounds predicted as A1R/A2AR–PDE10A multi-target ligands
In the next step docking and further substructure analysis were performed on compounds of the focused RECAP library, which were predicted as A1R/A2AR–PDE10A multi-target ligands from the ligand-based side in the previous step. 2563 compounds were predicted as actives against the three desired targets, and they were subsequently docked against a high resolution (1.8 Å) A2AR protein crystal structure (PDB ID: 4EIY) [33] its corresponding A1R homology model (see Methods for details), and PDE10A (PDB ID: 4DDL) [34].
Compounds which were carried forward to substructural analysis were selected when their docking score gave a value less than a pre-determined cut-off value computed from the docking scores. This cut-off value was evaluated as the docking score with the best F measure statistic obtained by docking a set of known actives and inactives against the protein crystal structures and the homology model (see Methods for details).
As a result, a distribution of RECAP compounds that were favorable as multi-target ligands by target prediction and docking was obtained, where 62.47% of the RECAP compounds that were predicted as A1R/A2AR–PDE10A multi-target ligands and docked against PDE10A exhibited docking scores lower than − 6.49 (the threshold of the best F measure discriminating between actives and inactives for known ligands). Out of the RECAP compounds which displayed docking scores lower than − 6.49 against PDE10A, 48.89 and 35.23% displayed docking scores lower than − 7.26 and − 8.49 against A1R and A2AR (the thresholds of the best F measures).
Substructure analysis of the compounds predicted as A1R/A2AR–PDE10A multi-target ligands
Substructure analysis was performed on compounds having a favorable assessment by target prediction and docking (i.e. those compounds whose docking scores were below the threshold for all three targets). The analysis revealed frequently occurring series, which shared the same core structure and which are shown in Fig. 2.

2563 compounds of the focused RECAP library were predicted as A1R/A2AR–PDE10A multi-target ligands, and docked against the A2AR protein crystal structure (PDB ID: 4EIY), A1R homology model, and the PDE10A protein crystal structure (PDB IB: 4DDL), the RECAP series which showed an agreement between the ligand-based and structure-based predictions were mainly a 6,7-alkoxyisoquinolines b [1,2,4] triazolo[1,5-c]quinazolines c 2-aminopyridine-3-carbonitriles d imidazo[1,5-a]quinoxalines
The chemical series were identified as [1,2,4]triazolo[1,5-c]quinazolines (50.4% of all positively predicted multi-target ligands by in silico target prediction as well as docking), imidazo[1,5-a]quinoxalines (14.4%), 6,7-alkoxyisoquinolines (10.6%), and 2-aminopyridine-3-carbonitriles (9.2%). These were in addition to various compounds containing the common and frequent heterocycles identified earlier (15.4%). Each series identified could be considered for synthesis, SAR studies and validation as A1R/A2AR–PDE10A multi-target ligands.
Synthesis of novel 2-aminopyridine-3-carbonitriles
Due to both ease of the reaction and anticipated yield, a one-pot synthetic scheme was selected for synthesizing one promising series, 2-aminopyridine-3-carbonitriles. The design resulted in 25 compounds for synthesis of which 21 were novel compounds and four (1, 2, 5, and 17) have previously been reported in the literature [35,36,37,38]. Compounds 1–25 were screened against PAINs (PAN Assay Interference Compounds) [39] using FAFDrug3 [40], and none of the compounds exihibited potential PAINs liability. Subsequently, their synthesis was performed as shown in Scheme 1, and all products were obtained with good yields, ranging from 46 to 85% (see Methods for details).

The one-pot synthetic route followed for the synthesis of novel 4,6-substituted 2-amino-pyridin-3-carbonitriles
Pharmacological evaluation of novel 2-aminopyridine-3-carbonitriles
Bioactivity testing was performed using A1 and A2A human adenosine receptors expressed in transfected CHO (A1) and HeLa (A2A) cells, as well as AD293 cells that were transiently transfected with human PDE10A. Table 1 includes the list of synthesized 4,6-substituted 2-amino-pyridin-3-carbonitriles, along with their Ki values against A1R, A2AR, and IC50 values against PDE10A. It can be seen that 15 compounds of the 25 synthesized 2-amino-pyridin-3-carbonitriles exhibited inhibitory activity against PDE10A below 10 μM. In addition, 13 compounds were adenosine receptor binders exhibiting selectivity towards A1R and A2AR, which has not been the case in the previous work reported by Mantri et al., where 2-amino-pyridin-3-carbonitriles were promiscuous towards the four adenosine receptor subtypes [36].
Given that the objective of this work is to find compounds displaying specific multi-target activity, compounds 8, 16, 21, and 25 were identified as A1R/A2AR–PDE10A multi-target ligands, inhibiting PDE10A with IC50 values of 2.4, 3.2, 10.0, and 5.1 µM respectively, and binding to A1R with Ki values of 294 and 34 nM (compounds 8 and 16, respectively), and to A2AR with Ki values of 41, 95, and 55 nM (compounds 16, 21, and 25, respectively). Notably, compound 16 exhibited the desired multi-target profile as a PDE10A inhibitor and a dual binder to A2AR and A1R.
It was previously reported that substituted pyridines exhibited PDE inhibitory activity [41, 42], and 2-amino-pyridin-3-carbonitriles are adenosine receptor ligands [36]. In this study we have now identified suitable compounds matching both criteria as A1R/A2AR–PDE10A multi-target ligands, satisfying the original compound design objective.
(SAR) structure–activity relationship analysis
The purpose of the SAR analysis was to rationalize the variation in activity of the newly discovered A1R/A2AR–PDE10A multi-target ligands against PDE10A, given that 2-amino-pyridin-3-carbonitriles have been discovered as a novel class of PDE10A inhibitors. Also due to the fact that compounds of this substructural class were documented as adenosine receptor ligands [36], computational SAR studies were focused on the PDE10A data, where the variation in potency was rationalized in relation to the physicochemical properties of the compounds (which were computed by FAFDrug3, Additional file 1: Table S1) [40].
A trend observed repeatedly in several cases was that when logP decreased, associated with an increase in tPSA, then this led to an improvement in the activity against PDE10A. Initial analysis concentrated on compounds 1–4, which have a phenyl substituent at position 4 of the pyridine ring. Compound 3 was the most potent PDE10A inhibitor with an IC50 of 2.0 µM, and a computed logP of 3.1 and tPSA of 103.9 Å2. Similarly, for compounds 5–7 having a phenyl substituent at position 6 of the pyridine ring, compound 6 was the most potent against PDE10A with an IC50 of 5.7 µM and a computed logP of 4.0 and tPSA of 81.2 Å2. For compounds 8–13, which have a cyclohexyl ring at position 4 of the pyridine ring, compound 12 displayed the most potent PDE10A inhibitory activity with an IC50 of 0.9 µM and a computed logP of 4.7 and tPSA of 90.9 Å2. For compounds 14–17, with a p-methoxyphenyl substituent at position 4 of the pyridine ring, compound 16 with the smallest predicted lipophilicity of 3.1 and tPSA of 85.1 Å2 displayed a good PDE10A inhibitory activity with an IC50 value equal to 3.2 µM, yet the most potent compound was 15 with an IC50 value of 1.5 µM and a computed logP of 4.4 and tPSA of 71.9 Å2. For compounds 19–22, with an o-methoxyphenyl substituent at position 4 of the pyridine ring, compound 22 displayed PDE10A inhibitory activity with the highest potency (IC50 value of 5.6 µM), and a computed logP of 3.7 and tPSA of 92.2 Å2. Finally a similar general trend is observed for the compounds 23 and 24 with a 4-hydroxyphenyl substituent at position 6 of the pyridine ring, where compound 24 was a more potent PDE10A inhibitor with an IC50 of 3.1 µM and computed logP of 3.4 and tPSA of 103.2 Å2. Hence, it could be deduced that in the majority of the series considered, where the substituents on a single position is varied, a decrease in computed lipophilicity associated with an increase in polarity generally improved the activity of compounds against PDE10A. This general trend can be attributed to the hydrophilic nature of the pocket, which favours the interactions between the ligand and the PDE10A protein by compounds exhibiting these properties.
Compound selectivity assessment
The selectivity of compounds 1–25 against the selected major off-targets A2BR, A3R, PDE7A, PDE7B, and PDE9A, was predicted using PIDGIN at a threshold for binding greater than or equal to 0.8, and subsequently tested experimentally. It is noted here that the IC50 values were determined for compounds with % inhibition at phosphodiesterases greater than 70%. As shown in Additional file 1: Table S2, the synthesized compounds are mostly inactive against those off-targets except for compounds 16, 17, 21, and 23 that exhibited IC50 values of 3.4, 3.5, 15.1 and 1.8 µM against PDE7A, and compounds 23 and 25, which exhibited IC50 values of 7.3 and 4.7 µM against PDE7B. Remarkably, compound 8 was found to exhibit selectivity over all tested off-targets using the above criterion, with the lowest selectivity measured for PDE7B (of 55% inhibition at 10 µM ligand concentration). This can be compared to the IC50 value of 8 at PDE10A, which is 2.4 μM (indicating approximately twofold selectivity for 8).
In general, the experimental results on off-target prediction for the synthesised 4,6-substituted 2-amino-pyridin-3-carbonitriles 1–25 agree with the predictions generated using PIDGIN utilised to bias the compound design towards selective compounds such as 8 (Additional file 1: Table S2). This compound would serve as a good starting point for analog modification to improve the selectivity of the synthesized ligands towards PDE10A.
Analysis of the molecular docking studies of the synthesized 2-aminopyridine-3-carbonitriles
The synthesized 2-aminopyridine-3-carbonitriles were docked against A2AR (PDB ID: 4EIY), A1R homology model, and PDE10A (PDB ID: 4DDL). Figure 3 shows the common predicted ligand-target interactions for representative multi-target ligands of A1R–PDE10A, A1R–A2AR, and A2AR–PDE10A, namely for compounds 8, 18, and 25.

