
Abstract
Background
Neonatal diabetes mellitus with hyperglycemia during the first 6 months of life is a rare disorder that can occur in all races and societies.
Case presentation
In this study, we introduced an Iranian (Persian) 65-day-old patient with neonatal diabetes mellitus with novel homozygous mutation in the pancreatic and duodenal homeobox 1, PDX1, gene, which is also known as IPF1 gene, located in exon 2. This case was a newborn boy born in Vali-Asr Hospital, Tehran; he was diagnosed as having hyperglycemia on 28th day. Genetic analysis detected a homozygous mutation on PDX1 gene on chromosome 13. It is a novel homozygous mutation in the PDX1 gene (NM_000209.3), p.Phe167Val. This mutation was confirmed by Sanger sequencing. There was no evidence of agenesis of the pancreas.
Conclusions
We reported a case of neonatal diabetes mellitus due to novel homozygous mutation in the PDX1 gene without exocrine pancreas manifestations.
Similar content being viewed by others

Introduction
Neonatal diabetes mellitus (NDM) is a monogenic form of diabetes [1] that is characterized by hyperglycemia and the need for insulin treatment within the first 6 months of life [2]. Symptoms of NDM include frequent urination and dehydration. NDM can be diagnosed by finding elevated levels of glucose in blood or urine. In severe cases, the deficiency of insulin may cause the body to produce an excess of acid, resulting in a potentially life-threatening condition called ketoacidosis [3]. NDM is a rare condition occurring in only one in 100,000 to 500,000 live births [3].
Clinically, NDM can be divided into three subgroups: (i) transient NDM (TNDM) in which insulin secretion is spontaneously recovered by several months of age (at a median age of 3 month); (ii) permanent NDM (PNDM) requiring lifelong medication; and (iii) PNDM existing as part of a syndrome (syndromic NDM). This phenotypic classification is useful, as the most common causative genetic abnormalities differ by each subtype, although overlap exists [1, 4].
Recent molecular analysis of NDM identified at least 12 responsible genes: chromosome 6q24, KCNJ11, ABCC8, INS, FOXP3, GCK, IPF1 which is also known as pancreatic and duodenal homeobox 1 (PDX1), PTF1A, EIF2AK3, GLUT2, HNF1β, and GLIS3 [4]. Defects on chromosome 6q24 (approximately 70%) and the KCNJ11 mutation have been recognized as the major causes of TNDM and PNDM, respectively, in Caucasians imprinting [1]. Early diagnosis as well as early insulin therapy initiation prevents the metabolic catastrophe of ketoacidosis and development of chronic and irreversible complications. Determination of clinical and molecular characteristics of this disorder helps to manage the short-term and long-term treatment, and, finally, it provides new achievements for future research. In this study, for the first time, we reported the case of an Iranian patient with NDM with novel homozygous mutation in PDX1 (IPF1) gene located in exon 2.
Case presentation
Clinical manifestations
Our patient was born by caesarean section in Vali-Asr Hospital, Tehran. He was an Iranian 65-day-old boy and was the second child of consanguineous healthy parents. It is noteworthy that his mother had a history of gestational diabetes and his parents’ relatives had diabetes mellitus type 2 history too (second-degree relatives). He was born at 38 weeks of gestation with birth weight of 1800 g (< third percentile) and length 46 cm (< third percentile). His blood pressure was 57–89.7 mmHg with failure to thrive (FTT). This newborn had normal anterior fontanelle, soft abdomen, and undescended testis. Other clinical examinations revealed heart rate (HR) 190 beats per minute, Saturation of Peripheral Oxygen (spO2) 5 minutes 87%, without respiratory distress syndrome (RDS).
Laboratory diagnostic methods
A drop of capillary blood (drawn from the heel) was sampled and applied to a test strip (Gluco Easy; Kyunggi, South Korea) to measure level of glucose. Before sampling, the baby’s heel was warmed up by hand massage followed by disinfecting the spot, and then a blood sample was taken.
Two ml of arterial blood was taken by a trained nurse. White blood cell (WBC) were counted with automated counters by Wright or May–Grünwald–Giemsa technique. Hemoglobin (HGB) and platelet (PLT) determinations were performed by an automated cell counter too. C-reactive protein (CRP) was measured in ethylenediaminetetraacetic acid (EDTA)-blood samples by a rapid immunometric method. A standard enzymatic test was used to measure alanine aminotransferase (ALT) and aspartate aminotransferase (AST). Bilirubin was measured by chromatography and amylase was measured by photometry (α-Amylase Kit).
Laboratory and imaging findings
An ultrasound of our patient revealed duodenal atresia, fatty liver, and normal spleen and pancreas. Echocardiography showed atrial septal defects (ASDs). Ultrasound of his brain revealed germinal matrix hemorrhage (GMH); electroencephalography was abnormal.
