
Abstract
Introduction
The objective of this study was to investigate the effects of tumor necrosis factor (TNF)-α inhibitors on circulating T helper-type 17 (Th17) cells and Th17-related cytokines in patients with rheumatoid arthritis (RA).
Methods
The frequencies of circulating Th17 cells and serum levels of Th17-related cytokines were determined using flow cytometry analysis and ELISA, respectively, in 48 RA patients both before (baseline) and six months after anti-TNF-α therapy. Therapeutic response was evaluated using European League Against Rheumatism (EULAR) response criteria.
Results
Significantly higher baseline frequencies of circulating Th17 cells and serum levels of interleukin (IL)-6, IL-17, IL-21, IL-23 and TNF-α were observed in active RA patients than in 12 healthy controls (all P < 0.001). After anti-TNF-α therapy, 36 patients (75%) were EULAR responders (20 good responders and 16 moderate responders) and 12 (25.0%) were non-responders. The mean levels of circulating Th17 cells and IL-17 significantly decreased (1.13% vs. 0.79%; 43.1 pg/ml vs. 27.8 pg/ml; respectively, both P < 0.001) in parallel with clinical remission in responders. Levels of IL-6, IL-21, IL-23 and TNF-α were significantly decreased after anti-TNF-α therapy in responders. In contrast, the mean levels of circulating Th17 cells and IL-17 significantly increased after anti-TNF-α therapy (2.94% vs. 4.23%; 92.1 pg/ml vs. 148.6 pg/ml; respectively, both P < 0.05) in non-responders. Logistic regression analysis identified a high baseline level of IL-17 as a significant predictor of poor therapeutic response.
Conclusions
The beneficial effect of anti-TNF-α therapy might involve a decrease in Th17-related cytokines in responders, whereas rising levels of circulating Th17-cells and IL-17 were observed in patients with an inadequate response to anti-TNF-α therapy.
Similar content being viewed by others


Introduction
Rheumatoid arthritis (RA) is characterized by the infiltration of macrophages and T cells into the joints, synovial hyperplasia, cartilage degradation and bone erosions [1]. Tumor necrosis factor (TNF)-α is a crucial inflammatory mediator in rheumatoid synovitis and subsequent tissue damage in RA [2, 3]. Although TNF-α inhibitors can be an effective and well-tolerated therapy for RA patients [4–6], a significant proportion of patients do not acquire advantageous effects [7]. In addition, the effect of TNF-α inhibitors on the immune response has not been fully explored.
T helper-type 17 (Th17) cells, a novel and distinct subset of Th cell, can secrete interleukin (IL)-17 in humans [8–10]. Interleukin-17 is a pleiotropic cytokine that participates in tissue inflammation and destruction by inducing the expression of pro-inflammatory cytokines and matrix metalloproteases [8, 11, 12]. The frequencies of Th17 cells were found to increase in peripheral blood mononuclear cells (PBMCs) of RA patients compared to healthy controls [13, 14]. An enhanced expression of IL-17 has been observed in the rheumatoid synovium [15] and synovial fluids of patients with early RA [16]. Interleukin-17 in vitro stimulates the production of TNF-α and IL-1β, and also synergizes with TNF-α to induce cartilage loss and promote osteoclastogenesis [17, 18]. A recent study showed that Th17 cells, but not Th1 cells, cooperate with synovial fibroblasts in a pro-inflammatory feedback loop that drives chronic destruction in RA [19]. Moreover, IL-17 has become a new therapeutic target for animal models with collagen-induced arthritis (CIA) and human RA [20–22]. These observations suggest that Th17 cells and IL-17 critically contribute to synovitis and bone destruction associated with RA.
Recently, TNF-α was shown in vitro to drive the production of IL-17 with the ability to differentiate T cells towards a Th17 phenotype [23]. In a psoriasis-like skin inflammation model, TNF-α enhanced the expression of Th17-related cytokine genes during priming but suppressed these cytokine transcripts when present during re-stimulation [24]. In CIA, TNF-α inhibitors reduced the number of Th17 cells in pathologic joints despite an increase of Th17 cells in inguinal lymph nodes [25]. Taken together, these findings show that TNF-α blockade has paradoxical effects on the expression of Th17-related cytokines in animal models of autoimmune diseases.