Docking studies predicted molecular interactions characteristic of the 4,6-substituted 2-amino-pyridin-3-carbonitriles with the A2AR protein crystal structure (PDB ID: 4EIY), A1R homology model, and PDE10A protein crystal structure (PDB ID: 4DDL), which are displayed for representative multi-target ligands with the following combinations: compound 8 (A1R–PDE10A), 18 (A1R–A2AR), and 25 (A2AR–PDE10A): a interactions with A2AR: the overlaid compounds 18 and 25 exhibit H-bonds via amino and carbonitrile groups with Asn253, and the pyridine rings are π-stacked with Phe168 b interactions with A1R: the overlaid compounds 8 and 18 exhibit H-bonds via amino and carbonitrile groups with Asn254, and the pyridine rings are π-stacked with Phe171 c interactions with PDE10A: the overlaid compounds 8 and 25 have the pyridine rings π-stacked with Phe686 and Phe719. The molecular interactions predicted for the active molecules are consistent with observed interactions between co-crystallised ligands and their corresponding protein crystal structures (PDB ID: 4EIY and 4DDL) [33, 34] and the interactions with the A1R homology model reported in the literature [51, 52]
It can be seen that compounds 8 and 25, with IC50 values of 2.4 and 5.1 µM respectively, share similarities in predicted binding modes, since their pyridine rings display π-stacking with Phe686 and Phe719 of PDE10A (Fig. 3). These are the type of interactions predicted to be exhibited by the majority of the synthesized ligands from this work, as well as the only existing interactions between co-crystallised PDE10A inhibitors discovered by fragment screening (PDB ID: 5C2E, 5C1W, 5C29, 5C2A ligands with Ki values of 2, 8, 700, 880, and 4.8 nM, respectively) [43]. It is noted that the ligand of 5C2A exhibits a considerable selectivity towards PDE10A over all the other PDEs (in the range of 100–1000 fold and greater over the majority of PDEs, with the least selectivity observed being in the range of 25–100 fold). This ligand exhibits only π-stacking interactions with Phe686 and Phe719, similar to the mode of interactions of compound 8 with PDE10A, which is relatively selective over all tested PDEs, with the lowest selectivity being measured for PDE7B (of 55% inhibition at 10 µM ligand concentration) and compound 25, which is selective against all tested PDEs except PDE7B (Table 1 and Additional file 1: Table S2). Additional interactions were seen in analogs discovered by fragment screening, namely hydrogen bonding with Gln716 and Tyr683 in the PDE10A selectivity pocket (PDB ID: 5C28 and 5C2H with Ki values of 2200 and 0.0082 nM respectively). [43] The ligand of 5C2H exhibits π-stacking with Phe686 and Phe719 and hydrogen bonding with Tyr683 in the PDE10A selectivity pocket. The 5C2H ligand showed a very high selectivity towards PDE10A, greater than 5000 fold, which emphasizes the consideration of compound 8 for analog modification to target the selectivity pocket in order to improve the folds of selectivity towards PDE10A. In addition, hydrogen bonding with Tyr683 in the PDE10A selectivity pocket is also seen in many other highly selective PDE10A inhibitors reported in the literature [44] (PDB ID: 5DH5, [45] 5B4L, [46] with Ki = 0.23 nM, and IC50 = 0.76 nM respectively), which further highlights the importance of analog modification to target the PDE10A selectivity pocket.
Moreover, it is noted that compounds 16 and 21 with IC50 values of 3.2 and 10.0 µM respectively (which are selective against all tested PDEs except PDE7A, Table 1 and Additional file 1: Table S2) were predicted to exhibit an additional type of interaction, H-bonding with Gln716 via their overlaid furan rings at position 6 of the pyridine ring (Additional file 1: Figure S3). In fact H-bonding with Gln716 was the only interaction, besides π-stacking with Phe686 and Phe719, which has been observed in many of the highly selective PDE10A ligands reported in the literature (PDB ID: 4DDL, [34] 3SN7, 3SNL, and 3SNI, [47] 5DH4 and 5DH5, [45] with IC50 values of 4.9, 0.7, 0.7, 11 nM and Ki = 0.23 nM respectively). As for other type of interactions generally exhibited by known PDE10A inhibitors such as hydrogen bonding with Gln726 and π-stacking with Phe729 (PDB ID: 5EDE) [48], none has been predicted for any of the compounds presented in this work.
Common predicted binding modes can also be observed for the synthesized compounds against the adenosine receptors A2AR and A1R. Figure 3 displays the interactions of two representative compounds 18 and 25, which exhibit Ki values of 948 and 55 nM respectively, and these are H-bonding of their pyridine rings with Asn253 and π-stacking of their amino and carbonitrile groups with Phe168 of A2AR. As for A1R, the overlaid compounds 8 and 18, with Ki values of 294 and 78 nM respectively, H-bond via their amino and carbonitrile groups with Asn254, and their pyridine rings are π-stacked against Phe171. It can be observed that the ligand/protein interactions predicted for the active compounds against the A2AR are also those seen in the co-crystallised ligand/protein crystal structures (PDB ID: 4EIY, [33] 3EML, [49] 5IU4, [50] with a Ki value of 0.8 nM for ZM241385, which is the common ligand for the three PDB IDs). Similar was the case for the reported interactions with the A1R homology model in the literature (with IC50 values of 2.9 and 6.2 nM for the reported ligands predicted to bind to the homology model of A1R) [51, 52].
Generally the compounds exhibited good selectivity towards A1R and A2AR (Table 1 and Additional file 1: Table S2) with a nanomolar range of binding affinities. As for the selectivity towards PDE10A, it could be improved by analog modification of compound 8, which favors the hydrogen bonding with Tyr683 in the PDE10A selectivity pocket. In addition, the potency of compounds against PDE10A could be optimized in itself, in order to achieve therapeutically relevant efficacy.
Computational assessment of CNS permeability
Compounds 8 and 16 exhibited the desired multi-target profile by inhibiting PDE10A and binding to A2AR and/or A1R. The physicochemical properties of these compounds were calculated by FAFDrug3 [40], and both compounds passed the Lipinski rule of 5 and the CNS filter, which takes into consideration the assessment of their ability to pass the blood brain barrier (Additional file 1: Figure S4) [53]. Hence, while further experimental work would be needed to establish the validity of those predictions, compounds 8 and 16 may serve as good starting points for further functional efficacy assessment and selectivity optimization towards PDE10A, A2AR and/or A1R for the subsequent consideration of multi-target drug development for the treatment of neurodegenerative diseases.
Conclusions
Here we report a successful computational strategy for designing the first A1R/A2AR–PDE10A multi-target ligands as a therapeutic prospect for neurodegenerative diseases. A retrosynthetic approach was employed using MOE/RECAP, followed by target prediction and docking of the resulting library against the desired targets. We have identified 2-aminopyridine-3-carbonitriles as a series that showed agreement between both the ligand- and structure-based predictions of activity against A1R, A2AR and PDE10A. The synthesis of this series via a one-pot synthetic scheme was pursued experimentally. As a result, compounds 8, 16, 21, and 25 were validated as A1R/A2AR–PDE10A multi-target ligands with IC50 values of 2.4, 3.2, 10.0, and 5.1 µM against PDE10A, and binding to A1R with Ki values of 294 and 34 nM (8 and 16 respectively), and to A2AR with Ki values of 41, 95, and 55 nM (16, 21, and 25 respectively). Furthermore, selectivity profiling of the synthesized 4,6-substituted 2-amino-pyridin-3-carbonitriles against other subtypes of both protein families showed that the multi-target ligand 8 exhibited a minimum of twofold selectivity over all tested off-targets. In addition, compounds 8 and 16 exhibited the desired multi-target profile against A1R, A2AR and PDE10A, which would serve as good starting points for further functional efficacy assessment and analog modification for the improvement of selectivity. In particular, this comprises investigating the signal transduction profiles of these compounds using techniques some of the authors have described before [51], as well as evaluating functional effects in cAMP assays to determine if these compounds do provide synergistic elevations in intracellular cAMP. One specific functional profile that would be of high interest and which is likely to elevate cAMP levels synergistically via the combination effect on multiple targets simultaneously, is the A1R antagonist/A2AR agonist, and PDE10A inhibitor.
In summary we have investigated a computational approach for the design of multi-target ligands that was validated experimentally via synthesis and pharmacological evaluation of 2-aminopyridine-3-carbonitriles as A1R/A2AR–PDE10A ligands. This approach is generally applicable to a wide range of multi-target ligand design problems, across disease areas and target families.
Experimental
Selecting reference molecules for the design of multi-target ligands
Using SQL (script provided in Additional file 1), human A1R (2860), A2AR (3566) ligands and PDE10A inhibitors (843) were extracted from the ChEMBL 20 database with Ki and IC50 values less than or equal to 1 μM respectively, and confidence scores of 8 or 9 [30]. Following extraction, the most frequent and common heterocycles between A1, A2A receptor ligands and PDE10A inhibitors were found by performing substructure analysis on each structure using the “Chemistry-> Analyze scaffolds” function in DataWarrior 4.2.2 [54]. Analysis of A1R, A2AR ligands and PDE10A inhibitors identified common and frequent heterocycles (pyridine, 1H-pyrazole, pyrimidine and 9H-purine for A1R and A2AR), and these were extracted from each set using RDKit, 9.1, Python [55]. It should be noted that compounds containing 9H-purine were also extracted from the original set even though this substructure is characteristic of A1R and A2AR only, since it is structurally similar to the common and frequent heterocycles identified (pyridine, 1H-pyrazole, and pyrimidine). Additional file 1: Figure S1 shows the most frequent heterocycles for the A1R, A2AR ligands, and PDE10A inhibitors and their relative frequencies in each set. It was found that they are furan, pyridine, xanthine, 1H-pyrazole, pyrimidine, piperazine, and 9H-purine. All of these heterocycles ranked among the top 30 for A1R, A2AR ligands and PDE10A inhibitors. This indicated their suitability for designing multi-target ligands at these protein targets, given the overlap in chemical (heterocyclic) space. In the case where no percentage is displayed for a particular target, this means that the heterocycle does not appear among the top 30 for the set of compounds involved.
