
Abstract
Tumor necrosis factor (TNF) is a pleiotropic cytokine involved in many aspects of immune regulation. Anti-TNF biological therapy has been considered a breakthrough in the treatment of chronic autoimmune diseases, such as rheumatoid arthritis (RA). In this review, because of the major involvement of T cells in RA pathogenesis, we discuss the effects of anti-TNF biotherapy on T-cell responses in RA patients. We also outline the potential fields for future research in the area of anti-TNF therapy in RA.
This could be useful to better understand the therapeutic efficiency and the side effects that are encountered in RA patients. Better targeting of T cells in RA could help set more specific anti-TNF strategies and develop prediction tools for response.
Similar content being viewed by others

Background
The discovery of the role of tumor necrosis factor (TNF) in the pathogenesis of rheumatoid arthritis (RA) has led to anti-TNF biological therapy as a breakthrough in the treatment of chronic autoimmune diseases, such as RA, Crohn’s disease, psoriatic arthritis, and spondyloarthritis [1]. Various anti-TNFs are currently used for the treatment of RA, including infliximab (IFX), a chimeric antibody, and two fully human antibodies adalimumab (ADA) and golimumab. Additionally, etanercept (ETA) is a human recombinant dimeric fusion protein consisting of two soluble p75 TNF-RII chains linked to a modified Fc portion of human IgG. Finally, certolizumab pegol (CZP) is a pegylated Fab’ fragment of a humanized anti-TNF antibody. Biosimilars of IFX and of ETA are already in use.
TNF is a pleiotropic cytokine involved in many aspects of immune regulation [2]. TNF is first synthesized as a biologically active transmembrane homotrimer (tmTNF), which is further released upon cleavage by tumor necrosis factor-alpha converting enzyme (TACE, also named ADAM17) protease. Soluble TNF binds to the receptors TNF-RI and TNF-RII, while tmTNF binds preferentially to TNF-RII. Anti-TNF biologics can block both soluble and tmTNF [3]. TNF can be produced by multiple cell types such as T and B cells and innate immune cells (dendritic cells, monocytes, neutrophils, mast cells). All these sources may contribute to the development of a pathological state of chronic inflammation, especially in RA. T cells are also targets of TNF either directly, like all cells that express TNF-Rs, or indirectly as a result of antigen presentation or costimulation. The immunomodulatory role of TNF-R2 on T-cell activity has been described in the collagen-induced arthritis (CIA) model of arthritis [4].
In this review, because of the major involvement of T cells in RA pathogenesis, we discuss the effects of anti-TNF biotherapy on T-cell responses in RA patients. This could be of help for the interpretation of the clinical effects (or lack thereof) of anti-TNF treatments, as well as being useful to better understand the side effects which are encountered in RA patients.
Role of T cells in RA
Much has been learned from mouse models in the understanding of RA, especially regarding the role of T cells. Collagen-induced, K/BxN, IL-1 RA-KO, and SKG models were shown to depend on T lymphocytes [5]. More specifically, the SKG model, depending on a mutation in ZAP 70 that affects the TcR-ζ chain signaling and T-cell selection, directly implicated the role of T cells in the development of experimental arthritis.
There has been an ongoing debate over the respective importance of macrophages and T cells. The presence of T cells in joints and the expansion of clonotypic T cells, as a result or a cause of inflammation, in the synovium of RA patients has fueled that debate. The role of the HLA-DR shared epitope in the development of RA is a strong indication for the role of T cells [6]. There is a T-cell response to citrullinated T-cell epitopes or PAD peptides [7] in patients who bear the RA susceptibility HLA-DR allele. A direct argument for the role of T lymphocytes in RA has been the successful use of CTLA4-Ig as a biotherapy that blocks the CD28-CD86/CD80 interaction [8]. Thus, the current view is that there is an interplay between pathogenic T cells, macrophages, and cytokines that contributes to the pathogenic imbalance in RA [9] and can be targeted with biologics.
Role of TNF in the development of the immune system
TNF has been shown to be essential in many stages of T-cell development. In the thymus, TNF promotes the apoptosis of triple-negative CD3/CD4/CD8 [10] and double positive CD4/CD8 thymocytes [11], as well as the development of single positive thymocytes [12]. Thus, it is expected that treatment of infants with anti-TNF might alter the development of their T cells. This needs further investigation.
Secondary lymphoid organs (SLO) are crucial for the development of efficient adaptive immune responses. Organized in well-demarcated T-cell zones and B-cell follicles, SLO bring the antigen that is trapped by various subsets of dendritic cells (DCs) in close contact with the immune cells, provide costimulatory signals from DCs, and thereby initiate an appropriate immune response.
TNF-mediated signaling is crucial for the development of some and for structural maintenance of most of the SLO. Distinct cellular sources and molecular forms of TNF contribute to the organization of SLO microarchitecture. TNF from B and T cells cooperates to maintain the structural integrity in lymph nodes, which are indispensable for the generation of efficient local immune responses.
