
Abstract
Background
Plasma amyloid-β (Aβ) levels are increasingly studied as a potential accessible marker of cognitive impairment and dementia. However, it remains underexplored whether plasma Aβ levels including the novel Aβ peptide 1–38 (Aβ1–38) relate to preclinical markers of neurodegeneration and risk of dementia. We investigated the association of plasma Aβ1–38, Aβ1–40, and Aβ1–42 levels with imaging markers of neurodegeneration and risk of dementia in a prospective population-based study.
Methods
We analyzed plasma Aβ levels in 458 individuals from the Rotterdam Study. Brain volumes, including gray matter, white matter, and hippocampus, were computed on the basis of 1.5-T magnetic resonance imaging (MRI). Dementia and its subtypes were defined on the basis of internationally accepted criteria.
Results
A total of 458 individuals (mean age, 67.8 ± 7.7 yr; 232 [50.7%] women) with baseline MRI scans and incident dementia were included. The mean ± SD values of Aβ1–38, Aβ1–40, and Aβ1–42 (in pg/ml) were 19.4 ± 4.3, 186.1 ± 35.9, and 56.3 ± 6.2, respectively, at baseline. Lower plasma Aβ1–42 levels were associated with smaller hippocampal volume (mean difference in hippocampal volume per SD decrease in Aβ1–42 levels, − 0.13; 95% CI, − 0.23 to − 0.04; p = 0.007). After a mean follow-up of 14.8 years (SD, 4.9; range, 4.1–23.5 yr), 79 persons developed dementia, 64 of whom were diagnosed with Alzheimer’s disease (AD). Lower levels of Aβ1–38 and Aβ1–42 were associated with increased risk of dementia, specifically AD (HR for AD per SD decrease in Aβ1–38 levels, 1.39; 95% CI, 1.00–2.16; HR for AD per SD decrease in Aβ1–42 levels, 1.35; 95% CI, 1.05–1.75) after adjustment for age, sex, education, cardiovascular risk factors, apolipoprotein E ε4 allele carrier status, and other Aβ isoforms.
Conclusions
Our results show that lower plasma Aβ levels were associated with risk of dementia and incident AD. Moreover, lower plasma Aβ1–42 levels were related to smaller hippocampal volume. These results suggest that plasma Aβ1–38 and Aβ1–42 maybe useful biomarkers for identification of individuals at risk of dementia.
Similar content being viewed by others


Background
Dementia due to Alzheimer’s disease (AD), constituting up to 70% of all dementias, is characterized by deposition of amyloid plaques and neurofibrillary tangles in the brain parenchyma [1]. Amyloid-β (Aβ) 1–40 (Aβ1–40) and Aβ1–42 peptides derived from amyloid precursor protein are present in these parenchymal plaques, and it is hypothesized that Aβ peptide abnormalities begin early in the neurodegenerative pathological cascade [2, 3]. According to this amyloid cascade hypothesis, an imbalance between the production and clearance of the Aβ peptides, especially the Aβ1–42 isoform, leads to their aggregation in the cortical tissue and vessels [3]. This Aβ deposition in the brain subsequently affects plasma concentrations, thus suggesting that circulating levels of Aβ could potentially be used as markers of disease risk [4].
Aβ1–40 and Aβ1–42 peptides are the two most studied AD biomarkers, which can be measured in plasma through minimally invasive techniques and thus can feasibly be determined in large samples. Several large community-based studies have reported on the association of high plasma Aβ1–40 and Aβ1–42 levels and lower Aβ1–42/Aβ1–40 ratios at baseline with risk of dementia and reduced cognitive measure scores [5,6,7]. Two studies have shown a link between higher levels of Aβ1–42 but not Aβ1–40 with risk of AD [8, 9]. Conversely, some studies reported an association of increased concentrations of Aβ1–42 and Aβ1–40 levels and higher Aβ1–42/Aβ1–40 ratio with reduced risk of dementia [10, 11]. Longitudinal studies with repeated measurements of Aβ have shown that individuals in preclinical stages of the disease [12] and persons with newly diagnosed AD [6] showed significant reductions in plasma Aβ1–42 concentrations over time. Though most of the study results are inconsistent, they nevertheless do suggest that plasma Aβ levels may be differentially associated with the risk for AD, possibly reflective of the stage of the disease and the complex pattern of production and clearance from the brain [4]. Limited data also suggest that increased circulating levels of Aβ1–40 and Aβ1–42 may induce cerebral atrophy, detected on magnetic resonance imaging (MRI) studies as cortical thinning in asymptomatic elderly persons through mechanisms such as synaptic deficits and neuronal loss [13,14,15] . More studies of a general population are needed to investigate the relationship between Aβ and markers of neurodegeneration as well as risk of dementia to further comprehend the underlying mechanisms in a subclinical phase.