Designing new multi-target ligands
A1R (2104), A2AR (2489) and PDE10A inhibitors (679) consisting of the common and frequent heterocycles, were subjected to RECAP analysis/synthesis in MOE [26]. The RECAP function electronically fragments and recombines molecules based on chemical knowledge of 11 chemical bond types derived from common chemical reactions [27]. As a result, 458,839 novel RECAP-derived compounds were formed. Finally the designed RECAP library was filtered using RDKit, Python according to the common and frequent heterocycles identified, which narrowed the list down to 22,233 compounds.
Target prediction
The SMILES of the designed RECAP library were standardized using the ChemAxon Command-Line Standardizer where the following options were selected: “Remove Fragment” (keep largest), “Neutralize”, “RemoveExplicitH”, “Clean2D”, “Mesomerize” and “Tautomerize” [56]. The standardized canonical SMILES were exported to CSV files, and subjected to enriched target prediction using PIDGIN 1.0 implementing the method developed by Liggi et al. [24, 31]. The target prediction for the designed RECAP library was performed using a recall probability threshold of 0.01 (which is a value consistent with greater confidence in the more positive predictions).
Enrichment calculations for the predicted targets of the designed RECAP library were performed to assess the likelihood of the active compounds against the targets of interest. In this procedure, the frequency of predicting A1R, A2AR and PDE10A targets for the designed RECAP library was compared with a background distribution of a diverse library covering a large chemical space and was assessed by two parameters: the estimation score and the average ratio. The cutoff selected for considering a target as sufficiently enriched required an estimation score less than or equal to 0.01 [31]. The statistical relevance of the prediction was assessed via a Chi squared test with yates correction in Scipy [32], using the contingency table of the RECAP library and background of randomly sampled PubChem compounds (Additional file 1: Figure S2).
Receptor preparation
Docking with Glide [57] was performed against the human A2AR protein crystal structure (PDB ID: 4EIY) bound to the antagonist ZM241385 and the PDE10A crystal structure (PDB ID: 4DDL) complexed with an inhibitor [33, 34]. Protein structures were prepared using the protein preparation wizard of maestro 9.3 [58], following the default protocol which accounts for energy refinement, hydrogen addition, pKa assignment, and side-chain rotational isomer refinement. Resolved water molecules were discarded, and the structure was centered using the co-crystallized ligand as the center of the receptor grid generated for each protein structure. The co-crystal structures of A2AR with 4-{2-[(7-amino-2-furan-2-yl[1, 2, 4]triazolo[1,5-a][1, 3, 5]triazin-5-yl)amino]ethyl}phenol (PDB ID: 4EIY), and PDE10A with 2-{1-[5-(6,7-dimethoxycinnolin-4-yl)-3-methylpyridin- 2-yl]piperidin-4-yl}propan-2-ol (PDB ID: 4DDL), were selected as target structures.
The A1R homology model (Additional file 2) was constructed according to the method reported by Yaziji et al. [59,60,61], where the protein sequence of the human A1R (accession number P30542) was aligned with the A2AR template of PDB ID: 4EIY.
Ligand preparation
The entire set of 2563 ligands was prepared for docking with LigPrep 2.5 [62] using the default settings and the Epik option which introduces energy penalties associated with ionization and tautomerization [63].
Cut-off generation for compound selection from docking models
In an attempt to validate the constructed A2AR, A1R, and PDE10A docking models, a set of known actives and inactives were docked against each target to ensure that they enriched actives. 81 A2AR receptor ligands reported in the literature were docked against the A2AR model [64, 65]. For consistency 81 ChEMBL actives were also selected (for each of the A1R and PDE10A proteins whose Ki and IC50 values are less than 10 µM), and these were docked against their respective target class. In addition, PubChem inactives (200 compounds) of each target class were docked.
A good separation was obtained for the medians of docking score distribution for actives versus inactives confirming that the actives are enriched. Additional file 1: Figure S5 shows the separation of the medians for the three docking models, − 6.93 (actives) versus − 5.64 (inactives) for the PDE10A docking model, − 7.66 (actives) versus − 6.01 (inactives) for the A2AR docking model, and − 7.60 (actives) versus − 5.66 (inactives) for the A1R docking model. Statistical analysis was performed with R using a Mann–Whitney test [66] on the active and inactive docking score distributions of each target. The differences in medians were significant with p values < 0.05 (script provided in Additional file 1).
The F1 score which is the harmonic mean of precision and recall, was computed (using a Python script, see Additional file 1) for all the docking scores of the ChEMBL actives and PubChem inactives for each model. A search was performed for a docking score threshold that gave the highest F1 score, in order to perform substructure analysis on compounds that were predicted as A1R/A2AR–PDE10A multi-target ligands by target prediction, and displayed docking scores that are lower than or equal to those with the highest F1 score for each of the three docking models (A1R, A2AR, and PDE10A, see Additional files 3, 4, and 5). Furthermore, the thresholds found are intended to serve as reference scores for any structure-based design problem at these target classes.
Docking
The RECAP compounds that were predicted as A1R/A2AR–PDE10A multi-target ligands were docked against the A2AR protein crystal structure (PDB ID: 4EIY) [33], the A1R homology model and the PDE10A protein crystal structure (PDB ID: 4DDL) [34] to investigate the molecular interactions. The Glide docking parameters used here are given in Additional file 1: Table S3. The parameters were deduced from docking experiments using known actives and inactives against each protein model.
Substructural analysis
Subsequently, substructure analysis was performed using DataWarrior 4.2.2, on the proposed A1R/A2AR–PDE10A multi-target ligands predicted by both ligand-based and structure-based techniques (considering docking scores less than or equal to the threshold of the best F measure for each docking model). The chemical series found were [1,2,4] triazolo[1,5-c]quinazolines (50.4%), imidazo[1,5-a]quinoxalines (14.4%), 6,7-alkoxyisoquinolines (10.6%), and 2-aminopyridine-3-carbonitriles (9.2%), in addition to various compounds consisting of the common and frequent heterocycles identified originally in the substructural analysis of the extracted ChEMBL compounds.
Synthesis of novel 4,6-substituted 2-amino-pyridin-3-carbonitriles
Due to both ease of the reaction and yield, a one-pot synthetic scheme was optimized for the purpose of synthesizing 2-aminopyridine-3-carbonitriles. For the other series, the synthetic routes were multi-step reactions, which due to synthetic complexity are not reported here.
The synthetic routes reported in the literature for the formation of derivatives of 6,7-alkoxyisoquinolines as selective PDE10A inhibitors involved multi-step reactions ranging from 3 to 13 steps [67, 68]. Whereas, the procedures for the synthesis of the imidazo[1,5-a]quinoxalines, known PDE10A inhibitors, consisted of 3–7 step reactions [69,70,71,72]. The [1,2,4]triazolo[1,5-c]quinazolines have been reported as potent and selective A2AR antagonists and PDE10A inhibitors, and their synthesis involved 4–7 step reactions [73,74,75].
Hence, given the fact that the 2-aminopyridine-3-carbonitriles were the only RECAP series that could be synthesized via a one-pot synthetic scheme [37, 76, 77], we have selected these for synthesis and subsequent validation as multi-target ligands. In particular, we selected compounds, which did not exihibit any potential PAINs liability upon screening with the FAFDrug3 ADME-Tox Filtering Tool [40].
Chemistry
Unless otherwise indicated, all starting materials, reagents and solvents were purchased and used without further purification. After extraction from aqueous phases, the organic solvents were dried over anhydrous sodium sulfate. The reactions were monitored by thin-layer chromatography (TLC) on 2.5 mm Merck silica gel GF 254 strips, and each of the purified compounds showed a single spot; unless stated otherwise, UV light and/or iodine vapor were used to detect compounds. The synthesis of the target compounds was performed in coated Kimble vials on a PLS (6 × 4) Organic Synthesizer with orbital stirring. Filtration and washing protocols for supported reagents were performed in a 12-channel vacuum manifold. The purity and identity of all tested compounds were established by a combination of HPLC, elemental analysis, mass spectrometry and NMR spectroscopy as described below. Purification of isolated products was carried out by column chromatography (Kieselgel 0.040–0.063 mm, E. Merck) or medium pressure liquid chromatography (MPLC) on a CombiFlash Companion (Teledyne ISCO) with RediSep pre-packed normal-phase silica gel (35–60 µm) columns followed by recrystallization. Melting points were determined on a Gallenkamp melting point apparatus and are uncorrected. The NMR spectra were recorded on Bruker AM300 and XM500 spectrometers. Chemical shifts are given as δ values against tetramethylsilane as internal standard and J values are given in Hz. Mass spectra were obtained on a Varian MAT-711 instrument. Analytical HPLC was performed on an Agilent 1100 system using an Agilent Zorbax SB-Phenyl, 2.1 mm × 150 mm, 5 µm column with gradient elution using the mobile phases (A) H2O containing 0.1% CF3COOH and (B) MeCN and a flow rate of 1 mL/min. The purity of all tested compounds was determined to be greater than or equal to 95%.
The synthesis of the 4,6-substituted 2-amino-pyridin-3-carbonitriles 1–25 was done via the one-pot synthetic route shown in Scheme 1. Varying both substituents on the ylidene malononitrile and the ketone reagents resulted in a variation of the substituents on positions 4 and 6 of the pyridine ring.