The requirement of TNF signaling for organized lymphoid structures in mice was confirmed by studies in humans. Rheumatoid arthritis patients receiving ETA lack germinal center development in their tonsils [13]. Similar experiments in mice showed that pharmacological inhibition of TNF by ETA leads to inhibition of follicular dendritic cell development and a subsequent decrease in germinal center response, as well as a reduction in the marginal zone [14]. However, the structure of B-cell follicles in the spleen remained unchanged, suggesting that some of the TNF-dependent features of splenic microarchitecture are not inhibited by ETA [14].
Altogether, TNF controls the development and organization of SLO structures and, thereby, influences the development of adaptive immune responses. This could be of importance during the follow-up of RA patients, especially children, treated with anti-TNF.
Role of TNF in T-cell differentiation, activation, and maturation: action of TNF inhibitors
Activation of naive T cells is initiated during their encounter with antigen peptide presented by mature DCs. This activation is dependent on coactivation mediated by the membrane interaction between members of the TNF/TNF-R family other than TNF cytokine on T cells and DCs. As a cytokine, TNF contributes to efficient antigen presentation by inducing DC maturation.
Interaction of T cells with antigen presenting cells leads to differentiation into effector and memory T cells (reviewed in [15]). To understand how anti-TNF treatment may exert an impact on the pathogenicity of T lymphocytes, we first need to overview the role of TNF in the activation of effector, memory, and regulatory T cells.
TNF is reported to negatively regulate the expansion of effector CD4+ and CD8+ T cells during viral infection through apoptosis, thus subsequently limiting the T cell memory compartment [16]. TNF, acting along with interleukin (IL)-33, transforming growth factor (TGF)-β and IL-15, induces resident memory T cells (TRM) with CD69 and CD103 expression [15]. These T cells do not recirculate and remain in the lymphoid tissue. Production of TNF by TRM in turn contributes to the maturation of DCs and efficient Ag presentation for recall T-cell activation. Anti-TNF biologics are thus expected to modulate the effector and the memory T-cell response during infections and vaccination (vide infra).
To invade inflamed tissue, T lymphocytes must have the capacity to traffic through endothelial cell junctions. This phenomenon, called diapedesis, has been shown to depend on TNF and interferon (IFN)-γ [17]. Thus, although this has never been tested formally, anti-TNF drugs have the capacity to reduce inflammation by interfering with diapedesis and migration of T cells to the joints.
However, TNF has a contrasting role in T-cell activation [18, 19]. The notion of long-term pathogenic effects of TNF in disease was pioneered by Maini and Feldmann, based on the observation of elevated TNF production in the joints of RA patients. They also reported that chronic exposure of cells to TNF impaired the T cell-specific recall response to tetanus toxoid. This inhibition was later shown to be due to attenuation of TcR signaling. At the molecular level, TNF appeared to inhibit CD3-ζ chain expression via Src-like adaptor protein (SLAP) degradation [20].
Toxicity of IFX for T cells is minimal and the metabolism of T cells is not significantly altered by anti-TNF [21]. However, T-cell subsets were not investigated, and this requires further studies. Regarding in-vitro T-cell activation, impairment of T cells from RA patients can be reversed by anti-TNF and, correspondingly, anti-TNF treatment of RA patients restores in-vitro proliferation in response to soluble antigens [22]. In a model of transmembrane expression of TNF in the Jurkat T-cell line (tm-Jurkat), Mitoma et al. [23] showed that IFX induces JNK activation and IL-10 production, and inhibits proliferation. Reverse signaling is a mechanism of signaling mediated by anti-TNF or TNF-R through binding to tmTNF [3]. Reverse signaling has been suggested to regulate inflammation in macrophages and T cells. However, the molecular mechanisms are not completely understood and demonstration of in-vivo reverse signaling has yet to be demonstrated.
Another possible mechanism of action of anti-TNF on T cells is the regulation of cell death. The action of anti-TNF drugs on cell death was tested using the Jurkat T-cell line transfected with tmTNF [24]. Due to the absence of the Fc fragment, CZP did not induce antibody-dependent cell-mediated cytotoxicity or complement-dependent cytotoxicity, whereas golimumab, IFX, and ADA did. CZP and ETA did not induce apoptosis in tmTNF Jurkat cells [24]. However, those data were obtained with cells overexpressing tmTNF and cannot be extrapolated to physiologic conditions. In tm-Jurkat T-cells, ADCC and CDC, induced by IFX and ADA, were of lower intensity than with ETA, and were not observed with CZP [24].
In patients with active RA, spontaneous apoptosis of CD4+CD25+ cells was evaluated at the start of treatment with IFX and after 3 months of treatment [25]. It was shown that spontaneous in-vitro apoptosis of CD4+CD25+ cells, which was increased in RA patients compared with healthy donors, was reduced after treatment with IFX [25].