In humans, an engineered p75 TNFRII dimer, etanercept, suppressed the gene expression levels of Th17-related cytokines including IL-6 and IL-23 in cutaneous lesions of psoriasis [26]. Kageyama et al. also reported a significant decrease in serum levels of IL-23 at three and six months after etanercept therapy in RA patients [27]. TNF-α inhibitor, adalmumab, reduced the frequency of circulating Th17 cells and serum IL-6 levels in RA patients [28]. However, a recent study showed that an increased frequency of circulating Th17 cells after TNF-α blockade is accompanied by a decrease in Th17-specific chemokine receptor expression in RA [29]. When taken together, these results reveal conflicting effects of TNF-α inhibitors on Th17 cells and IL-17 in humans.
In the present study, we attempted to determine whether or not the clinical response to anti-TNF-α therapy of RA patients led to changes in the levels of circulating Th17 cells and Th17-related cytokines, and we also investigated their clinical implication.
Materials and methods
Patients
A total of 48 consecutive patients (39 females and 9 males; mean age ± SD 50.1 ± 13.5 years), who fulfilled the 1987 revised criteria of the American College of Rheumatology for RA [30], were evaluated before and six months after anti-TNF-α therapy. All patients remained in active disease in spite of treatment with methotrexate (MTX) and other disease-modifying anti-rheumatic drugs (DMARDs), for whom anti-TNF-α therapy was initiated based on the British Society for Rheumatology guidelines [31]. Fourteen patients received etanercept at a dose of 25 mg twice weekly and 34 patients received adalimumab at a dose of 40 mg every other week in combination with a stable dose of MTX of 7.5 to 15 mg weekly. Corticosteroids (≦10 mg/day) and non-steroid anti-inflammatory drugs (NSAIDs) were allowed but were given at stable doses for at least four weeks before and during the six-month anti-TNF-α therapy. Disease activity was assessed by the 28-joint disease activity score (DAS28) [32]. The therapeutic response was evaluated six months after anti-TNF-α therapy was started, according to the European League Against Rheumatism (EULAR) response criteria [33]. The patients were categorized into good, moderate or non-responders based on the amount of change in the DAS28 and the level of DAS28 reached. Good responders are defined as patients who have a decrease in DAS28 from baseline (ΔDAS28) of >1.2 and a DAS28 at the sixth month of < 3.2; moderate responders have either ΔDAS28 of >1.2 and a DAS28 at the sixth month of ≧3.2 or ΔDAS28 of 0.6 to 1.2 and a DAS28 at the sixth month of < 5.1; and non-responders are those who have either ΔDAS28 of < 0.6 or a DAS28 at the sixth month of ≧5.1 [33]. To obtain a better analysis, we combined good responders and moderate responders into EULAR responders. Twelve age- and sex-matched healthy volunteers (10 females and 2 males, 47.6 ± 8.1 years), who had no rheumatic disease, were used as normal controls. Blood samples were collected at baseline (before starting anti-TNF-α therapy) and six months after anti-TNF-α therapy. The Ethics Committee of Taichung Veterans General Hospital approved this study and the written consent of each participant was obtained.