Besides Aβ1–40 and Aβ1–42 peptides, which have been investigated extensively, another isoform—Aβ1–38—is of interest because it is reported to be elevated in the cerebrospinal fluid (CSF) of patients with sporadic AD [16]. Moreover, it is suggested to be a sensitive and specific marker for diagnosing AD over the range of other non-AD dementias, and subsequent studies confirmed the suitability of Aβ1–38 as an additional biomarker for differential diagnosis of dementia [17, 18]. Despite this ongoing research, little is known regarding whether plasma Aβ levels, including the novel Aβ1–38, are associated with preclinical markers of neurodegeneration, such as gray matter, white matter, and hippocampal atrophy, and the risk of dementia in a large population-based setting. We examined the association of plasma Aβ1–38, Aβ1–40, and Aβ1–42 levels with neurodegenerative markers and risk of dementia in a subsample of the Rotterdam Study.
Methods
Study population
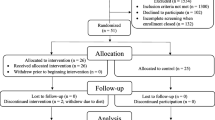
The Rotterdam Study is a population-based prospective cohort study of middle-aged and elderly persons living in the Ommoord district in the city of Rotterdam, the Netherlands. All participants in this study undergo reexamination every 3–4 years and are being followed continuously for events, including occurrence of dementia. This study is embedded within the first cohort, which was initiated in 1990 with 7983 participants at baseline (aged ≥ 55 yr). Persons were randomly selected (n = 563) during the second visit of the first cohort (1995–1996) and were invited to undergo neuroimaging [19]. Blood samples of the same individuals were drawn in the 1998 and 1999. After individuals with insufficient plasma (n = 22), no MRI scans (n = 73), and prevalent dementia (n = 10) were removed, the final sample size consisted of 458 persons [20].
Plasma assessment
Blood samples were drawn into ethylenediaminetetraacetic acid (EDTA) tubes for plasma collection. After centrifugation (2500 × g, + 4 °C for 20 min), plasma samples were stored at − 80 °C within 60 minutes of collection. Plasma levels of Aβ1–38, Aβ1–40, and Aβ1–42 were quantified by EUROIMMUN β-Amyloid 1–38, 1–42, and 1–40 plasma enzyme-linked immunosorbent assays (EUROIMMUN, Lübeck, Germany), which have been validated and described in more detail previously [21, 22]. For quality control (QC) purposes, QC samples were produced by pooling of EDTA plasma samples from individual participants. After aliquoting, samples were stored at − 80 °C. The samples were coded QC1, QC2, QC8, QC9, and QC10 and were used in the three plasma amyloid assays. QC in the three assays was ± 2 SD of each amyloid concentration (in pg/ml) across study participants, with the ranges of concentration values detectable being 5.9–18.7 pg/ml for Aβ1–38, 67.6–161.8 pg/ml for Aβ1–40, and 46.6–55.6 pg/ml for Aβ1–42. The average coefficients of variation of measurement of Aβ1–38, Aβ1–40, and Aβ1–42 in QC plasma samples during the study were 11.04%, 5.72%, and 8.70%, respectively [22].
Brain imaging
Brain MRI was performed using a 1.5-T MRI system (VISION MR; Siemens AG, Erlangen, Germany) to obtain T1-weighted, proton density, T2-weighted, and high-resolution inversion recovery double-contrast three-dimensional half-Fourier-acquired single-shot turbo spin echo (HASTE) sequences [23]. Image preprocessing and the tissue classification algorithm have been described elsewhere [23]. Briefly, the k-nearest neighbors brain tissue classifier technique was used to classify voxels into CSF, gray matter, normal white matter, and white matter hyperintensities. Intracranial volume was the sum of the CSF, gray matter, normal white matter, and white matter hyperintensities. We used a validated nonrigid registration algorithm to map brain regions to the template scan. Hippocampal volumes were manually outlined on coronal HASTE sequences perpendicular to the long axis of the hippocampus [24].