Effects of anti-TNF on T-helper cell subset differentiation
There is now a growing literature in RA patients on increased T helper (Th)1 [26,27,28,29] and Th17 [26, 27, 29, 30] responses following TNF blockade. Th17 and shifting to nonclassic Th1 have been described as potential components of the pathophysiology of RA, but their overall significance is debated [31].
Hull et al. reported that patients responding to ADA or ETA had an increase in circulating Th17 [30]. Conversely, an increase in Th17 has been reported in patients not responding to TNF inhibitors [27, 32, 33]. Along similar lines, a good response was correlated with low levels of Th17 and was shown to be controlled by regulatory T cells (Tregs) in patients treated with ADA, not in those treated with ETA [34]. Th1 compartments were also reported to be increased in patients not responding to IFX [27] and, conversely, in patients in remission in response to ADA [26].
Furthermore, all the anti-TNF drugs IFX, ADA, CZP, and ETA induce IL-17+CD4+ T cells expressing IL-10 in RA patients [35]. The induction of IL-10 in association with IL-17 by Th17 suggests a modulatory role of those cells, but this needs to be demonstrated.
In conclusion, Th17 and Th1 compartments are increased in response to TNF inhibitors but a definitive answer as to whether they are linked to good or poor responses is needed. This is likely to depend on Th CD4+ T-cell phenotyping techniques, on the biologic administered, and the methodology used.
STAT6, which is associated with the Th2 response, was also induced in T cells from patients treated with ADA [36]. This would suggest a role for ADA in modifying T-cell polarization. Modifications of macrophage polarization induced by anti-TNF (our unpublished data) could also lead to changes in T-cell polarization.
The development of paradoxical psoriasis as a side effect of anti-TNF (ETA, IFX, or ADA) treatment in RA patients has been observed. The mechanism has been shown to involve IFN-α produced by plasmacytoid dendritic cells whose maturation is inhibited by anti-TNF [37] and not to the emergence of Th17 cells during treatment as previously suggested [38]. Recently, a new population of CD4+ T cells, called T peripheral helper (Tph) cells, has been identified in the synovial membrane of RA patients using mass cytometry technology [39]. Tph cells are CD4+ T cells that express high levels of the checkpoint protein PD-1 and, contrary to T-follicular helper cells (Tfh), do not express CXCR5. Tph cells induce the differentiation of plasma cells through IL-21. The inhibition of Tph by anti-TNFs [33] may prevent the differentiation of plasmablasts [39].
Anti-TNF treatments affect Tregs in RA
There are 2 types of CD4CD25 FoxP3-positive Tregs, inducible (iTregs) and natural (nTregs). Inducible Tregs depend on TNF-R2 as exemplified by the observation that TNF-R2 is critical for stabilization and homeostasis of Tregs [40]. TNF has been reported to be either an activator or inhibitor of Tregs depending on the study, as reviewed in [41]. TNF was reported to inhibit both the phosphorylation of FoxP3 and the development of Tregs in correlation with an increase in IL-17- and IFN-γ-producing CD4+ T cells [42]. However, it was shown that Tregs did not lose their suppressive activity in the presence of TNF. Because TNF has costimulatory effects [18], T-effector cells (Teff) may appear resistant to the effect of Tregs [43], and this may have led to previous misinterpretation of the negative role of TNF on Tregs. It was first shown that Tregs from RA patients are present but defective and their function can be restored by IFX treatment. An induced population of iTregs, whose activity is mediated through IL-10 and TGF-β, is restored under the action of IFX, whereas defective nTregs are not [44]. This can be explained by a new mechanism of action with binding of ADA to tmTNF, which is strongly expressed by monocytes from RA patients. ADA induces higher levels of tmTNF in those monocytes and promotes interaction with TNF-R2-expressing iTregs, which subsequently expand [45]. Such a phenomenon is not observed with ETA. Thus, anti-TNF antibody, but not soluble receptor, induces iTregs through increased expression of tmTNF.
On the T lymphocyte side, the role of soluble versus tmTNF has been explored in several models. T lymphocyte-monocyte contact is important in inflammation. This involves, in part, tmTNF interaction with TNF-R2 on adjacent cells [46]. Blocking tmTNF on T lymphocytes impairs the production of TNF by monocytes [46], and tmTNF expressed by T cells is responsible for the modulation of IL-10 production by monocytes [47].
In T cells, IFX but not ETA induces IL-10 production through reverse signaling, showing disparity in the efficacy of biologics regarding molecular mechanisms [23] but suggesting a possible regulatory role for reverse signaling depending on the biologic used.