Synthetic procedure
Substituted ylidene malononitrile (1.0 mmol), ketone (1.0 mmol) and ammonium acetate (5.0 mmol) in a 1:1 toluene/EtOH mixture (7 mL) were stirred in a coated Kimble vial at 120 °C for 12–24 h. After reaction completion (TLC control), distilled water was added and the mixture was extracted with ethyl acetate (3 × 10 mL). The organic phase was dried (Na2SO4) and evaporated under reduced pressure to afford an oily residue that was purified by column chromatography using n-hexane-ethyl acetate in 2:1 mixture.
2-amino-6-(4-fluorophenyl)-4-phenylpyridine-3-carbonitrile (1)
Purified by column chromatography (n-hexane-ethyl acetate 2:1) and then recrystallized from EtOH to give 0.246 g, 85% yield (97% purity by HPLC). MP 226–228 °C. 1H NMR (300 MHz, CDCl3), δ (ppm) 8.08–7.95 (m, 2H), 7.69–7.58 (m, 2H), 7.60–7.47 (m, 3H), 7.23–7.09 (m, 3H), 5.34 (s, 2H). MS (EI) m/z (%): 289.07 (M+, 100), 262.07 (7). Analysis calculated for C18H12FN3: C, 74.73; H, 4.18; F, 6.57; N 14.52. Found: C, 74.70; H, 4.19; F, 6.55; N, 14.54.
2-amino-6-(4-hydroxyphenyl)-4-phenylpyridine-3-carbonitrile (2)
Purified by column chromatography (n-hexane-ethyl acetate 2:1) and then recrystallized from EtOH to give 0.227 g, 79% yield (96% purity by HPLC). MP 241–243 °C. 1H NMR (300 MHz, CDCl3), δ (ppm) 9.92 (s, 1H), 7.99 (d, J = 8.6 Hz, 2H), 7.78–7.59 (m, 2H), 7.58–7.47 (m, 3H), 7.15 (s, 1H), 6.88 (s, 2H), 6.83 (d, J = 8.7 Hz, 2H). MS (EI) m/z (%): 287.04 (M+, 100), 259.89 (10). Analysis calculated for C18H13N3O: C, 75.25; H, 4.56; N, 14.63; O, 5.57. Found: C, 75.27; H, 4.54; N, 14.62; O, 5.59.
2-amino-4-phenyl-6-(1,3-thiazol-2-yl)pyridine-3-carbonitrile (3)
Purified by column chromatography (n-hexane-ethyl acetate 2:1) and then recrystallized from EtOH to give 0.172 g, 62% yield (95% purity by HPLC). MP 154–156 °C. 1H NMR (300 MHz, CDCl3), δ (ppm) 7.95 (d, J = 3.0 Hz, 1H), 7.72 (s, 1H), 7.66–7.65 (m, 2H), 7.52–7.50 (m, 4H), 5.30 (s, 2H). MS (EI) m/z (%): 278.03 (M+, 100), 276.97 (45). Analysis calculated for C15H10N4S: C, 64.73; H, 3.62; N, 20.13; S, 11.52. Found: C, 64.85; H, 3.48; N, 20.25; S, 11.42.
2-amino-6-(1-methyl-1H-pyrrol-2-yl)-4-phenylpyridine-3-carbonitrile (4)
Purified by column chromatography (n-hexane-ethyl acetate 2:1) and then recrystallized from EtOH to give 0.189 g, 69% yield (98% purity by HPLC). MP 152–153 °C. 1H NMR (300 MHz, CDCl3), δ (ppm) 7.67–7.54 (m, 2H), 7.56–7.42 (m, 3H), 7.30 (s, 1H), 6.91 (s, 1H), 6.66–6.59 (m, 2H), 5.23 (s, 2H), 3.70 (s, 3H). MS (EI) m/z (%): 274.14 (M+, 100). Analysis calculated for C17H14N4: C, 74.43; H, 5.14; N, 20.42. Found: C, 74.57; H, 5.12; N, 20.30.
2-amino-4-(2-methoxyphenyl)-6-phenylpyridine-3-carbonitrile (5)
Purified by column chromatography (n-hexane-ethyl acetate 2:1) and then recrystallized from EtOH to give 0.238 g, 79% yield (97% purity by HPLC). MP 199–200 °C. 1H NMR (300 MHz, CDCl3), δ (ppm) 8.03–7.93 (m, 2H), 7.52–7.41 (m, 4H), 7.31 (dd, J1 = 7.5 Hz, J2 = 1.8 Hz, 1H), 7.17 (s, 1H), 7.11–7.02 (m, 2H), 5.27 (s, 2H), 3.88 (s, 3H). MS (EI) m/z (%): 301.16 (M+, 100), 270.12 (7), 120.10 (16.3). Analysis calculated for C19H15N3O: C, 75.73; H, 5.02; N, 13.94; O, 5.31. Found: C, 75.76; H, 5.04; N, 13.92; O, 5.33.
2-amino-4-(2,4-dimethoxyphenyl)-6-phenylpyridine-3-carbonitrile (6)
Purified by column chromatography (n-hexane–ethyl acetate 2:1) and then recrystallized from EtOH to give 0.238 g, 72% yield (99% purity by HPLC). MP 155–157 °C. 1H NMR (300 MHz, CDCl3), δ (ppm) 8.02-7.90 (m, 2H), 7.52–7.38 (m, 3H), 7.32–7.22 (m, 1H), 7.16 (s, 1H), 6.69–6.55 (m, 2H), 5.25 (s, 2H), 3.88 (s, 3H), 3.86 (s, 3H). MS (EI) m/z (%): 331.14 (M+, 100), 165.51 (9), 120.16 (11.3). Analysis calculated for C20H17N3O2: C, 72.49; H, 5.17; N, 12.68; O, 9.66. Found: C, 72.50; H, 5.19; N, 12.71; O, 9.70.
2-amino-4-(2H-1,3-benzodioxol-5-yl)-6-phenylpyridine-3-carbonitrile (7)
Purified by column chromatography (n-hexane-ethyl acetate 2:1) and then recrystallized from EtOH to give 0.236 g, 75% yield (96% purity by HPLC). MP 220–221 °C. 1H NMR (300 MHz, CDCl3), δ (ppm) 8.12–7.86 (m, 2H), 7.56–7.38 (m, 3H), 7.20–7.08 (m, 3H), 6.95 (d, J = 8.0 Hz, 1H), 6.06 (s, 2H), 5.33 (s, 2H). MS (EI) m/z (%): 315.11 (M+, 100), 157.52 (5). Analysis calculated for C19H13N3O2: C, 72.37; H, 4.16; N, 13.33; O, 10.15. Found: C, 72.45; H, 4.06; N, 13.49; O, 10.00.
2-amino-4-cyclohexyl-6-phenylpyridine-3-carbonitrile (8)
Purified by column chromatography (n-hexane-ethyl acetate 2:1) and then recrystallized from EtOH to give 0.216 g, 78% yield (98% purity by HPLC). MP 125–126 °C. 1H NMR (300 MHz, CDCl3) δ (ppm): 7.95–7.92 (m, 1H), 7.53–7.43 (m, 3H), 7.05 (s, 1H), 6.73 (s, 1H), 5.22 (s, 2H), 2.90–2.85 (m, 2H), 1.90–1.78 (m, 4H), 1.52–1.39 (m, 4H), 1.33–1.25 (m, 1H). MS (EI) m/z (%): 277.25 (M+, 74), 246.15 (56), 222.15 (100). Analysis calculated for C18H19N3: C, 77.95; H, 6.90; N, 15.15. Found: C, 78.03; H, 6.96; N, 15.01.
2-amino-4-cyclohexyl-6-(2-fluorophenyl)pyridine-3-carbonitrile (9)
Purified by column chromatography (n-hexane-ethyl acetate 2:1) and then recrystallized from EtOH to give 0.186 g, 63% yield (95% purity by HPLC). MP 126–127 °C. 1H NMR (300 MHz, CDCl3), δ (ppm) 7.89 (td, J = 7.8, 1.9 Hz, 1H), 7.47–7.31 (m, 1H), 7.25–7.03 (m, 3H), 5.18 (s, 2H), 2.98–2.67 (m, 1H), 1.99–1.73 (m, 5H), 1.53–1.16 (m, 5H). MS (EI) m/z (%): 295.15 (M+, 98.05), 263.05 (23.28), 251.00 (12), 240.00 (100). Analysis calculated for C18H18FN3: C, 73.20; H, 6.14; F, 6.43; N, 14.23. Found: C, 73.22; H, 6.17; F, 6.44; N, 14.25.
2-amino-4-cyclohexyl-6-(2-methylphenyl)pyridine-3-carbonitrile (10)
Purified by column chromatography (n-hexane-ethyl acetate 2:1) and then recrystallized from EtOH to give 0.236 g, 81% yield (97% purity by HPLC). MP 120–121 °C. 1H NMR (300 MHz, CDCl3), δ (ppm) 7.73–7.10 (m, 4H), 6.71 (s, 1H), 5.20 (s, 2H), 2.95–2.77 (m, 1H), 2.35 (s, 3H), 2.01–1.69 (m, 5H), 1.56–1.34 (m, 4H), 1.34–1.18 (m, 1H). MS (EI) m/z (%): 291.14 (M+, 100), 236.12 (48), 208.10 (91.7). Analysis calculated for C19H21N3: C, 78.32; H, 7.26; N, 14.42. Found: C, 78.48; H, 7.18; N, 14.34.