Assessment of dementia
Participants were screened for dementia at baseline and at follow-up examinations [20]. Screening was performed using the Mini Mental State Examination (MMSE) and the Geriatric Mental State Schedule (GMS) organic level. Individuals with screen-positive results (MMSE < 26 or GMS organic level > 0) subsequently underwent an examination and informant interview with the Cambridge Examination for Mental Disorders of the Elderly. Additionally, the whole cohort was continuously monitored for dementia through computerized linkage of the study database and digitized medical records of general practitioners and the Regional Institute for Outpatient Mental Health Care. When required and available, neuroimaging was used to facilitate dementia diagnosis. A consensus panel led by a consultant neurologist established the final diagnosis according to standard criteria for dementia (Diagnostic and Statistical Manual of Mental Disorders, Third Edition–Revised). The diagnosis of AD was made using the National Institute of Neurological and Communicative Disorders and Stroke-Alzheimer’s Disease and Related Disorders Association [25] and National Institute of Neurological Disorders and Stroke-Association Internationale pour la Recherché et l’Enseignement en Neurosciences criteria for vascular dementia [26]. The first cohort was followed for dementia until 15 years (based on maximum follow-up) after baseline examination (i.e., January 2015). Follow-up for dementia was complete for 99.5% of potential person-years in this cohort.
Covariate assessment
Data on demographics and medical history were recorded on the same day of dementia screening. Blood pressure was measured in two readings using a random zero sphygmomanometer in a sitting position, and the mean of both measurements was calculated. Mean arterial blood pressure was calculated as two-thirds of the diastolic blood pressure plus one-third of the systolic blood pressure. Serum total cholesterol levels were measured using an automated enzymatic procedure. Diabetes mellitus was defined as fasting blood glucose ≥ 7 mmol/L or receiving treatment for diabetes. Smoking was categorized into ever versus never smokers. Education was treated as the number of years of formal education. Apolipoprotein E (APOE) genotype was determined using PCRs on coded DNA samples. Distribution of APOE genotype and allele frequencies were in Hardy-Weinberg equilibrium. APOE-ε4 carrier status was defined by the presence of at least one ε4 allele.
Statistical analysis
Plasma Aβ levels and brain tissue volumes were standardized (by subtracting each variable by population mean divided by SD). Plasma Aβ levels were expressed as per-SD decrease. The Aβ1–40/Aβ1–42 ratio was calculated using raw values of Aβ1–40 and Aβ1–42 levels. We first performed linear regression models to determine the association between Aβ levels and brain tissue volumes (total brain volumes, white matter volume, gray matter volume, and hippocampal volume). The models were adjusted for age, sex, intracranial volume, mean arterial blood pressure, total cholesterol, diabetes, smoking, and APOE-ε4 carrier status. We tested the independent effects of Aβ levels with brain tissue volumes by adding all three isoforms together in the regression models.
Using Cox proportional hazards models, we calculated HRs with corresponding 95% CIs for dementia and its subtypes with per-SD decrease in Aβ level. Participants were censored within the follow-up period at date of event diagnosis, death, or loss to follow-up, whichever came first. The proportional hazards assumption was tested by adding the interaction terms of Aβ1–38, Aβ1–40, Aβ1–42, and Aβ1–40/Aβ1–42 ratio with follow-up time in different models. All Cox proportional hazards models were initially adjusted for age, sex, and education and subsequently for mean arterial blood pressure, total cholesterol, diabetes, smoking, APOE-ε4 carrier status, and the other Aβ isoforms using a similar approach to the one described above. We also investigated whether the association between plasma Aβ and dementia was different in carriers and noncarriers of the APOE-ε4 allele. The level of significance was set to 5%, and all tests were two-sided. Statistical analyses were performed using IBM SPSS Statistics version 24 software (IBM, Armonk, NY, USA).