A magnetic resonance imaging (MRI) study demonstrated hypogenesis of the corpus callosum. A lumbar puncture culture was negative.
Blood examination on the first day (day of birth) showed disseminated intravascular coagulation (DIC), anemia (Hb, 6/4), metabolic acidosis, thrombocytopenia (PLT, 116), hyperbilirubinemia, and neonatal cholestasis. The laboratory findings after the first 24 hours of birth are presented in Table 1.
Genetic findings
Genetic analysis detected a homozygous mutation on PDX1 gene on chromosome 13: 27,920,020-27,926,231 (c.499 T>G) (Fig. 1) (Additional file 1: Figures S1 and S2). (GRCh38): 13:27,919,981-27,926,313 with cytogenetic location: 13q12.2 and genomic coordinates (GRCh38): 13:27,919,981-27,926,313.

The sequenced data showing mutation in c.499 T>G of PDX1 gene
This transcript has two exons, is annotated with 18 domains and features, and associated with 214 variations and maps to 38 oligo probes. This gene is a member of the human consensus coding sequence (CCDS) set. It is a novel homozygous mutation in the PDX1 (IPF1) gene located in exon 2 (NM_000209.3), p.Phe167Val. This mutation was confirmed by Sanger sequencing. Some PDX1 mutations and their phenotypes shown in Table 2.
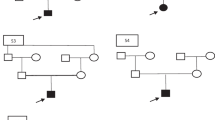
Pedigree analyses demonstrated diabetes mellitus type 2 in our patient’s mother and some maternal relatives.
Follow-up and treatment
On the 28th day, he was diagnosed as having hyperglycemia (serum glucose level was 496 mg/dL) by blood examination and treatment with regular daily insulin injections (1.0 unit/kg per day) was started immediately. The insulin infusion rates were regularly titrated according to pre-prandial blood sugar levels; the type of lactation was breast with fortified formula. Weight and height trend in relation to initiation and follow-up of insulin injection is presented in Table 3. Glibenclamide was added later (at 47 days of age) and stopped at the age of 60 days. However, the baby continued to get regular isophane insulin (NPH).
Other treatment operations included infant oxygen hood (oxyhood), nasal continuous positive airway pressure (nCPAP), broad-spectrum antibiotics prescription, granulocyte colony-stimulating factor (G-CSF), human intravenous immunoglobulin (IVIG), fresh frozen plasma (FFP), cryoprecipitate, surgery of duodenum atresia (at 28 days of age), phototherapy, ursodeoxycholic acid (UDCA), fat-soluble vitamins prescription, and total parenteral nutrition (TPN) for 65 days. He was discharged at 65-days old against medical advice, transferred to the city of residence, and was supervised by a gynecologist.
A summary of relevant past interventions and their outcomes is presented in Table 1.
Discussion and conclusions
Here, we report a syndromic case of NDM with mutation in PDX1 gene. This was a novel homozygous mutation, p.Phe167Val, is located in the gene on chromosome 13 (c.499 T>G). His mother had suffered from gestational diabetes and some second-degree relatives had diabetes mellitus type 2. There was no evidence of agenesis of the pancreas. Various single gene and chromosomal abnormalities have been identified that cause different manifestations of NDM [4].
NDM is permanent in nearly half of the patients and may be caused by mutations affecting genes that play a critical role in cell development, cell survival, or cell function. Recently, monogenic causes were recognized in 50% of cases of permanent insulin-dependent diabetes occurring before the age of 6 months [5]. Syndromic NDM is most commonly a result of mutations in FOXP3, EIF2AK3, PTF1A GLIS3, NEUROD1, HNF1B, immunodysregulation polyendocrinopathy enteropathy X-linked (IPEX) syndrome, and PDX1. PDX1 also termed IPF1 encodes insulin promoter factor1 with additional features of agenesis of the pancreas [4].
PDX1 has been reported as a main factor in pancreas development and function [6, 7]. Mutations in PDX1 may be involved in several disorders, including agenesis of the pancreas or congenital pancreatic hypoplasia (PAGEN1; Online Mendelian Inheritance in Man 260370) and diabetes mellitus (for example, Online Mendelian Inheritance in Man 222100; MODY, Type 1, Online Mendelian Inheritance in Man 125850) [8]. Clearly, if a patient presents with features consistent with a syndrome, testing for the relevant mutation should be carried out first. However, if negative, testing for the most prevalent causes in descending order should be carried out. With the exception of IPEX and HNF1B-related diabetes, the syndromic causes of NDM are autosomal recessive, carrying a 25% recurrence risk in subsequent children. A correct diagnosis that allows the proper treatment to be selected should lead to better glucose control and improved health in the long term. Testing of other family members may also be indicated to determine whether they are at risk for diabetes. According to studies, the same defect that causes PNDM or TNDM can be present in parents or other first-degree relatives and be diagnosed as type 1 diabetes, monogenic diabetes of youth, or type 2 diabetes mellitus as subsequently detailed. Current evidence suggests that the mutation is likely to be pathogenic. The first cases (three patients from two families) with biallelic PDX1 mutations that had complete pancreatic agenesis were found by Nicolino et al. (2010) [9]. Later, De Franco et al. (2013) identified three cases with permanent neonatal diabetes (2.9%). One proband and his affected brother were compound heterozygotes for a frameshift and a novel missense mutation (p.A34fsX191; c.98dupC and p.P87L; c.260C>T). The other two probands were homozygous for novel PDX1 missense mutations (p.A152G; c.455C>G and p.R176Q; c.527G>A) [5].