Quantitation of circulating Th17cells using flow cytometry analysis
In order to detect circulating Th17 cells, phycoerythrin (PE)-conjugated anti-IL-17 (eBioscience, San Diego, CA, USA) and Phycoerythrin-Cyanin 5 (PC5)-conjugated anti-CD4 (Beckman Coulter, Marseilles, France) were quantified using flow cytometry according to the manufacturer's protocol and a technique previously described [34, 35]. Briefly, aliquots of 1,000 μl of the sterile heparinized whole blood were stimulated with a combination of 25 ng/ml of phorbol myristate acetate and 1 μg/ml of ionomycin (Sigma, Deisenhofen, Germany) and cultured for one hour at 37°C in a humidified 5% CO2 incubator. Whole blood was treated with 10 μg/ml of Brefeldin A (Sigma, Germany) to inhibit intracellular protein transport. Activated cultures of blood samples were washed in wash buffer (phosphate buffered saline, 5% foetal bovine serum, 0.1% sodium azide; Merck, Darmstadt, Germany) and then stained with 20 μl of PC5-conjugated CD4-specific monoclonal antibody (mAb) (Beckman Coulter, Marseilles, France) for 15 minutes at room temperature (RT). Erythrocytes were lysed by adding 2 ml of fluorescence-activated cell sorter (FACS) lysing solution (Becton Dickinson, Lincoln Park, NJ, USA). After five minutes of incubation, the samples were centrifuged and washed with 0.1% BSA-PBS, and subsequently fixed with 100 μl Reagent 1 (Beckman Coulter, Marseilles, France) for 10 minutes. After washing, the pellet was incubated with 100 μl Reagent 2, saponin (Beckman Coulter, Marseilles, France) for five minutes at RT in the dark. The samples were washed twice with 0.1% BSA-PBS and then incubated with PE-conjugated IL-17-specific mAb (eBiosciences, San Diego, CA, USA) for 30 minutes at RT in the dark. An isotype control IgG1-PE (eBiosciences, USA) was used for the IL-17 staining at RT in the dark. After staining, the cells were washed and immediately analysed using flow cytometry (Beckman Coulter, USA). Lymphocytes were gated on the basis of forward- and side- scatter properties and at least 10,000 CD4+ cells were analysed. The results were analysed using Expo32 software (Beckman Coulter, Miami, FL, USA).
Determination of serum levels of Th17-related cytokines by ELISA
Serum levels of IL-6, IL-17, IL-21, IL-23 and TNF-α were determined in 48 RA patients at baseline and after six months of anti-TNF-α therapy, and in 12 healthy controls using enzyme-linked immunosorbent assay (ELISA) according to the manufacturer's instructions (eBiosciences, USA).
Determination of serum levels of anti-cyclic citrullinated peptide (anti-CCP) antibody and rheumatoid factor (RF)-IgM
Determination of the anti-CCP antibody was performed by ELISA using a commercial kit (INOVA Diagnostics Inc., San Diego, CA, USA). A result was considered positive for anti-CCP antibodies if the titer was above 20 IU/ml. Serum levels of RF-IgM were measured by nephelometry (Dade Behring Inc., Newark, DE, USA). A result was considered positive for RF when the concentration was above 15 IU/ml.
Statistical analysis
The results are presented as the mean ± SD or median (interquartile range, IQR). The non-parametric Mann-Whitney U test was used for between-group comparisons of serum levels of IL-6, IL-17, IL-21, IL-23 and TNF-α, and for the percentages of circulating Th17 cells. The correlation coefficient was obtained by the non-parametric Spearman's rank correlation test. The Wilcoxon signed rank test was used to compare the percentages of circulating Th17 cells and serum levels of Th17-related cytokines during follow-up for the RA patients after anti-TNF-α therapy. A probability of less than 0.05 was considered significant.
Results
Baseline characteristics of RA patients with a different therapeutic response
As illustrated in Table 1, the majority of RA patients were female and all patients had active disease (DAS28, mean ± SD, 7.22 ± 0.79) before starting anti-TNF-α therapy. After anti-TNF-α therapy, 36 (75.0%) patients were EULAR responders, including 20 (41.7%) good responders and 16 (33.3%) moderate responders, whereas 12 (25.0%) were non-responders. Although non-responders had higher erythrocyte sedimentation rates (ESR) and RF titers compared to responders, the differences did not reach statistical significance. There were also no significant differences in the baseline demographic data, positive rate of RF and anti-CCP antibodies, daily dose of corticosteroids, or the proportion of patients previously using DMARDs between responders and non-responders.