Results
Baseline characteristics of the participants are shown in Table 1. The mean age of the participants was 67.8 ± 7.7 years, and 232 (50.7%) were women. Diabetes was present in 55 (12.1%) of the study population, whereas the frequency of ever smokers was 317 (69.7%). Almost 30% (n = 137) persons were APOE-ε4 carriers. The mean ± SD values of Aβ1–38, Aβ1–40, and Aβ1–42 were 19.4 ± 4.3, 186.1 ± 35.9, and 56.3 ± 6.2, whereas for white matter volume, gray matter volume, and hippocampal volume, the respective values were 354.1 ± 85.4, 522.7 ± 55.8, and 6.4 ± 0.9. The correlation between Aβ1–38 and Aβ1–40 was 0.81 (Pearson’s correlation coefficient, R); between Aβ1–38 and Aβ1–42, R = 0.24, and between Aβ1–40 and Aβ1–42, R = 0.25.
Table 2 shows the cross-sectional analysis between Aβ levels and brain tissue volumes adjusted for age, sex, vascular risk factors, APOE-ε4 carrier status, intracranial volume, and other Aβ isoforms, when appropriate. Lower Aβ1–38, Aβ1–40, and Aβ1–42 levels and lower Aβ1–40/Aβ1–42 ratio were not associated with total brain, gray, and white matter volumes. A significant association was observed between lower plasma Aβ1–42 levels and smaller hippocampal volume (mean difference in hippocampal volume per SD decrease in Aβ1–42 levels, − 0.13; 95% CI, − 0.23 to − 0.04; p = 0.007).
During a mean follow-up of 14.8 years (SD, 4.9; range, 4.1–23.5 yr), 241 individuals died. In the same follow-up, 79 persons developed dementia. Of these 79 individuals with dementia, 64 were diagnosed with AD and 15 with vascular dementia. The interaction terms of Aβ levels with follow-up time in all models were nonsignificant (p value for interaction between Aβ1–38 and time, p = 0.832; between Aβ1–40 and time, p = 0.820; between Aβ1–42 and time, p = 0.998; and between Aβ1–40/Aβ1–42 ratio and time, p = 0.984). In the multivariable analysis, lower levels of Aβ1–38 and Aβ1–42 were associated with increased risk of dementia (HR for dementia per SD decrease in Aβ1–38, 1.33; 95% CI, 1.01–1.89; HR for dementia per SD decrease in Aβ1–42, 1.27; 95% CI, 1.02–1.58) (Table 3). No association was observed between Aβ1–40 and Aβ1–40/Aβ1–42 ratio and dementia. When the analysis was performed separately for dementia subtypes, lower levels of Aβ1–38 and Aβ1–42 were associated with increased risk of AD (HR for AD per SD decrease in Aβ1–38, 1.39; 95% CI, 1.00–2.16; HR for AD per SD decrease in Aβ1–42, 1.35; 95% CI, 1.05–1.75). The HRs for AD and vascular dementia were closely similar but insignificant in cases of vascular dementia (Table 3). The association between lower levels of Aβ and risk of dementia among APOE-ε4 carriers and noncarriers appeared similar, such that lower levels of Aβ1–38, Aβ1–40, and Aβ1–42 were associated with increased risk of dementia in both strata. A stronger association was observed for Aβ1–38 with increased risk of dementia among APOE-ε4 carriers (HR for dementia per SD decrease in Aβ1–38, 1.58; 95% CI, 1.01–2.89), whereas for Aβ1–42, this association was observed only among APOE-ε4 noncarriers (HR for dementia per SD decrease in Aβ1–42, 1.47; 95% CI, 1.09–1.99) (Table 4).
Discussion
In this study, we showed that lower levels of plasma Aβ were not associated with preclinical markers of neurodegeneration (i.e., total gray matter and white matter volumes), except for Aβ1–42, which was associated with smaller hippocampal volume in elderly individuals. Individuals with lower levels of Aβ1–38 and Aβ1–42 had an independent increased risk of dementia, specifically AD. These findings suggest that Aβ1–38 and Aβ1–42 may be involved in different pathways leading to dementia.