Consequences of anti-TNF treatments on T-cell control of infections
Reactivation of tuberculosis during anti-TNF therapy by monoclonal antibodies and, to a lesser extent, by ETA has been a major drawback of biotherapies of rheumatic diseases [48]. Production of IFN-γ is, along with TNF, a major element of the T-cell immune response against tuberculosis. Nowadays, recommendations are to test for prior tuberculosis infection before anti-TNF treatments using interferon-gamma release assays (IGRAs) that detect specific T-cell response. Antituberculosis antibiotic prophylaxis has considerably reduced the risks of reactivation.
IFX triggers a reduction in CD8+ terminally differentiated effector memory CD45RA+ T cells (TEMRA cells) with antimicrobial activity against mycobacterium tuberculosis and is responsible for impairing the T-cell defense against microbes [49].
CD4+ T-cell proliferation and IFN-γ production against tuberculosis PPD and CFP-10 antigens were shown to be impaired by a 14-week treatment with anti-TNF in patients with a positive test for prior tuberculosis infection [50]. The inhibition was more pronounced in vitro with antibodies than with ETA.
CD8+-derived TNF is essential for antilisteria activity in mice. Patients treated with IFX are at higher risk for infections with listeria, another intracellular bacteria, than those treated with ETA [51].
Viral infections are controlled at least in part by CD4+ and CD8+ T lymphocytes through their cytotoxic activity and their release of cytokines such as TNF and IFN-γ [52]. Anti-TNF biotherapies have been shown to induce disparate changes in the antivirus immunity which may be due to modifications of SLO and/or direct inhibition of the antiviral effect of TNF. There is no clear evidence for a risk of varicella zoster virus (VZV) and cytomegalovirus (CMV) reactivation in patients undertaking biotherapies [53] and we have shown that the anti-CMV CD4+ response in RA patients treated with IFX, ADA, or ETA is conserved [54]. However, caution is required with respect to the safety of anti-TNF in patients with those viral infections.
Hepatitis B infections are controlled by T lymphocytes. Depletion of the T-cell response by anti-TNF treatments may explain the resurgence of hepatitis B chronic infections which may occur more frequently with antibodies than with ETA [55]. There is an increased risk of viral reactivation in patients with chronic HBV. Antiviral prophylaxis is required in these patients. It is not known whether the risks with different anti-TNFs are similar or not. Patients with past infection have no particular risk [56].
Recommendations with regard to hepatitis B and C infections in patients treated with anti-TNF have been proposed [57]. Caution is required in patients treated with anti-TNF regarding the follow-up of chronic infection or active infection. In any case, TNF inhibitors can be discontinued as they do not induce irreversible inhibition of TNF production [58].
Consequences of anti-TNF on T-dependent B-cell responses
TNF is involved in T cell-dependent B-cell responses. Resting memory CD45RO+ T cells activated by cytokines, among them TNF, can provide help to B cells for the production of IgM, IgG, and IgA [59]. CD4+ T cells expressing tmTNF provide a costimulatory signal for B cells [60].
Regarding response to vaccines, clinical studies performed with influenza and pneumococcal [61] vaccination reported only modest decreases in antibody titers in patients treated with ADA and safe immunization. Vaccination recommendations for the physician are provided in a recent article [62].
A proportion of RA patients treated with anti-TNF biologics develop antidrug antibodies that can hamper the efficiency of treatments [63]. Due to its structure, ETA has lower immunogenicity than anti-TNF antibodies and anti-ETA antibodies seem to be non-neutralizing [63]. Antigen presenting cells take up anti-TNF antibodies as antigens and present epitopes to CD4+ T cells. Such immunogenicity of anti-TNF antibodies in RA patients suggests that there is no profound decay of T cell-mediated B-cell immunity. Thus, from a functional point of view, only a partial decrease of the B-cell response is observed in RA patients treated with anti-TNF. Although cumbersome, a way of reducing the immunogenicity of anti-TNF antibodies would be to identify T cell epitopes and to modify them accordingly. From a clinical point of view, prescribing anti-TNF with methotrexate, an immunosuppressive drug that reduces the production of Th1 cytokines [64], decreases the risk of antidrug antibodies.
Conclusion
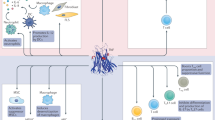
Figure 1 summarizes the consequences of anti-TNF on T-cell homeostasis. Anti-TNF can regulate the T-cell responses in many ways. By inducing iTregs through TNF-RII and restoring T-cell function, these biologics contribute to reducing the autoimmune process. Although apparently contradictory, the induction of iTregs and the restoration of T-cell effector functions suggest that anti-TNF acts on multiple aspects of T-cell homeostasis. Although T cell-dependent B-cell activation is decreased, the risks of immunization resulting in anti-antibodies hampers the efficiency of treatment. In this regard, ETA induces less antidrug antibodies.