This study reported a novel mutation related to NDM with mutation in PDX1 gene. Mutation in this gene is rare but detection of it is vital. Correctly distinguishing monogenic NDM from type 1 diabetes presenting in infancy critically impacts treatment decisions, surveillance of complications and associated conditions, and has important genetic implications for siblings and offspring of affected individuals.
Conclusion
The PDX1 gene in mutation screening for syndromic NDM is introduced as a genetic diagnosis even in the absence of pancreas appearances.
Availability of data and materials
All data generated or analyzed during this study are included in this published article. Data sets will be available if requested.
Abbreviations
- Hb:
-
Hemoglobin
- MODY:
-
Maturity-onset diabetes of the young
- NDM:
-
Neonatal diabetes mellitus
- PDX1 :
-
Pancreatic and duodenal homeobox 1
- PLT:
-
Platelet
- PNDM:
-
Permanent neonatal diabetes mellitus
- TNDM:
-
Transient neonatal diabetes mellitus
References
Nagashima K, Tanaka D, Inagaki N. Epidemiology, clinical characteristics, and genetic etiology of neonatal diabetes in Japan. Pediatr Int. 2017;59(2):129–33.
Takagi M, Takeda R, Yagi H, Ariyasu D, Fukuzawa R, Hasegawa T. Case of transient neonatal diabetes due to a novel mutation in ABCC8. Clin Pediatr Endocrinol. 2016;25(4):139–41.
Monogenic Forms of Diabetes: Neonatal Diabetes Mellitus and Maturity-onset Diabetes of the Young. U.S. Department of Health and Human Services. National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), NIH Publication No. 07–6141 March 2007. Accessed in https://www.niddk.nih.gov/health-information/diabetes/overview/what-is-diabetes/monogenic-neonatal-mellitus-mody
Naylor RN, Greeley CAW, Bell GI, Philipson LH. Genetics and pathophysiology of neonatal diabetes mellitus. J Diabetes Investig. 2011;2(3):158–69.
De Franco E, Shaw-Smith C, Flanagan SE, Edghill EL, Wolf J, Otte V, Ebinger F, Varthakavi P, Vasanthi T, Edvardsson S, Hattersley AT, Ellard S. Biallelic PDX1 (insulin promoter factor1) mutations causing neonatal diabetes without exocrine pancreatic insufficiency. Diabet Med. 2013;30(5):e197–200.
Babu DA, Deering TG, Mirmira RG. A feat of metabolic proportions: Pdx1 orchestrates islet development and function in the maintenance of glucose homeostasis. Mol Genet Metab. 2007;92:43–55.
Oliver-Krasinski JM, Stoffers DA. On the origin of the beta cell. Genes Dev. 2008;22:1998–2021.
OMIM. Pancreas/duodenum homeobox protein 1; PDX1. https://www.omim.org/entry/600733.
Nicolino M, Claiborn KC, Senée V, Boland A, Stoffers DA, Julier C. A novel hypomorphic PDX1 mutation responsible for permanent neonatal diabetes with subclinical exocrine deficiency. Diabetes. 2010;59(3):733–40.
Acknowledgements
We thank Dr Vafa Ghorban Sabbagh from the Maternal-Fetal and Neonatal Research Center, Tehran University of Medical Science for cooperation.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
MA and LS carried out the molecular genetic studies, participated in the sequence alignment and drafted the manuscript.HD, NN, EA, TE and NF have collaborated in care of the patient throughout hospitalization, treatment and follow-up. LS and HD participated in the design of the study and performed the statistical analysis. NF,NN,EA and TE conceived of the study and participated in its design and coordination. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Institutional Ethical Board Committee at Tehran university of medical sciences; R.TUMS.IKHC.REC.1396.4166.
Consent for publication
Written informed consent was obtained from the patient’s legal guardians for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Figures S1 and S2: Molecular genetic laboratory reports. (ZIP 2090 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Sahebi, L., Niknafs, N., Dalili, H. et al. Iranian neonatal diabetes mellitus due to mutation in PDX1 gene: a case report. J Med Case Reports 13, 258 (2019). https://doi.org/10.1186/s13256-019-2149-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13256-019-2149-x