Previous research has shown that higher plasma Aβ levels (1–40 and 1–42 peptides) at baseline were associated with cognitive dysfunction and faster cognitive decline, regardless of dementia status at follow-up, supporting the notion that plasma Aβ may induce a variety of brain pathologies (including cortical atrophy) earlier in life [27]. A recent study consisting of 100 participants also reported that higher plasma Aβ1–42 levels were associated with thinner temporal cortex in cognitively normal elderly persons [13]. However, apart from the latter study, no studies have yet combined the structural brain changes with plasma Aβ levels to assess the differences in cognitively normal elderly individuals. Moreover, the effects of novel Aβ1–38 in addition to Aβ1–40 and 1–42 on cerebral atrophy and whether it is an important fluid biomarker for neurodegeneration and dementia has not been explored previously. Contrary to what we expected, our findings showed that lower plasma Aβ levels (including Aβ1–38) were not related to brain tissue volumes, except for hippocampal volume, where a significant association was observed between lower Aβ1–42 levels and smaller hippocampal volume in the elderly. The possible reason for the lack of association between Aβ1–38 and brain atrophy could be that plasma Aβ1–38 levels may reflect vascular disease in the brain rather than neurodegeneration. On one hand, this is supported by our previous study in which we showed that Aβ1–38 reflected microvascular damage in the brain and possibly induced adverse changes by inflammation, imbalance of oxygen free radicals, and apoptosis [22]. On the other hand, the association between Aβ1–42 and the hippocampus in the present study supports the notion that as Aβ1–42 starts to deposit in the brain after the age of 60 years, lower plasma Aβ levels that follow this deposition relate to hippocampal atrophy. A recent study has shown that accumulation of Aβ and tau pathologies in the brain were related to a decrease in hippocampal volume, including its critical subcompartments (i.e., CA1 and subiculum) in the earliest stages of AD prodromes [28]. Moreover, another study with healthy control individuals and persons with subjective cognitive complaints reported greater cortical thickness at intermediate levels of Aβ pathology (measured using CSF) [29]. By contrast, our data suggest that lower plasma Aβ levels (which are thought to indirectly reflect accumulation of amyloid in brain) do not necessarily correlate with universal volumetric decline in all structures (gray matter and white matter volumes), which is supported by some recent data [30].
Several lines of evidence suggest that Aβ levels in the CSF and plasma are in dynamic equilibrium with each other and that increased Aβ production in the brain gives rise to raised levels in the plasma [31]. It is further reported that as Aβ starts to deposit in the brain in the form of plaques, this in turn leads to lower plasma Aβ levels, which has been related to a higher risk of dementia [32]. Previous cross-sectional studies examining the association between Aβ levels and dementia have been mainly inconsistent [33,34,35,36]. It has been further reported that because plasma Aβ levels tend to change over the course of the dementia process, longitudinal studies are more useful in assessing the link between Aβ levels and risk of dementia in asymptomatic individuals [7]. Thus far, limited data have shown that plasma Aβ1–42 levels significantly decline in concentration in persons with newly diagnosed AD compared with individuals with prevalent AD and control subjects [12]. It is also suggested that plasma Aβ1–42 levels decline at an average rate of 12% per year among individuals with mild cognitive impairment [6]. Our results add to the previous reports by showing that lower levels of baseline plasma Aβ1–38, in addition to Aβ1–40 and Aβ1–42, were associated with a reduced risk of dementia. The association of Aβ1–38 and Aβ1–42 with dementia persisted when all the isoforms were added together in the model, indicating an independent link with increased dementia risk. These observations further support the findings that Aβ1–38 and Aβ1–42 may be generated independently by γ-secretase and that the production of these peptides is not coordinately regulated [16]. This was further confirmed in in vitro experiments involving γ-secretase modulators (sulindac sulfide), where Aβ1–38 levels were increased upon treatment with sulindac sulfide with no concurrent effect on Aβ1–42 levels, thus arguing against a precursor–product relationship [37, 38]. Though no association was observed between Aβ1–38 levels and neurodegenerative markers in this study, a link was still observed between lower levels of Aβ1–38 and increased risk of dementia. As mentioned before, the Aβ1–38 isoform is a marker of vascular pathology, and a higher level may indicate the activation of different inflammatory cascades (cytokines, cluster of differentiation 40 ligand, and tumor necrosis factor α) [22], which may increase the vulnerability to dementia, but such a relationship requires further clarification. Moreover, the relationship of plasma Aβ levels and brain amyloid deposition is suggested to be further complicated by dynamics of the blood-brain barrier and other possible sources of Aβ materials outside the central nervous system, including platelets and skeletal muscle cells [32].