The frequencies of circulating Th17 cells in RA patients before anti-TNF-α therapy
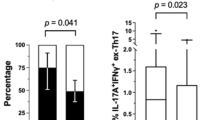
Representative examples of flow cytometric dot-plots of intracellular IL-17 staining in Th cells obtained from the PB of one RA patient and from a healthy control are shown in Figure 1 A. Significantly higher baseline frequencies of circulating Th17 cells were observed in RA patients (median 1.11%, IQR 0.50% to 2.05%) than in healthy controls (median 0.12%, IQR 0.05% to 0.18%; P < 0.001, Figure 1B). Among the RA patients, significantly higher baseline frequencies of circulating Th17 cells were observed in EULAR non-responders than in responders (P < 0.01, Figure 2A).

The comparison of circulating levels of Th17-cells and Th17-related cytokines between RA patients and HC. (A) Flow cytometric dot-plots of intracellular IL-17 production in Th cells obtained from peripheral blood of one representative patient with rheumatoid arthritis and healthy control. The comparison of circulating Th17 cells frequencies is shown (B) between 48 RA patients before starting anti-TNF-α therapy (baseline) and 12 HC. The comparison of baseline levels of serum IL-17 (C), IL-6 (D), IL-21 (E), and IL-23 (F) is shown between RA patients and HC. The horizontal line indicates median value. P-value was assessed by Mann-Whitney U test. HC, healthy control; IL, interleukin; RA, rheumatoid arthritis; Th17 cells, T helper-type 17 cells; TNF-α, tumor necrosis factor alpha.

The change in circulating levels of Th17-cells and Th17-related cytokines in EULAR responders and non-responders. The changes in (A) the frequencies of circulating Th17-cells (T helper-type 17 cells) and serum levels of (B) interleukin (IL)-17, (C) IL-6, (D) IL-21, (E) IL-23, and (F) tumor necrosis factor (TNF)-α in 36 responders and 12 non-responders to anti-TNF-α therapy according to European League Against Rheumatism (EULAR) response criteria. Bars represent the mean value and SEM. *P < 0.05, **P < 0.005, ***P < 0.001, versus before starting anti-TNF-α therapy, determined by the Wilcoxon signed rank test.#P < 0.01, non-responders versus responders.
Serum levels of Th17-related cytokines in RA patients before anti-TNF-α therapy
As shown in Figure 1, the baseline serum levels of Th17-related cytokines including IL-17, IL-6, IL-21 and IL-23, were significantly higher in RA patients than in healthy controls. Among the RA patients, significantly higher IL-17 levels were observed in non-responders than in responders (P < 0.01, Figure 2 and Table 2). Although non-responders seemed to have higher levels of IL-6 and TNF-α when compared to the levels in responders, the differences did not reach statistical significance. There was also no significant difference in the levels of Th17-related cytokines between the etanercept-treated group and the adalimumab-treated group (data not shown).
Correlation between the frequencies of circulating Th17 cells and disease activity or serum levels of Th17-related cytokines in RA patients
The frequencies of circulating Th17 cells were positively correlated with DAS28 scores (r = 0.294, P < 0.05), IL-17 levels (r = 0.737, P < 0.001), IL-6 levels (r = 0.347, P < 0.05), and TNF-α levels (r = 0.495, P < 0.001) in RA patients before the anti-TNF-α therapy was started. Serum IL-6 levels were also positively correlated with IL-17 levels (r = 0.303, P < 0.05) in RA patients. After six months of anti-TNF-α therapy, the frequencies of circulating Th17 cells remained positively correlated with DAS28 scores (r = 0.375, P < 0.01), IL-17 levels (r = 0.727, P < 0.001), IL-6 levels (r = 0.311, P < 0.05), and TNF-α levels (r = 0.362, P < 0.05).