Summary of anti-TNF impact on T cells in RA and possible topics of interest for future investigations. Targets of antitumor necrosis factor (TNF) presented in this figure are developed in the main text. Questions raised, and possible topics of future research, are indicated: What is the mechanism of the increase of transmembrane (tm)TNF expression on macrophages that leads to expansion of inducible regulatory T cells (iTregs)? Are T helper (Th)17 cells definitely not responsible for paradoxical psoriasis? What is the role of interleukin (IL)-17/IL-10 producing T cells in the control of rheumatoid arthritis (RA)? Are anti-TNFs other than ETA modifying maturation of thymus and SLO? Are anti-TNFs modifying T-cell metabolism? Are anti-TNFs modifying T-cell diapedesis? What are the molecular mechanisms of reverse signaling? Is there a significant role for in-vivo reverse signaling? Is PD-1 a therapeutic target in RA? Do T peripheral helper (Tph) cells have specific migratory properties? Are plasmablasts induced by Tph pathogenic? Do they produce anti-CCP antibodies? How to modulate immunization against anti-TNF? How to improve targeted anti-TNF biotherapy?
Differences between antibodies (IFX and ADA), and monovalent CZP, and ETA were outlined in the present review. They are summarized in Table 1. It appears that ETA and CZP induce less cell death and apoptosis than IFX and ADA. Alteration of the immune response to infections is less pronounced with ETA than with IFX and ADA but control of bacterial and viral infections is decreased by anti-TNF, and assessment of the infection and vaccine status is required. Vaccinations are recommended but not those using attenuated viruses or bacteria.
The mode of action, especially on T cells, of TNF-inhibitors is still not completely understood. For example, reverse signaling induced by TNF inhibitors must be explored in more detail. Future therapeutic strategy for RA should still take TNF inhibitors into account despite the availability of other biologics targeting other cytokines such as IL-6 and the more recent advent of JAKi. The choice of molecule should depend on better knowledge of the mode of action of the various TNF inhibitors. Nonspecific effects of anti-TNF antibodies on the immune system plead for a more targeted action such as bispecific antibodies targeting cells on the one hand and proinflammatory cytokine on the other [65].
Abbreviations
- ADA:
-
Adalimumab
- CMV:
-
Cytomegalovirus
- CZP:
-
Certolizumab pegol
- DC:
-
Dendritic cell
- ETA:
-
Etanercept
- IFX:
-
Infliximab
- IFN:
-
Interferon
- IL:
-
Interleukin
- iTreg:
-
Inducible regulatory T cell
- RA:
-
Rheumatoid arthritis
- SLO:
-
Secondary lymphoid organs
- Th:
-
T helper
- TGF:
-
Transforming growth factor
- tm:
-
Transmembrane
- TNF:
-
Tumor necrosis factor
- Tph:
-
T peripheral helper
- Treg:
-
Regulatory T cell
- TRM :
-
Resident memory T cells
- VZV:
-
Varicella zoster virus
References
Feldmann M, Maini RN. Anti-TNF therapy, from rationale to standard of care: what lessons has it taught us? J Immunol. 2010;185:791–4.
Kalliolias GD, Ivashkiv LB. TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat Rev Rheumatol. 2016;12:49–62.
Horiuchi T, Mitoma H, Harashima S, Tsukamoto H, Shimoda T. Transmembrane TNF-alpha: structure, function and interaction with anti-TNF agents. Rheumatology (Oxford). 2010;49:1215–28.
McCann FE, Perocheau DP, Ruspi G, Blazek K, Davies ML, Feldmann M, et al. Selective tumor necrosis factor receptor I blockade is antiinflammatory and reveals immunoregulatory role of tumor necrosis factor receptor II in collagen-induced arthritis: immunoregulatory role of TNFRII in CIA. Arthritis Rheumatol. 2014;66:2728–38.
Benson RA, McInnes IB, Garside P, Brewer JM. Model answers: rational application of murine models in arthritis research. Eur J Immunol. 2018;48:32–8.
Gourraud P-A, Dieudé P, Boyer J-F, Nogueira L, Cambon-Thomsen A, Mazières B, et al. A new classification of HLA-DRB1 alleles differentiates predisposing and protective alleles for autoantibody production in rheumatoid arthritis. Arthritis Res Ther. 2007;9:R27.
Roudier J, Balandraud N, Auger I. HLA-DRB1 polymorphism, anti-citrullinated protein antibodies, and rheumatoid arthritis. J Biol Chem. 2018;293:7038.
Kremer JM, Westhovens R, Leon M, Di Giorgio E, Alten R, Steinfeld S, et al. Treatment of rheumatoid arthritis by selective inhibition of T-cell activation with fusion protein CTLA4Ig. N Engl J Med. 2003;349:1907–15.
Firestein GS. The T cell cometh: interplay between adaptive immunity and cytokine networks in rheumatoid arthritis. J Clin Invest. 2004;114:471–4.