With respect to the subtypes of dementia, similar associations were observed between plasma Aβ1–38 and Aβ1–42 levels with AD and vascular dementia. Though the significant results were observed only in cases of AD, this finding might be related to the fact that most of the individuals diagnosed with incident dementia had AD (72%). Also, in an elderly population, mixed pathology is commonly observed, which might explain the similar estimates for AD and vascular dementia. Moreover, the association of lower levels of Aβ1–38 with increased risk of dementia was more significant in carriers of the APOE-ε4 allele than in noncarriers. Though an opposite link was observed between Aβ1–42 and increased dementia risk in APOE-ε4 noncarriers, the similarity in the direction of effect estimates further suggests a role of mixed pathology in the development of dementia, regardless of APOE carrier status.
Strengths of our study include its prospective design, the population-based setting, volumetric quantification of the brain tissues, and virtually complete follow-up for dementia in the older cohort. There are some potential limitations of our study. First, because plasma samples were collected after MRI acquisition with a 3-year time window, the effect estimates calculated in this study may represent over- or underestimations. Second, plasma Aβ levels provide an indirect measure of brain-specific Aβ pathology, and an in vivo analysis of brain-specific Aβ burden (such as with amyloid positron emission tomography) could allow more accurate measures of Aβ burden. Finally, we lacked repeated measurement of plasma Aβ concentrations, which limits its ability to better estimate the trajectory of plasma levels over time in relation to risk of dementia. However, this could be a subject for further studies.
Conclusions
This study provides evidence that lower plasma Aβ levels, specifically Aβ1–38 and Aβ1–42, are associated with increased risk of dementia, specifically AD. Future studies should examine whether inclusion of novel plasma Aβ1–38 levels as an additional biomarker can provide further information on risk of developing dementia and AD dementia.
Abbreviations
- AD:
-
Alzheimer’s disease
- APOE:
-
Apolipoprotein E
- Aβ:
-
Amyloid-β
- CSF:
-
Cerebrospinal fluid
- EDTA:
-
Ethylenediaminetetraacetic acid
- GMS:
-
Geriatric Mental State Schedule
- HASTE:
-
Half-Fourier-acquired single-shot turbo spin echo
- MMSE:
-
Mini Mental State Examination
- MRI:
-
Magnetic resonance imaging
- QC:
-
Quality control
References
Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–6.
Jack CR Jr, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, et al. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010;9:119–28.
Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–6.
Shah NS, Vidal JS, Masaki K, Petrovitch H, Ross GW, Tilley C, et al. Midlife blood pressure, plasma β-amyloid, and the risk for Alzheimer disease: the Honolulu Asia Aging Study. Hypertension. 2012;59:780–6.
Graff-Radford NR, Crook JE, Lucas J, Boeve BF, Knopman DS, Ivnik RJ, et al. Association of low plasma Aβ42/Aβ40 ratios with increased imminent risk for mild cognitive impairment and Alzheimer disease. Arch Neurol. 2007;64:354–62.
Mayeux R, Honig LS, Tang MX, Manly J, Stern Y, Schupf N, et al. Plasma Aβ40 and Aβ42 and Alzheimer’s disease: relation to age, mortality, and risk. Neurology. 2003;61:1185–90.
van Oijen M, Hofman A, Soares HD, Koudstaal PJ, Breteler MM. Plasma Aβ1–40 and Aβ1–42 and the risk of dementia: a prospective case-cohort study. Lancet Neurol. 2006;5:655–60.
Lopez OL, Kuller LH, Mehta PD, Becker JT, Gach HM, Sweet RA, et al. Plasma amyloid levels and the risk of AD in normal subjects in the Cardiovascular Health Study. Neurology. 2008;70:1664–71.
Schupf N, Tang MX, Fukuyama H, Manly J, Andrews H, Mehta P, et al. Peripheral Aβ subspecies as risk biomarkers of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2008;105:14052–7.
Lambert JC, Schraen-Maschke S, Richard F, Fievet N, Rouaud O, Berr C, et al. Association of plasma amyloid-β with risk of dementia: the prospective Three-City Study. Neurology. 2009;73:847–53.