Effects of TNF-α inhibitors on the levels of Th17 cells and Th17-related cytokines
As illustrated in Figure 2 and Table 2, the mean levels of circulating Th17 cells and IL-17 significantly declined after six-month anti-TNF-α therapy (1.13% vs. 0.79%; 43.1 pg/ml vs. 27.8 pg/ml; respectively, both P < 0.001), in parallel with the decrease in DAS28 (7.18 vs. 5.21, P < 0.001) and in anti-CCP titers (58.7 ± 9.6 vs. 51.8 ± 8.7 IU/ml, P < 0.005) in responders. Serum levels of IL-6, IL-21, IL-23 and TNF-α were also significantly decreased after anti-TNF-α therapy in responders. In contrast, the mean levels of circulating Th17 cells and IL-17 significantly increased (2.94% vs. 4.23%; 92.1 pg/ml vs. 148.6 pg/ml; respectively, both P < 0.05), while TNF-α levels decreased after anti-TNF-α therapy (29.2 pg/ml vs. 15.2 pg/ml, P < 0.005) in non-responders. There were no significant changes in RF titers after anti-TNF-α therapy in either responders or non-responders.
Multiple logistic regression analysis of the effects of Th17 cell frequencies and serum Th17-related cytokines on therapeutic efficacy
Multiple logistic regression analysis showed that only a high baseline IL-17 level (≧40.0 pg/ml) could significantly predict a poor response to anti-TNF-α therapy (P < 0.01), with a medium level of specificity (83.3%) and sensitivity (66.7%).
Discussion
Consistent with the findings of previous reports [36, 37], our results showed that 36 (75.0%) patients have moderate or good EULAR response to the six-month anti-TNF-α therapy and 12 (25.0%) were non-responders. Although non-responders seem to have higher baseline ESR, titers of RF and anti-CCP antibodies, and serum levels of TNF-α and IL-6, when compared to responders, the differences do not reach statistical significance. Our findings suggest that an inadequate response to anti-TNF-α therapy is not merely a reflection of high disease activity.
The present study is the first attempt to investigate the effects of TNF-α inhibitors on the levels of circulating Th17 cells and Th17-related cytokines in RA patients with a different therapeutic response. In order to obtain a better reflection of in vivo cytokine patterns than is achievable with PBMCs, whole blood was stimulated with mitogens and lymphocytes double-stained with IL-17 and CD4 were analysed using flow cytometry. Our results showed significantly higher baseline frequencies of circulating Th17 cells in active RA patients compared with healthy controls, confirming the findings of previous studies [13, 14, 29]. We also showed that the baseline levels of circulating Th17 cells were positively correlated with DAS28 scores in RA patients. Our data and the findings of previous studies [15, 16] suggest a potential role of Th17 cells in the pathogenesis of RA.
Similar to the results of recent studies [28], our findings showed a significant decrease in the frequencies of circulating Th17 cells after anti-TNF-α therapy, in parallel with the decrease in DAS28 in EULAR responders. Although the diseases studied were different, our results were also consistent with the findings of previous studies which showed that the TNF-α inhibitor reduced Th17 cell responses with the amelioration of psoriatic skin lesions [38] and that the TNFR1 inhibitor suppressed the Th17 response with clinical benefits in experimental autoimmune encephalomyelitis mice [39]. In contrast, our results showed a significant increase in circulating Th17 cell frequencies in non-responders. Our findings in non-responders were similar to the results of a recent study that showed an increased frequency of circulating Th17 cells after TNF-α blockade in RA patients [29]. The divergent effects on Th17 cells in vivo after anti-TNF-α therapy for RA patients suggest that there may be different subsets of the immune response to TNF-α inhibitors. It is tempting to speculate that the change in frequencies of circulating Th17 cells may help to differentiate between EULAR responders and non-responders because these changes occur within the first six months of anti-TNF-α therapy.
The Th17 cells have a specific role in immune function through the production of effector cytokines. Through the secretion of IL-17, Th17 cells act on the differentiation of osteoclasts and bone resorption [40], and can stimulate the monocytes to produce pro-inflammatory cytokines, thus amplifying the inflammatory cascade [11, 17, 18]. In the present study, we showed that serum IL-17 levels were significantly elevated and correlated with DAS28 in active RA patients. Our data were consistent with the results of recent studies describing an increase in IL-17 levels in RA patients [14, 41], and they also supported the results of clinical trials showing the therapeutic benefits of IL-17 blockade [20–22]. During a longitudinal follow-up of RA patients who received anti-TNF-α therapy, we found that serum IL-17 levels significantly decreased, in parallel with the clinical remission in the responders, whereas increasing IL-17 levels were found in non-responders. Whether or not this imbalance of Th17 cells and IL-17 between PB and affected joints contributes to the change in Th17-related cytokines after anti-TNF-α therapy remains unclear.