Baseta JG, Stutman O. TNF regulates thymocyte production by apoptosis and proliferation of the triple negative (CD3-CD4-CD8-) subset. J Immunol. 2000;165:5621–30.
Guevara Patiño JA, Ivanov VN, Lacy E, Elkon KB, Marino MW, Nikolic-Zugić J. TNF-alpha is the critical mediator of the cyclic AMP-induced apoptosis of CD8+4+ double-positive thymocytes. J Immunol. 2000;164:1689–94.
Webb LV, Ley SC, Seddon B. TNF activation of NF-κB is essential for development of single-positive thymocytes. J Exp Med. 2016;213:1399–407.
Anolik JH, Ravikumar R, Barnard J, Owen T, Almudevar A, Milner ECB, et al. Cutting edge: anti-tumor necrosis factor therapy in rheumatoid arthritis inhibits memory B lymphocytes via effects on lymphoid germinal centers and follicular dendritic cell networks. J Immunol. 2008;180:688–92.
Tumanov AV, Grivennikov SI, Kruglov AA, Shebzukhov YV, Koroleva EP, Piao Y, et al. Cellular source and molecular form of TNF specify its distinct functions in organization of secondary lymphoid organs. Blood. 2010;116:3456–64.
Chang JT, Wherry EJ, Goldrath AW. Molecular regulation of effector and memory T cell differentiation. Nat Immunol. 2014;15:1104–15.
Suresh M, Singh A, Fischer C. Role of tumor necrosis factor receptors in regulating CD8 T-cell responses during acute lymphocytic choriomeningitis virus infection. J Virol. 2005;79:202–13.
Jaczewska J, Abdulreda MH, Yau CY, Schmitt MM, Schubert I, Berggren P-O, et al. TNF-α and IFN-γ promote lymphocyte adhesion to endothelial junctional regions facilitating transendothelial migration. J Leukoc Biol. 2014;95:265–74.
Kim EY, Priatel JJ, Teh S-J, Teh H-S. TNF receptor type 2 (p75) functions as a costimulator for antigen-driven T cell responses in vivo. J Immunol. 2006;176:1026–35.
Chen X, Oppenheim JJ. Contrasting effects of TNF and anti-TNF on the activation of effector T cells and regulatory T cells in autoimmunity. FEBS Lett. 2011;585:3611–8.
Érsek B, Molnár V, Balogh A, Matkó J, Cope AP, Buzás EI, et al. CD3ζ-chain expression of human T lymphocytes is regulated by TNF via Src-like adaptor protein-dependent proteasomal degradation. J Immunol. 2012;189:1602–10.
Chimenti MS, Tucci P, Candi E, Perricone R, Melino G, Willis AE. Metabolic profiling of human CD4+ cells following treatment with methotrexate and anti-TNF-α infliximab. Cell Cycle. 2013;12:3025–36.
Cope AP, Londei M, Chu NR, Cohen SB, Elliott MJ, Brennan FM, et al. Chronic exposure to tumor necrosis factor (TNF) in vitro impairs the activation of T cells through the T cell receptor/CD3 complex; reversal in vivo by anti-TNF antibodies in patients with rheumatoid arthritis. J Clin Invest. 1994;94:749–60.
Mitoma H, Horiuchi T, Hatta N, Tsukamoto H, Harashima S-I, Kikuchi Y, et al. Infliximab induces potent anti-inflammatory responses by outside-to-inside signals through transmembrane TNF-alpha. Gastroenterology. 2005;128:376–92.
Ueda N, Tsukamoto H, Mitoma H, Ayano M, Tanaka A, Ohta S, et al. The cytotoxic effects of certolizumab pegol and golimumab mediated by transmembrane tumor necrosis factor α. Inflamm Bowel Dis. 2013;19:1224–31.
Toubi E, Kessel A, Mahmudov Z, Hallas K, Rozenbaum M, Rosner I. Increased spontaneous apoptosis of CD4+CD25+ T cells in patients with active rheumatoid arthritis is reduced by infliximab. Ann N Y Acad Sci. 2005;1051:506–14.
Aerts NE, De Knop KJ, Leysen J, Ebo DG, Bridts CH, Weyler JJ, et al. Increased IL-17 production by peripheral T helper cells after tumour necrosis factor blockade in rheumatoid arthritis is accompanied by inhibition of migration-associated chemokine receptor expression. Rheumatology (Oxford). 2010;49:2264–72.
Talotta R, Berzi A, Atzeni F, Batticciotto A, Clerici M, Sarzi-Puttini P, et al. Paradoxical expansion of Th1 and Th17 lymphocytes in rheumatoid arthritis following infliximab treatment: a possible explanation for a lack of clinical response. J Clin Immunol. 2015;35:550–7.
Szalay B, Vásárhelyi B, Cseh A, Tulassay T, Deák M, Kovács L, et al. The impact of conventional DMARD and biological therapies on CD4+ cell subsets in rheumatoid arthritis: a follow-up study. Clin Rheumatol. 2014;33:175–85.