Chouraki V, Beiser A, Younkin L, Preis SR, Weinstein G, Hansson O, et al. Plasma amyloid-β and risk of Alzheimer’s disease in the Framingham Heart Study. Alzheimers Dement. 2015;11:249–57.
Pomara N, Willoughby LM, Sidtis JJ, Mehta PD. Selective reductions in plasma Aβ 1–42 in healthy elderly subjects during longitudinal follow-up: a preliminary report. Am J Geriatr Psychiatry. 2005;13:914–7.
Llado-Saz S, Atienza M, Cantero JL. Increased levels of plasma amyloid-β are related to cortical thinning and cognitive decline in cognitively normal elderly subjects. Neurobiol Aging. 2015;36:2791–7.
Zahs KR. Ashe KH. β-Amyloid oligomers in aging and Alzheimer’s disease. Front Aging Neurosci. 2013;5:28.
Selkoe DJ. Soluble oligomers of the amyloid-β-protein impair synaptic plasticity and behavior. Behav Brain Res. 2008;192:106–13.
Reinert J, Martens H, Huettenrauch M, Kolbow T, Lannfelt L, Ingelsson M, et al. Aβ38 in the brains of patients with sporadic and familial Alzheimer’s disease and transgenic mouse models. J Alzheimers Dis. 2014;39:871–81.
Welge V, Fiege O, Lewczuk P, Mollenhauer B, Esselmann H, Klafki HW, et al. Combined CSF tau, p-tau181 and amyloid-β 38/40/42 for diagnosing Alzheimer’s disease. J Neural Transm. 2009;116:203–12.
Wiltfang J, Esselmann H, Bibl M, Smirnov A, Otto M, Paul S, et al. Highly conserved and disease-specific patterns of carboxyterminally truncated Aβ peptides 1–37/38/39 in addition to 1–40/42 in Alzheimer’s disease and in patients with chronic neuroinflammation. J Neurochem. 2002;81:481–96.
Ikram MA, Vrooman HA, Vernooij MW, van der Lijn F, Hofman A, van der Lugt A, et al. Brain tissue volumes in the general elderly population. The Rotterdam Scan Study. Neurobiol Aging. 2008;29:882–90.
de Bruijn RF, Bos MJ, Portegies ML, Hofman A, Franco OH, Koudstaal PJ, et al. The potential for prevention of dementia across two decades: the prospective, population-based Rotterdam Study. BMC Med. 2015;13:132.
Lerche S, Brockmann K, Pilotto A, Wurster I, Sünkel U, Hobert MA, et al. Prospective longitudinal course of cognition in older subjects with mild parkinsonian signs. Alzheimers Res Ther. 2016;8:42.
Hilal S, Akoudad S, van Duijn CM, Niessen WJ, Verbeek MM, Vanderstichele H, et al. Plasma amyloid-β levels, cerebral small vessel disease, and cognition: the Rotterdam Study. J Alzheimers Dis. 2017;60:977–87.
Ikram MA, van der Lugt A, Niessen WJ, Koudstaal PJ, Krestin GP, Hofman A, et al. The Rotterdam Scan Study: design update 2016 and main findings. Eur J Epidemiol. 2015;30:1299–315.
den Heijer T, Geerlings MI, Hoebeek FE, Hofman A, Koudstaal PJ, Breteler MM. Use of hippocampal and amygdalar volumes on magnetic resonance imaging to predict dementia in cognitively intact elderly people. Arch Gen Psychiatry. 2006;63:57–62.
McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR Jr, Kawas CH, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:263–9.
Roman GC, Tatemichi TK, Erkinjuntti T, Cummings JL, Masdeu JC, Garcia JH, et al. Vascular dementia: diagnostic criteria for research studies: report of the NINDS-AIREN international workshop. Neurology. 1993;43:250–60.
Cosentino SA, Stern Y, Sokolov E, Scarmeas N, Manly JJ, Tang MX, et al. Plasma β-amyloid and cognitive decline. Arch Neurol. 2010;67:1485–90.
Tardif CL, Devenyi GA, Amaral RSC, Pelleieux S, Poirier J, Rosa-Neto P, et al. Regionally specific changes in the hippocampal circuitry accompany progression of cerebrospinal fluid biomarkers in preclinical Alzheimer’s disease. Hum Brain Mapp. 2018;39:971–84.