2-amino-4-cyclohexyl-6-(thiophen-2-yl)pyridine-3-carbonitrile (11)
Purified by column chromatography (n-hexane-ethyl acetate 2:1) and then recrystallized from EtOH to give 0.167 g, 59% yield (98% purity by HPLC). MP 160–162 °C. 1H NMR (300 MHz, CDCl3), δ(ppm) 7.63–7.62(m, 1H), 7.44 (d, J = 4.5 Hz, 1H), 7.12–7.09 (m, 1H), 6.96 (s, 1H), 5.14 (s, 2H), 2.82–2.79 (m, 1H), 1.90–1.78 (m, 5H), 1.55–1.43 (m, 4H), 1.30–1.19 (m, 1H). MS (EI) m/z (%): 283.04 (M+, 100), 251.99 (19), 228.02 (92). Analysis calculated for C16H17N3S: C, 67.81; H, 6.05; N, 14.83; S, 11.31. Found: C, 67.89; H, 6.13; N, 14.77; S, 11.21.
2-amino-4-cyclohexyl-6-(thiophen-3-yl)pyridine-3-carbonitrile (12)
Purified by column chromatography (n-hexane-ethyl acetate 2:1) and then recrystallized from EtOH to give 0.147 g, 52% yield (96% purity by HPLC). MP 145–146 °C. 1H NMR (300 MHz, CDCl3), δ(ppm) 7.94 (dd, J = 3.0, 1.3 Hz, 1H), 7.59 (dd, J = 5.1, 1.3 Hz, 1H), 7.38 (dd, J = 5.1, 3.0 Hz, 1H), 6.93 (s, 1H), 5.14 (s, 2H), 2.95–2.73 (m, 1H), 2.06–1.73 (m, 5H), 1.56–1.37 (m, 4H), 1.38–1.19 (m, 1H). MS (EI) m/z (%):(%): 283.07 (M+, 100), 228.04 (93), 214.96 (52).Analysis calculated for C16H17N3S: C, 67.81; H, 6.05; N, 14.83; S, 11.31. Found: C, 67.91; H, 6.09; N, 14.67; S, 11.33.
2-amino-4-cyclohexyl-6-(furan-2-yl)pyridine-3-carbonitrile (13)
Purified by column chromatography (n-hexane-ethyl acetate 2:1) and then recrystallized from EtOH to give 0.174 g, 65% yield (98% purity by HPLC). MP 177–178 °C. 1H NMR (300 MHz, CDCl3), δ(ppm) 7.55 (dd, J = 1.7, 0.8 Hz, 1H), 7.06 (dd, J = 3.4, 0.8 Hz, 1H), 7.03 (s, 1H), 6.54 (dd, J = 3.5, 1.8 Hz, 1H), 5.15 (s, 2H), 3.01–2.68 (m, 1H), 2.04–1.74 (m, 5H), 1.55–1.39 (m, 4H), 1.34–1.20 (m, 1H). MS (EI) m/z (%): 267.11 (M+, 100), 212.02 (69). Analysis calculated for C16H17N3O: C, 71.89; H, 6.41; N, 15.72; O, 5.98. Found: C, 71.91; H, 6.43; N, 15.71.
2-amino-6-(2-fluorophenyl)-4-(4-methoxyphenyl)pyridine-3-carbonitrile (14)
Purified by column chromatography (n-hexane-ethyl acetate 2:1) and then recrystallized from EtOH to give 0.188 g, 59% yield (97% purity by HPLC). MP 180–181 °C. 1H NMR (300 MHz, CDCl3), δ (ppm) 7.96 (td, J1 = 7.8, J2 = 1.9 Hz, 1H), 7.65–7.58 (m, 2H), 7.47–7.37 (m, 1H), 7.31–7.23 (m, 2H), 7.23–7.09 (m, 1H), 7.09–6.98 (m, 2H), 5.32 (s, 2H), 3.88 (s, 3H). MS (EI) m/z (%): 319.12 (M+, 100), 304.18 (12), 249.13 (16). Analysis calculated for C19H14FN3O: C, 71.46; H, 4.42; F, 5.95; N, 13.16; O, 5.01. Found: C, 71.48; H, 4.44; F, 5.97; O, 5.05.
2-amino-4-(4-methoxyphenyl)-6-(2-methylphenyl)pyridine-3-carbonitrile (15)
Purified by column chromatography (n-hexane-ethyl acetate 2:1) and then recrystallized from EtOH to give 0.205 g, 65% yield (95% purity by HPLC). MP 151–152 °C. 1H NMR (300 MHz, CDCl3), δ (ppm) 7.61 (d, J = 8.3 Hz, 2H), 7.40 (d, J = 7.3 Hz, 1H), 7.37–7.27 (m, 3H), 7.03 (d, J = 8.2 Hz, 2H), 6.86 (s, 1H), 5.32 (s, 2H), 3.87 (s, 3H), 2.42 (s, 3H). MS (EI) m/z (%): 314.10 (M+, 100), 271.06 (7), 208.11 (52). Analysis calculated for C20H17N3O: C, 76.17; H, 5.43; N, 13.32; O, 5.07. Found: C, 76.31; H, 5.33; N, 13.52; O, 4.84.
2-amino-6-(furan-2-yl)-4-(4-methoxyphenyl)pyridine-3-carbonitrile (16)
Purified by column chromatography (n-hexane-ethyl acetate 2:1) and then recrystallized from EtOH to give 0.198 g, 68% yield (99% purity by HPLC). MP 205–207 °C. 1H NMR (300 MHz, CDCl3) δ (ppm): 7.65–7.54 (m, 3H), 7.16 (s, 1H), 7.11 (d, J = 3.5 Hz, 1H), 7.03 (d, J = 8.8 Hz, 2H), 6.62–6.51 (m, 1H), 5.30 (s, 2H), 3.88 (s, 3H). MS (EI) m/z (%): 291.12 (M+, 100), 145.63 (5). Analysis calculated for C17H13N3O2: C, 70.09; H, 4.50; N, 14.42; O, 10.98. Found: C, 70.21; H, 4.38; N, 14.68, O, 10.73.
2-amino-6-(4-hydroxyphenyl)-4-(4-methoxyphenyl)pyridine-3-carbonitrile (17)
Purified by column chromatography (n-hexane- ethyl acetate 2:1) and then recrystallized from EtOH to give 0.222 g, 70% yield (99% purity by HPLC). MP 248–250 °C. 1H NMR (300 MHz, CDCl3), δ (ppm) 9.89 (s, 1H), 7.98 (d, J = 8.7 Hz, 2H), 7.61 (d, J = 8.7 Hz, 2H), 7.11–7.06 (m, 3H), 6.84–6.81 (m, 4H), 3.82 (s, 3H). MS (EI) m/z (%): 317.17 (M+, 100), 302.04 (6), 158.50 (14). Analysis calculated for C19H15N3O2: C, 71.91; H, 4.76; N, 13.24; O, 10.08. Found: C, 71.94; H, 4.79; N, 13.25; O, 10.11.
2-amino-4,6-bis(2-fluorophenyl)pyridine-3-carbonitrile (18)
Purified by column chromatography (n-hexane-ethyl acetate 2:1) and then recrystallized from EtOH to give 0.219 g, 73% yield (98% purity by HPLC). MP 180–181 °C. 1H NMR (300 MHz, CDCl3), δ (ppm) 8.05–7.90 (m, 1H), 7.56–7.41 (m, 2H), 7.33–7.06 (m, 6H), 5.34 (s, 2H). MS (EI) m/z (%): 307.06 (M+, 100), 279.99 (8). Analysis calculated for C18H11F2N3: C, 70.35; H, 3.61; F, 12.36, N, 13.67. Found: C, 70.37; H, 3.63; F, 12.33; N, 13.66.
2-amino-6-(2-fluorophenyl)-4-(2-methoxyphenyl)pyridine-3-carbonitrile (19)
Purified by column chromatography (n-hexane-ethyl acetate 2:1) and then recrystallized from EtOH to give 0.245 g, 78% yield (97% purity by HPLC). MP 187–188 °C. 1H NMR (300 MHz, CDCl3), δ (ppm) 7.97 (td, J = 7.8, 1.9 Hz, 1H), 7.52–7.35 (m, 2H), 7.31 (td, J = 7.2, 1.5 Hz, 1H), 7.26–7.19 (m, 2H), 7.17–6.95 (m, 3H), 5.27 (s, 2H), 3.88 (s, 3H). MS (EI) m/z (%): 319.12 (M+, 100), 290.14 (7), 138.01 (14). Analysis calculated for C19H14N3FO: C, 71.46; H, 4.42; F, 5.95; N, 13.16; O, 5.01. Found: C, 71.44; H, 4.43; F, 5.92; O, 5.04.
2-amino-4-(2-methoxyphenyl)-6-(2-methylphenyl)pyridine-3-carbonitrile (20)
Purified by column chromatography (n-hexane-ethyl acetate 2:1) and then recrystallized from EtOH to give 0.186 g, 64% yield (98% purity by HPLC). MP 181–183 °C. 1H NMR (300 MHz, CDCl3) δ (ppm): 7.47–7.40 (m, 2H), 7.32–7.28 (m, 4H), 7.09–7.02 (m, 2H), 6.86 (s, 1H), 5.29 (s, 2H), 3.88 (s, 3H), 2.43 (s, 3H). MS (EI) m/z (%): 315.13 (M+, 100), 298.16 (12), 284.09 (18), 208.10 (81.6). Analysis calculated for C20H17N3O: C, 76.17; H, 5.43; N, 13.32; O, 5.07. Found: C, 76.19; H, 5.41; N, 13.36; O, 5.03.
2-amino-6-(furan-2-yl)-4-(2-methoxyphenyl)pyridine-3-carbonitrile (21)
Purified by column chromatography (n-hexane-ethyl acetate 2:1) and then recrystallized from EtOH to give 0.244 g, 77% yield (96% purity by HPLC). MP 187–188 °C. 1H NMR (300 MHz, CDCl3), δ (ppm): 7.55 (s, 1H), 7.44 (t, J = 8.1 Hz, 1H), 7.30 (dd, J = 7.4, 1.7 Hz, 1H), 7.15–6.98 (m, 4H), 6.54 (dd, J = 3.3, 1.7 Hz, 1H), 5.24 (s, 2H), 3.87 (s, 3H). MS (EI) m/z (%): 291.10 (M+, 100), 262.14 (10). Analysis calculated for C17H13N3O2: C, 70.09; H, 4.50; N, 14.42; O, 10.98. Found: C, 70.11; H, 4.51; N, 14.41; O, 11.01.