Dulic S, Vásárhelyi Z, Sava F, Berta L, Szalay B, Toldi G, et al. T-cell subsets in rheumatoid arthritis patients on long-term anti-TNF or IL-6 receptor blocker therapy. Mediat Inflamm. 2017;2017:6894374.
Hull DN, Cooksley H, Chokshi S, Williams RO, Abraham S, Taylor PC. Increase in circulating Th17 cells during anti-TNF therapy is associated with ultrasonographic improvement of synovitis in rheumatoid arthritis. Arthritis Res Ther. 2016;18:303.
Cosmi L, Liotta F, Maggi E, Romagnani S, Annunziato F. Th17 and non-classic Th1 cells in chronic inflammatory disorders: two sides of the same coin. Int Arch Allergy Immunol. 2014;164:171–7.
Chen D-Y, Chen Y-M, Chen H-H, Hsieh C-W, Lin C-C, Lan J-L. Increasing levels of circulating Th17 cells and interleukin-17 in rheumatoid arthritis patients with an inadequate response to anti-TNF-α therapy. Arthritis Res Ther. 2011;13:R126.
Alzabin S, Abraham SM, Taher TE, Palfreeman A, Hull D, McNamee K, et al. Incomplete response of inflammatory arthritis to TNFα blockade is associated with the Th17 pathway. Ann Rheum Dis. 2012;71:1741–8.
McGovern JL, Nguyen DX, Notley CA, Mauri C, Isenberg DA, Ehrenstein MR. Th17 cells are restrained by Treg cells via the inhibition of interleukin-6 in patients with rheumatoid arthritis responding to anti-tumor necrosis factor antibody therapy. Arthritis Rheum. 2012;64:3129–38.
Evans HG, Roostalu U, Walter GJ, Gullick NJ, Frederiksen KS, Roberts CA, et al. TNF-α blockade induces IL-10 expression in human CD4+ T cells. Nat Commun. 2014;5:3199.
Aerts NE, Ebo DG, Bridts CH, Stevens WJ, De Clerck LS. T cell signal transducer and activator of transcription (STAT) 4 and 6 are affected by adalimumab therapy in rheumatoid arthritis. Clin Exp Rheumatol. 2010;28:208–14.
Conrad C, Di Domizio J, Mylonas A, Belkhodja C, Demaria O, Navarini AA, et al. TNF blockade induces a dysregulated type I interferon response without autoimmunity in paradoxical psoriasis. Nat Commun. 2018;9:25.
Wendling D, Prati C. Paradoxical effects of anti-TNF-α agents in inflammatory diseases. Expert Rev Clin Immunol. 2014;10:159–69.
Rao DA, Gurish MF, Marshall JL, Slowikowski K, Fonseka CY, Liu Y, et al. Pathologically expanded peripheral T helper cell subset drives B cells in rheumatoid arthritis. Nature. 2017;542:110–4.
Chen X, Wu X, Zhou Q, Howard OMZ, Netea MG, Oppenheim JJ. TNFR2 is critical for the stabilization of the CD4+Foxp3+ regulatory T cell phenotype in the inflammatory environment. J Immunol. 2013;190:1076–84.
Byng-Maddick R, Ehrenstein MR. The impact of biological therapy on regulatory T cells in rheumatoid arthritis. Rheumatology (Oxford). 2015;54:768–75.
Nie H, Zheng Y, Li R, Guo TB, He D, Fang L, et al. Phosphorylation of FOXP3 controls regulatory T cell function and is inhibited by TNF-α in rheumatoid arthritis. Nat Med. 2013;19:322–8.
Zaragoza B, Chen X, Oppenheim JJ, Baeyens A, Gregoire S, Chader D, et al. Suppressive activity of human regulatory T cells is maintained in the presence of TNF. Nat Med. 2016;22:16–7.
Nadkarni S, Mauri C, Ehrenstein MR. Anti-TNF-alpha therapy induces a distinct regulatory T cell population in patients with rheumatoid arthritis via TGF-beta. J Exp Med. 2007;204:33–9.
Nguyen DX, Ehrenstein MR. Anti-TNF drives regulatory T cell expansion by paradoxically promoting membrane TNF-TNF-RII binding in rheumatoid arthritis. J Exp Med. 2016;213:1241–53.
Rossol M, Meusch U, Pierer M, Kaltenhäuser S, Häntzschel H, Hauschildt S, et al. Interaction between transmembrane TNF and TNFR1/2 mediates the activation of monocytes by contact with T cells. J Immunol. 2007;179:4239–48.
Parry SL, Sebbag M, Feldmann M, Brennan FM. Contact with T cells modulates monocyte IL-10 production: role of T cell membrane TNF-alpha. J Immunol. 1997;158:3673–81.