Fortea J, Sala-Llonch R, Bartres-Faz D, Llado A, Sole-Padulles C, Bosch B, et al. Cognitively preserved subjects with transitional cerebrospinal fluid β-amyloid 1–42 values have thicker cortex in Alzheimer’s disease vulnerable areas. Biol Psychiatry. 2011;70:183–90.
Jack CR Jr, Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS, et al. Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013;12:207–16.
Ghersi-Egea JF, Gorevic PD, Ghiso J, Frangione B, Patlak CS, Fenstermacher JD. Fate of cerebrospinal fluid-borne amyloid-β-peptide: rapid clearance into blood and appreciable accumulation by cerebral arteries. J Neurochem. 1996;67:880–3.
Kawarabayashi T, Younkin LH, Saido TC, Shoji M, Ashe KH, Younkin SG. Age-dependent changes in brain, CSF, and plasma amyloid β protein in the Tg2576 transgenic mouse model of Alzheimer’s disease. J Neurosci. 2001;21:372–81.
Mehta PD, Pirttila T, Mehta SP, Sersen EA, Aisen PS, Wisniewski HM. Plasma and cerebrospinal fluid levels of amyloid-β proteins 1–40 and 1–42 in Alzheimer disease. Arch Neurol. 2000;57:100–5.
Iwatsubo T. Amyloid-β protein in plasma as a diagnostic marker for Alzheimer’s disease. Neurobiol Aging. 1998;19:161–3.
Blennow K, Mattsson N, Schöll M, Hansson O, Zetterberg H. Amyloid biomarkers in Alzheimer’s disease. Trends Pharmacol Sci. 2015;36:297–309.
Blennow K, Hampel H, Weiner M, Zetterberg H. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat Rev Neurol. 2010;6:131–44.
Page RM, Baumann K, Tomioka M, Perez-Revuelta BI, Fukumori A, Jacobsen H, et al. Generation of Aβ38 and Aβ42 is independently and differentially affected by familial Alzheimer disease-associated presenilin mutations and γ-secretase modulation. J Biol Chem. 2008;283:677–83.
Czirr E, Cottrell BA, Leuchtenberger S, Kukar T, Ladd TB, Esselmann H, et al. Independent generation of Aβ42 and Aβ38 peptide species by γ-secretase. J Biol Chem. 2008;283:17049–54.
Acknowledgements
We thank Andrea Vermeij and Jeannette Vergeer for plasma analysis in this study.
Funding
This study is part of the Cerebral Amyloid Angiopathy Vascular Imaging and fluid markers of Amyloid deposition (CAVIA) project (no. 733050202), which has been made possible by ZonMW. The CAVIA project is part of ‘Memorabel’, the research and innovation programme for dementia, as part of the Dutch national ‘Deltaplan for Dementia’ (zonmw.nl/dementia research). The CAVIA project is a consortium of Radboudumc, Leiden University Medical Center (LUMC), Erasmus Medical Center (EMC), Vrije Universiteit Medical Center (VUmc), ADX Neurosciences, Philips Healthcare, Stony Brook University and Massachusetts General Hospital. The sponsor had no role in study design; the collection, analysis and interpretation of data; or in the writing of the manuscript.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
SH participated in data acquisition and performed statistical analysis as well as drafting and revision of the manuscript. FJW, MMV, HV, MKI and ES participated in data acquisition and revised the manuscript for intellectual content. MAI and MWV were responsible for the study concept and design, obtaining funding, and drafting and revising the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The Rotterdam Study was approved by the medical ethics committee according to the Population Study Act Rotterdam Study, as well as by the Ministry of Health, Welfare and Sports of the Netherlands (PG/OGZ 3098756). Written informed consent was obtained from all participants prior to study recruitment.
Consent for publication
Not applicable
Competing interests
SH, FJW, MMV, HV, MKI, MAI and MWV report no competing interests. ES is a shareholder of ADx NeuroSciences NV, Gent, Belgium, which provided the kits for plasma assessment of amyloid-β levels.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Hilal, S., Wolters, F.J., Verbeek, M.M. et al. Plasma amyloid-β levels, cerebral atrophy and risk of dementia: a population-based study. Alz Res Therapy 10, 63 (2018). https://doi.org/10.1186/s13195-018-0395-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13195-018-0395-6