2-amino-6-(4-hydroxyphenyl)-4-(2-methoxyphenyl)pyridine-3-carbonitrile (22)
Purified by column chromatography (n-hexane-ethyl acetate 2:1) and then recrystallized from EtOH to give 0.193 g, 60% yield (96% purity by HPLC). MP 210–212 °C. 1H NMR (300 MHz, DMSO-d 6 ), δ (ppm): 9.91 (s, 1H), 7.93 (d, J = 9.0 Hz, 2H), 7.45 (t, J = 7.8 Hz, 1H), 7.29 (dd, J = 7.4, 1.7 Hz, 1H), 7.16 (d, J = 8.3 Hz, 1H), 7.07 (d, J = 7.5 Hz, 1H), 7.03 (s, 1H), 6.82 (d, J = 8.9 Hz, 2H), 6.77 (s, 2H), 3.77 (s, 3H). MS (EI) m/z (%): 317.13 (M+, 100), 300.09 (8), 286.11 (6).Analysis calculated for C19H15N3O2: C, 71.91; H, 4.76; Cl, 13.24; O, 10.08. Found: C, 71.92; H, 4.74; Cl, 13.27; O, 10.05.
2-amino-4-(2-chlorophenyl)-6-(4-hydroxyphenyl)pyridine-3-carbonitrile (23)
Purified by column chromatography (n-hexane-ethyl acetate 2:1) and then recrystallized from EtOH to give 0.179 g, 59% yield (98% purity by HPLC). MP 215–217 °C. 1H NMR (300 MHz, DMSO-d 6 ), δ (ppm): 9.90 (s, 1H), 8.16–7.22 (m, 2H), 7.69–7.30 (m, 4H), 7.16–6.50 (m, 5H). MS (EI) m/z (%): 320.99 (M+, 100), 286.04 (5). Analysis calculated for C18H12ClN3O: C, 67.19; H, 3.76; Cl, 11.02; N, 13.06; O, 4.97. Found: C, 67.37; H, 3.94; Cl, 11.18; N, 12.88; O, 4.63.
2-amino-4,6-bis(4-hydroxyphenyl)pyridine-3-carbo-nitrile (24)
Purified by column chromatography (n-hexane–ethyl acetate 2:1) and then recrystallized from EtOH to give 0.151 g, 53% yield (97% purity by HPLC). MP 299–300 °C. 1H NMR (300 MHz, DMSO-d 6 ), δ (ppm) 9.92 (s, 2H), 8.19–7.79 (m, 2H), 7.68–7.37 (m, 2H), 7.42–6.99 (m, 1H), 7.01–6.62 (m, 6H). MS (EI) m/z (%): 303.06 (M+, 100), 184.01 (6). Analysis calculated for C18H13N3O2: C, 71.28; H, 4.32; N, 13.85; O, 10.55. Found: C, 71.40; H, 4.54; N, 13.75; O, 10.31.
2-amino-4-(furan-2-yl)-6-(thiophen-3-yl)pyridine-3-carbonitrile (25)
Purified by column chromatography (n-hexane-ethyl acetate 2:1) and then recrystallized from EtOH to give 0.123 g, 46% yield (95% purity by HPLC). MP 156–157 °C. 1H NMR (300 MHz, CDCl3) δ(ppm): 8.01 (dd, J = 3.0, 1.2 Hz, 1H), 7.66 (dd, J = 5.1, 1.2 Hz, 1H), 7.62 (dd, J = 1.8, 0.6 Hz, 1H), 7.48 (dd, J = 3.6, 0.6 Hz, 1H), 7.45 (s, 1H), 7.40 (dd, J = 5.1, 3.0 Hz, 1H), 7.40 (dd, J = 5.1, 3.0 Hz, 1H), 6.61 (dd, J = 3.6, 1.8 Hz, 1H), 5.26 (s, 2H). MS (EI) m/z (%): 267.06 (M+, 100), 237.98 (6), 210.99 (7). Analysis calculated for C14H9N3OS: C, 62.91; H, 3.39; N, 15.72; O, 5.99; S, 11.99. Found: C, 63.11; H, 3.47; N, 15.58; O, 5.97; S, 11.87.
Pharmacological evaluation of novel 4,6-substituted 2-amino-pyridin-3-carbonitriles
Pharmacological evaluation was performed in a radioligand binding competition assay, using A1, A2A, A2B, and A3 human receptors expressed in transfected CHO (A1), HeLa (A2A and A3), and HEK-293 (A2B) according to the procedure reported by Bosch et al. [78].
The activity measurements against the phosphodiesterases PDE7A, PDE7B, PDE9A and PDE10A were performed using AD293 cells that were transiently and separately transfected with human PDE7A, PDE7B, PDE9A, and PDE10A following the procedure described by Shipe et al. [43]. The IC50 values were obtained by fitting the data with non-linear regression using Prism 2.1 software (GraphPad, San Diego, CA) [79], and the reported results are the mean of 3 experiments (n = 3) each performed in duplicate.
Abbreviations
- AR:
-
adenosine receptor
- A1R:
-
A1 adenosine receptor
- A2AR:
-
A2A adenosine receptor
- A2BR:
-
A2B adenosine receptor
- A3R:
-
A3 adenosine receptor
- PDE10A:
-
cAMP and cAMP-inhibited cGMP 3′,5′-cyclic phosphodiesterase 10A
- cAMP:
-
cyclic adenosine monophosphate
- PDE7A:
-
high affinity cAMP-specific 3′,5′-cyclic phosphodiesterase 7A
- PDE7B:
-
cAMP-specific 3′,5′-cyclic phosphodiesterase 7B
- PDE9A:
-
high affinity cGMP-specific 3′,5′-cyclic phosphodiesterase 9A
- PDGFR:
-
platelet-derived growth factor receptor
- VEGFR:
-
vascular endothelial growth factor receptor
- ABL1:
-
tyrosine-protein kinase ABL1
- SRC:
-
proto-oncogene tyrosine-protein kinase Src
- EGFR:
-
epidermal growth factor receptor
- HER2:
-
receptor tyrosine-protein kinase erbB-2
- MAO-A:
-
amine oxidase [flavin-containing] A
- MAO-B:
-
amine oxidase [flavin-containing] B
- AChE:
-
acetylcholinesterase
- BuChE:
-
butyrylcholinesterase
- H3-R:
-
histamine H3 receptor
- HMT:
-
histone methyltransferases
- hCB2R:
-
human cannabinoid receptor 2
- EtOH:
-
ethanol
References
Kovacs GG (2014) Current concepts of neurodegenrative diseases. Eur Med J 1:78–86
Lang AE, Obeso JA (2004) Personal view challenges in Parkinson’s disease: restoration of the nigrostriatal dopamine system is not enough. Lancet Neurol 3:309–316
Latini S, Pedata F (2001) Adenosine in the central nervous system: release mechanisms and extracellular concentrations. J Neurochem 79:463–484
Shook BC, Rassnick S, Wallace N, Crooke J, Ault M, Chakravarty D, Barbay JK, Wang A, Powell MT, Leonard K, Alford V, Scannevin RH, Carroll K, Lampron L, Westover L, Lim H, Russell R, Branum S, Wells KM, Damon S, Youells S, Li X, Beauchamp DA, Rhodes K, Jackson PF (2012) Design and characterization of optimized adenosine A2A/A1 receptor antagonists for the treatment of Parkinson’s disease. J Med Chem 55:1402–1417
Li M, Wang X, Meintzer MKAY, Laessig T, Birnbaum MJ, Heidenreich KIMA (2000) Cyclic AMP promotes neuronal survival by phosphorylation of glycogen synthase kinase 3 β. Mol Cell Biol 20:9356–9363
Schmidt CJ, Chapin DS, Cianfrogna J, Corman ML, Hajos M, Harms JF, Hoffman WE, Lebel LA, McCarthy SA, Nelson FR, Proulx-LaFrance C, Majchrzak MJ, Ramirez AD, Schmidt K, Seymour PA, Siuciak JA, Tingley FD, Williams RD, Verhoest PR, Menniti FS (2008) Preclinical characterization of selective phosphodiesterase 10A inhibitors: a new therapeutic approach to the treatment of schizophrenia. J Pharmacol Exp Ther 325:681–690
Giampà C, Patassini S, Borreca A, Laurenti D, Marullo F, Bernardi G, Menniti FS, Fusco FR (2009) Phosphodiesterase 10 inhibition reduces striatal excitotoxicity in the quinolinic acid model of Huntington’s disease. Neurobiol Dis 34:450–456
Niccolini F, Foltynie T, Reis Marques T, Muhlert N, Tziortzi AC, Searle GE, Natesan S, Kapur S, Rabiner EA, Gunn RN, Piccini P, Politis M (2015) Loss of phosphodiesterase 10A expression is associated with progression and severity in Parkinson’s disease. Brain 138:3003–3015
Stiles GL (1992) Adenosine receptors. JBiolChem 267:6451–6454
Rickles RJ, Pierce LT, Giordano TP, Tam WF, Mcmillin DW, Delmore J, Laubach JP, Borisy AA, Richardson PG, Lee MS (2010) Adenosine A2A receptor agonists and PDE inhibitors: a synergistic multitarget mechanism discovered through systematic combination screening in B-cell malignancies. Blood 116:593–603
Gomes CV, Kaster MP, Tomé AR, Agostinho PM, Cunha RA (2011) Adenosine receptors and brain diseases: neuroprotection and neurodegeneration. Biochim Biophys Acta 1808:1380–1399
Arooj M, Sakkiah S, Cao GP, Lee KW (2013) An innovative strategy for dual inhibitor design and its application in dual inhibition of human thymidylate synthase and dihydrofolate reductase enzymes. PLoS ONE 8:1–15
Morphy R, Kay C, Rankovic Z, Morphy R (2004) From magic bullets to designed multiple ligands. Drug Discov Today 9:641–651
Csermely P, Ágoston V, Pongor S (2005) The efficiency of multi-target drugs: the network approach might help drug design. Trends Pharmacol Sci 26:178–182
Khan M, Maryam A, Qazi JI, Ma T (2015) Targeting apoptosis and multiple signaling pathways with Icariside II in cancer cells. Int J Biol Sci 11:1100–1112
Amelio I, Landré V, Knight RA, Lisitsa A, Melino G, Antonov AV (2015) Polypharmacology of small molecules targeting the ubiquitin-proteasome and ubiquitin-like systems. Oncotarget 6:9646–9656
Peters J (2013) Polypharmacology—foe or friend? J Med Chem 56:8955–8971
Besnard J, Ruda GF, Setola V, Abecassis K, Ramona M, Huang X, Norval S, Sassano MF, Shin AI, Webster A, Simeons FRC, Stojanovski L, Prat A, Seidah NG, Constam DB, Bickerton GR, Read KD, Wetsel WC, Ian H, Roth BL, Hopkins AL (2012) Automated design of ligands to polypharmacological profiles. Nature 492:1–17