Wallis RS. Infectious complications of tumor necrosis factor blockade. Curr Opin Infect Dis. 2009;22:403–9.
Bruns H, Meinken C, Schauenberg P, Härter G, Kern P, Modlin RL, et al. Anti-TNF immunotherapy reduces CD8+ T cell-mediated antimicrobial activity against Mycobacterium tuberculosis in humans. J Clin Invest. 2009;119:1167–77.
Hamdi H, Mariette X, Godot V, Weldingh K, Hamid AM, Prejean M-V, et al. Inhibition of anti-tuberculosis T-lymphocyte function with tumour necrosis factor antagonists. Arthritis Res Ther. 2006;8:R114.
Slifman NR, Gershon SK, Lee J-H, Edwards ET, Braun MM. Listeria monocytogenes infection as a complication of treatment with tumor necrosis factor alpha-neutralizing agents. Arthritis Rheum. 2003;48:319–24.
Kaech SM, Wherry EJ, Ahmed R. Effector and memory T-cell differentiation: implications for vaccine development. Nat Rev Immunol. 2002;2:251–62.
Kim SY, Solomon DH. Tumor necrosis factor blockade and the risk of viral infection. Nat Rev Rheumatol. 2010;6:165–74.
Davignon J-L, Boyer J-F, Jamard B, Nigon D, Constantin A, Cantagrel A. Maintenance of cytomegalovirus-specific CD4pos T-cell response in rheumatoid arthritis patients receiving anti-tumor necrosis factor treatments. Arthritis Res Ther. 2010;12:R142.
Carroll MB, Bond MI. Use of tumor necrosis factor-alpha inhibitors in patients with chronic hepatitis B infection. Semin Arthritis Rheum. 2008;38:208–17.
Nard FD, Todoerti M, Grosso V, Monti S, Breda S, Rossi S, et al. Risk of hepatitis B virus reactivation in rheumatoid arthritis patients undergoing biologic treatment: extending perspective from old to newer drugs. World J Hepatol. 2015;7:344–61.
Viganò M, Degasperi E, Aghemo A, Lampertico P, Colombo M. Anti-TNF drugs in patients with hepatitis B or C virus infection: safety and clinical management. Expert Opin Biol Ther. 2012;12:193–207.
Balog A, Klausz G, Gál J, Molnár T, Nagy F, Ocsovszky I, et al. Investigation of the prognostic value of TNF-alpha gene polymorphism among patients treated with infliximab, and the effects of infliximab therapy on TNF-alpha production and apoptosis. Pathobiology. 2004;71:274–80.
Unutmaz D, Pileri P, Abrignani S. Antigen-independent activation of naive and memory resting T cells by a cytokine combination. J Exp Med. 1994;180:1159–64.
Aversa G, Punnonen J, de Vries JE. The 26-kD transmembrane form of tumor necrosis factor alpha on activated CD4+ T cell clones provides a costimulatory signal for human B cell activation. J Exp Med. 1993;177:1575–85.
Kaine JL, Kivitz AJ, Birbara C, Luo AY. Immune responses following administration of influenza and pneumococcal vaccines to patients with rheumatoid arthritis receiving adalimumab. J Rheumatol. 2007;34:272–9.
Friedman MA, Winthrop KL. Vaccines and disease-modifying antirheumatic drugs: practical implications for the rheumatologist. Rheum Dis Clin N Am. 2017;43:1–13.
van Schouwenburg PA, Rispens T, Wolbink GJ. Immunogenicity of anti-TNF biologic therapies for rheumatoid arthritis. Nat Rev Rheumatol. 2013;9:164–72.
Cutolo M, Sulli A, Pizzorni C, Seriolo B, Straub RH. Anti-inflammatory mechanisms of methotrexate in rheumatoid arthritis. Ann Rheum Dis. 2001;60:729–35.
Drutskaya MS, Efimov GA, Kruglov AA, Nedospasov SA. Can we design a better anti-cytokine therapy? J Leukoc Biol. 2017;102:783–90.
Nesbitt A, Fossati G, Bergin M, Stephens P, Stephens S, Foulkes R, et al. Mechanism of action of certolizumab pegol (CDP870): in vitro comparison with other anti-tumor necrosis factor alpha agents. Inflamm Bowel Dis. 2007;13:1323–32.
Acknowledgements
We thank Sergei A. Nedospasov for critical reading of the manuscript.
Funding
This work was supported by CNRS “CoopInter” and a Pfizer “Passerelle” grant.
Author information
Authors and Affiliations
Contributions
All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Davignon, JL., Rauwel, B., Degboé, Y. et al. Modulation of T-cell responses by anti-tumor necrosis factor treatments in rheumatoid arthritis: a review. Arthritis Res Ther 20, 229 (2018). https://doi.org/10.1186/s13075-018-1725-6
Published:
DOI: https://doi.org/10.1186/s13075-018-1725-6