Ma XH, Shi Z, Tan C, Jiang Y, Go ML, Low BC, Chen YZ (2010) In-silico approaches to multi-target drug discovery computer aided multi-target drug design, multi-target virtual screening. Pharm Res 27:739–749
Huang S (2002) Rational drug discovery: what can we learn from regulatory networks? Drug Discov Today 7:163–169
Nikolic K, Mavridis L, Djikic T, Vucicevic J, Agbaba D, Yelekci K, Mitchell JBO (2016) Drug design for CNS diseases: polypharmacological profiling of compounds using cheminformatic, 3D-QSAR and virtual screening methodologies. Front Neurosci 10:1–21
Laura M, Cavalli A (2016) Multitarget drug discovery and polypharmacology. ChemMedChem 11:1190–1192
Koutsoukas A, Simms B, Kirchmair J, Bond PJ, Whitmore AV, Zimmer S, Young MP, Jenkins JL, Glick M, Glen RC, Bender A (2011) From in silico target prediction to multi-target drug design: current databases, methods and applications. J Proteomics 74:2554–2574
Mervin LH, Afzal AM, Drakakis G, Lewis R, Engkvist O, Bender A (2015) Target prediction utilising negative bioactivity data covering large chemical space. J Cheminform 7:1–16
Dolles D, Nimczick M, Scheiner M, Ramler J, Stadtmüller P, Sawatzky E, Drakopoulos A, Sotriffer C, Wittmann H, Strasser A, Decker M (2016) Aminobenzimidazoles and structural isomers as templates for dual-acting butyrylcholinesterase inhibitors and hCB2R ligands to combat neurodegenerative disorders. ChemMedChem 11:1270–1283
Molecular Operating Environment (MOE) (2013) Chemical Computing Group Inc., 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2016
Lewell XQ, Judd DB, Watson SP, Hann MM (1998) RECAP-retrosynthetic combinatorial analysis procedure: a powerful new technique for identifying privileged molecular fragments with useful applications in combinatorial chemistry. J Chem Inf Comput Sci 38:511–522
Weber L (2002) The application of multi-component reactions in drug discovery. Curr Med Chem 9:2085–2093
Musonda CC, Gut J, Rosenthal PJ, Yardley V, Carvalho de Souza RC, Chibale K (2006) Application of multicomponent reactions to antimalarial drug discovery. Part 2: new antiplasmodial and antitrypanosomal 4-aminoquinoline γ- and δ-lactams via a ‘catch and release’ protocol. Bioorganic Med Chem 14:5605–5615
Gaulton A, Bellis LJ, Bento AP, Chambers J, Davies M, Hersey A, Light Y, McGlinchey S, Michalovich D, Al-Lazikani B, Overington JP (2011) ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res 40:1–8
Liggi S, Drakakis G, Hendry AE, Hanson KM, Brewerton SC, Wheeler GN, Bodkin MJ, Evans DA, Bender A (2013) Extensions to in silico bioactivity predictions using pathway annotations and differential pharmacology analysis: application to xenopus laevis phenotypic readouts. Mol Inform 32:1009–1024
Jones E, Oliphant T, Peterson P (2001) SciPy: open source scientific tools for python. http://www.scipy.org/. Accessed 27 Feb 2017
Liu W, Chun E, Thompson AA, Chubukov P, Xu F, Han GW, Roth CB, Heitman LH, Ijzerman AP, Cherezov V, Stevens RC (2012) Structural basis for allosteric regulation of GPCRs by sodium ions. Science 337:232–236
Hu E, Kunz RK, Rumfelt S, Chen N, Bürli R, Li C, Andrews KL, Zhang J, Chmait S, Kogan J, Lindstrom M, Hitchcock SA, Treanor J (2012) Discovery of potent, selective, and metabolically stable 4-(pyridin-3-yl) cinnolines as novel phosphodiesterase 10A (PDE10A) inhibitors. Bioorg Med Chem Lett 22:2262–2265
Ch OS, Cn IC, Ali M, Kiafar M, Yarie M, Taherpour A (2016) Experimental and Theoretical Studies of the nanostructured {Fe3O4@SiO2@(CH2)3Im}C(CN)3 catalyst for 2-amino-3-cyanopyridine preparation via an anomeric based oxidation. RSC Adv 6:50100–50111
Mantri M, De Graaf O, Van Veldhoven J, Go A, Mulder-krieger T, Link R, De Vries H, Beukers MW, Brussee J, Ijzerman AP (2008) 2-amino-6-furan-2-yl-4-substituted nicotinonitriles as A2A adenosine receptor antagonists. J Med Chem 51:4449–4455
Khalili D (2016) Graphene oxide: a reusable and metal-free carbocatalyst for the one-pot synthesis of 2-amino-3-cyanopyridines in water. Tetrahedron Lett 57:1721–1723
Safari J, Hossein S, Dehghan S (2012) Ultrasound-promoted an efficient method for one-pot synthesis of 2-amino-4,6-diphenylnicotinonitriles in water: a rapid procedure without catalyst. Ultrason Sonochem 19:1061–1069
Baell JB, Holloway GA (2010) New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem 53:2719–2740
Lagorce D, Sperandio O, Baell JB, Miteva MA, Villoutreix BO (2015) FAF-Drugs3: a web server for compound property calculation and chemical library design. Nucleic Acids Res 43:W200–W207
Lingam VSP, Dahale Dnyaneshwar H, Rathi Vijay E, Shingote YB, Thakur RR, Mindhe AS, Kummari S, Khairatkar-joshi N, Bajpai M, Shah DM, Sapalya RS, Gullapalli S, Gupta PK, Gudi GS, Jadhav SB, Pattem R, Thomas A (2015) Design, synthesis, and pharmacological evaluation of 5,6- disubstituted pyridin-2(1H)-one derivatives as phosphodiesterase 10A (PDE10A) antagonists. J Med Chem 58:8292–8308
Abadi AH, Ibrahim TM, Abouzid KM, Lehmann J, Tinsley HN, Gary BD, Piazza GA (2009) Design, synthesis and biological evaluation of novel pyridine derivatives as anticancer agents and phosphodiesterase 3 inhibitors. Bioorg Med Chem 17:5974–5982
Shipe WD, Sharik SS, Barrow JC, Mcgaughey GB, Theberge CR, Uslaner JM, Yan Y, Renger JJ, Smith SM, Coleman PJ, Cox CD (2015) Discovery and optimization of a series of pyrimidine-based phosphodiesterase 10A (PDE10A) inhibitors through fragment screening, structure-based design, and parallel synthesis. J Med Chem 58:7888–7894
Meegalla SK, Huang H, Illig CR, Parks D, Chen J, Lee Y, Wilson K, Patel S, Cheung W, Lu T, Kirchner T, Askari H, Geisler J, Patch R, Gibbs A, Rady B, Connelly M, Player M (2016) Discovery of novel potent imidazo [1,2-b] pyridazine PDE10A inhibitors. Bioorg Med Chem Lett 26:4216–4222
Raheem IT, Schreier JD, Fuerst J, Gantert L, Hostetler E, Huszar S, Joshi A, Kandebo M, Kim S, Li J, Ma B, McGaughey G, Sharma S, Shipe W, Uslaner J, Vandeveer G, Yan Y, Renger J, Smith S, Coleman P, Cox C (2016) Discovery of pyrazolopyrimidine phosphodiesterase 10A inhibitors for the treatment of schizophrenia. Bioorg Med Chem Lett 26:126–132
Yoshikawa M, Hitaka T, Hasui T, Fushimi M, Kunitomo J, Kokubo H, Oki H, Nakashima K, Taniguchi T (2016) Design and synthesis of potent and selective pyridazin-4(1H)-one-based PDE10A inhibitors interacting with Tyr683 in the PDE10A selectivity pocket. Bioorg Med Chem 24:3447–3455
Malamas MS, Ni Y, Erdei J, Stange H, Schindler R, Lankau H, Grunwald C, Fan K, Parris K, Langen B, Egerland U, Hage T, Marquis K, Grauer S, Brennan J, Navarra R, Graf R, Harrison B, Robichaud A, Kronbach T, Pangalos M, Hoefgen N, Brandon N (2011) Highly potent, selective, and orally active phosphodiesterase 10A inhibitors. J Med Chem 54:7621–7638
Kuhn B, Guba W, Banner D, Bissantz C, Ceccarelli S, Haap W, Körner M, Kuglstatter A, Lerner C, Mattei P, Neidhart W, Pinard E, Rudolph M, Schulz-gasch T, Woltering T, Stahl M (2016) A real-world perspective on molecular design. J Med Chem 59:4087–4102
Jaakola V, Griffith M, Hanson M, Cherezov V, Chien E, Lane J, Ijzerman A, Stevens R (2008) The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science 322:1211–1218
Segala E, Guo D, Cheng RKY, Bortolato A, Deflorian F, Doré A, Errey J, Heitman L, Ijzerman A, Marshall F, Cooke R (2016) Controlling the dissociation of ligands from the adenosine A2A receptor through modulation of salt bridge strength. J Med Chem 59:6470–6479
Knight A, Hemmings JL, Win I, Leuenberger M, Frattini E, Frenguelli BG, Dowell SJ, Lochner M, Ladds G (2016) Discovery of novel adenosine receptor agonists that exhibit subtype selectivity. J Med Chem 59:947–964
Nguyen ATN, Baltos J, Thomas T, Nguyen T, Munoz L, Gregory K, White P, Sexton P, Christopoulos A, May L (2016) Extracellular loop 2 of the adenosine A1 receptor has a key role in orthosteric ligand affinity and agonist efficacy. Mol Pharmacol 90:703–714
Jeffrey P, Summer S (2010) Neurobiology of disease assessment of the blood–brain barrier in CNS drug discovery. Neurobiol Dis J 37:33–37
Sander T, Freyss J, von Korff M, Rufener C (2015) DataWarrior: an open-source program for chemistry aware data visualization and analysis. J Chem Inf Model 55:460–473
RDKit: Cheminformatics and Machine Learning Software (2013) http://www.rdkit.org. Accessed 27 Feb 2017
ChemAxon Standardizer. https://www.chemaxon.com/products/standardizer. Accessed 27 Feb 2017
Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL (2004) Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem 47:1750–1759
Madhavi Sastry G, Adzhigirey M, Day T, Annabhimoju R, Sherman W (2013) Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des 27:221–234
Yaziji V, Rodriguez D, Gutierrez-de-Terran H, Coelho A, Caamano O, Garcia-Mera X, Brea J, Loza MI, Cadavid MI, Sotelo E (2011) Pyrimidine derivatives as potent and selective A3 adenosine receptor antagonists. J Med Chem 54:457–471
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Iii CLB, Dixon JS, Moult J, Stevens RC (2009) Community-wide assessment of GPCR structure modelling and ligand docking: GPCR dock 2008. Nat Rev Drug Discov 8:455–463
Schrödinger Release 2016-4 (2016) LigPrep, Schrödinger, LLC, New York
Shelley JC, Cholleti A, Frye LL, Greenwood JR, Timlin MR, Uchimaya M (2007) Epik: a software program for pKa prediction and protonation state generation for drug-like molecules. J Comput Aided Mol Des 21:681–691
de Lera Ruiz M, Lim Y-H, Junying Z (2013) Adenosine A2A receptor as a drug discovery target. J Med Chem 57:3623–3650
Drabczyńska A, Yuzlenko O, Köse M, Paskaleva M, Schiedel AC, Karolak-Wojciechowska J, Handzlik J, Karcz T, Kuder K, Müller CE, Kieć-Kononowicz K (2011) Synthesis and biological activity of tricyclic cycloalkylimidazo-, pyrimido- and diazepinopurinediones. Eur J Med Chem 46:3590–3607
R Core Team (2016) R: A language and environment for statistical computing (version 3.2.4)
Khunnawutmanotham N, Sahakitpichan P (2015) Divergent total syntheses to azafluoranthene and dehydroaporphine alkaloids. Eur J Org Chem 28:6324–6332
Molina P, Alajarin M, Vidal A (1990) Synthesis of isoquinoline derivatives. J Org Chem 55:6140–6147
Wagner S, Scheunemann M, Dipper K, Egerland U, Hoefgen N, Steinbach J, Burst P (2016) Development of highly potent phosphodiesterase 10A (PDE10A) inhibitors: synthesis and in vitro evaluation of 1, 8-dipyridinyl- and 1-pyridinyl-substituted imidazo [1, 5-a] quinoxalines. Eur J Med Chem 107:97–108
Chen P, Doweyko AM, Norris D, Gu HH, Spergel SH, Das J, Moquin RV, Lin J, Wityak J, Iwanowicz EJ, Mcintyre KW, Shuster DJ, Behnia K, Chong S, De Fex H, Pang S, Pitt S, Shen DR, Thrall S, Stanley P, Kocy OR, Witmer MR, Kanner SB, Schieven GL, Barrish JC (2004) Imidazoquinoxaline Src-family kinase p56Lck inhibitors: SAR, QSAR, and the discovery of (S)-N-(2-chloro-6-methylphenyl)-2-(3-methyl-1-piperazinyl)Imidazo-[1,5-a]pyrido[3,2-e] pyrazin-6-amine (BMS-279700) as a Potent and Orally active inhibitor with excellent in vivo antiinflammatory activity. J Med Chem 47:4517–4529
Bartolome JM, De Diego SAA, Artola M, Delgado F, Delgado O, Mart CM, Pena MA, Tong HM, Van Gool M, Alonso JM, Fontana A, Macdonald GJ, Megens A, Langlois X, Somers M, Vanhoof G, Conde-ceide S (2015) Identification of a novel orally bioavailable phosphodiesterase 10A (PDE10A) inhibitor with efficacy in animal models of schizophrenia. J Med Chem 58:978–993
Malamas MS, Ni Y, Erdei J, Stange H, Schindler R, Lankau H, Grunwald C, Fan KY, Parris K, Langen B, Egerland U, Hage T, Marquis KL, Grauer S, Brennan J, Navarra R, Graf R, Harrison BL, Robichaud A (2011) Highly potent, selective, and orally active phosphodiesterase 10A inhibitors. J Med Chem 54:7621–7638
Neustadt BR, Hao J, Lindo N, Greenlee WJ, Stamford AW, Tulshian D, Ongini E, Hunter J, Monopoli A, Bertorelli R, Foster C, Arik L, Lachowicz J, Feng K (2007) Potent, selective, and orally active adenosine A2A receptor antagonists: arylpiperazine derivatives of Pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidines. Bioorg Med Chem Lett 17:1376–1380
Bolcato C, Cusan C, Pastorin G, Spalluto G, Cacciari B, Klotz KN, Morizzo E, Moro S (2008) Pyrazolo-triazolo-pyrimidines as adenosine receptor antagonists: effect of the N-5 bond type on the affinity and selectivity at the four adenosine receptor subtypes. Purinergic Signal 4:39–46
Kehler J, Ritzen A, Langgård M, Leth S, Farah MM, Bundgaard C, Tornby C, Nielsen J, Paul J (2011) Triazoloquinazolines as a novel class of phosphodiesterase 10A (PDE10A) inhibitors. Bioorg Med Chem Lett 21:3738–3742
Zadpour M, Behbahani FK (2015) Iron (III) phosphate as a green and reusable catalyst for the synthesis of 4,6-disubstituted 2-aminopyridine-3-carbonitriles. Monatsh Chem 146:1865–1869
Patoliya PU, Gohel VP, Purohit DM, Patolia VN (2015) Synthesis and biological evaluation of some new cyano pyridine derivatives. J Chem Pharm Res 7:182–186
Bosch MP, Campos F, Niubo I, Rosell G, Diaz JL, Brea J, Loza MI, Guerrero A (2004) Synthesis and biological activity of new potential agonists for the human adenosine A2A receptor. J Med Chem 47:4041–4053
Motulsky H, Christopoulos A (2004) Fitting models to biological data using linear and nonlinear regression. A practical guide to curve fitting
Authors’ contributions
LK and CV contributed equally to the work. LK developed the computational workflow for designing multi-target ligands at A1R, A2AR, and PDE10A and wrote this manuscript. CV validated experimentally the designed ligands. FS contributed in the validation of the docking models used, AZ helped in implementing Mann–Whitney test and LM in applying the Chi square test. AB and ES conceived the main theme on which the work was performed and ensured that the scientific aspect of the study was rationally valid. RG revised and edited the manuscript. All authors read and approved the final manuscript.
Acknowledgements
Dr. Hugo Gutiérrez-de-Terán is thanked for providing the A1R homology model for molecular docking studies.
Competing interests
The authors declared that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Funding sources
LK thanks the IDB Cambridge International Scholarship for support. This work was financially supported by the ERC Starting Grant to AB (No. 336159), the Consellería de Cultura, Educación e Ordenación Universitaria of the Galician Government: (Grant: GPC2014/03), Centro singular de investigación de Galicia accreditation 2016–2019 (ED431G/09) and the European Regional Development Fund (ERDF).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding authors
Additional files
Additional file 1.
Supplementary data describing substructural analysis of extracted ChEMBL compounds, statistical analysis of enriched target prediction of RECAP compounds, separation in medians of active/inactive docking score distributions for the docking models, computed logP and tPSA values and selectivity profiling data for compounds 1–25, docking parameters used, scripts for compound extraction from the ChEMBL database, computation of Mann–Whitney test and F1 scores.
Additional file 2.
Coordinates of the A1R homology model.
Additional file 3.
CSV file of computed F1 scores of the A1R docking model.
Additional file 4.
CSV file of computed F1 scores of the A2AR docking model.
Additional file 5.
CSV file of computed F1 scores of the PDE10A docking model.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Kalash, L., Val, C., Azuaje, J. et al. Computer-aided design of multi-target ligands at A1R, A2AR and PDE10A, key proteins in neurodegenerative diseases. J Cheminform 9, 67 (2017). https://doi.org/10.1186/s13321-017-0249-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13321-017-0249-4