Accumulating evidence indicates that IL-6 can enhance Th17 cell differentiation by promoting the sequential engagement of IL-21/IL-23 pathways and that it plays a critical role in Th17-dependent autoimmune diseases [42]. In the present study, we showed that the high baseline levels of IL-6 were positively correlated with the frequencies of circulating Th17 cells in RA patients. After anti-TNF-α therapy, a significant decrease in the levels of circulating Th17 cells and IL-17, in parallel with the decrease in DAS28 and serum IL-6 levels was observed in responders. These observations support the role of IL-6 in the differentiation of Th17 cells [42–44], and the therapeutic benefits of IL-6 receptor inhibitors in RA patients [45].
Interleukin-21 is required to reinforce differentiation of Th17 cells, and it plays a critical role in Th17-dependent autoimmune diseases [46]. In contrast to IL-12, IL-23 does not promote the development of Th1 cells, but it is crucial for the expansion and maintenance of Th17 cells [47]. In the present study, we showed that the baseline levels of IL-21 and IL-23 were significantly elevated in active RA patients, supporting their role in the pathogenesis of this disease [42, 48]. In addition, we found that serum levels of IL-21 and IL-23 significantly decreased, in parallel with the clinical remission in responders after anti-TNF-α therapy. Consistent with our data, the results of the study by Kageyama et al. also showed a significant decrease in serum IL-23 levels at three and six months after anti-TNF-α (etanercept) therapy in RA patients [27].
The mechanisms of the therapeutic effects of TNF-α inhibitors on RA have not been fully explored. Lundy et al. showed that TNF-α has a positive feedback effect on Th17 cells, causing a vicious cycle of synovitis [49]. TNF-α also promotes IL-17 production by inducing dendritic cells to direct the differentiation of Th cells towards the Th17 phenotype [23], and by the administration of TNF-like ligand 1A enhancing Th17 differentiation with IL-17 production [50]. Our results showed a significant decrease in the levels of circulating Th17 cells and TNF-α after effective therapy with TNF-α inhibitors, suggesting that the down-regulation of both Th17-related cytokines and TNF-α may be one of the mechanisms of anti-TNF-α therapy [19]. Our findings support the hypothesis that anti-TNF-α therapy could reduce Th17 responses by blocking the TNF-α-dependent positive feedback system [23]. Another explanation may be the inhibition of emigration of Th17 cells from lymphoid organs after anti-TNF-α therapy [24].
In the present study, we showed that one-fourth of RA patients have an inadequate response to anti-TNF-α therapy despite a significant decrease in serum TNF-α level, suggesting that neutralization of TNF-α cannot fully explain the therapeutic effect of TNF-α inhibitors in non-responders. Additionally, significantly higher baseline levels of circulating Th17-cells and IL-17 were observed in non-responders when compared to responders. During a longitudinal follow-up of RA patients undergoing anti-TNF-α therapy, we found a significant increase in circulating levels of Th17-cells and IL-17 in non-responders. Although the cut-off value for baseline IL-17 (40 pg/mL) used in our study was not absolute, a multivariate logistic regression analysis showed that only a high baseline level of IL-17 could be a significant predictor of poor therapeutic response to TNF-α inhibitors. Our observations suggest that an inadequate response to anti-TNF-α therapy may reflect TNF-independent but Th17-dominant inflammatory process, which has been observed in previous studies [15, 16].
This was a preliminary study that enrolled a limited number of active RA patients who were followed up for six months. Because we mainly investigate the effects of anti-TNF-α therapy on Th17 cells and Th17-related cytokines, we did not include a group of RA patients without anti-TNF-α therapy as a disease control. Because the patients enrolled in our study were not an early RA population, our results might not be directly applicable to early RA patients undergoing anti-TNF-α therapy. Therefore, a long-term study enrolling a larger group of patients, including an additional early RA population, or a control group using DMARDs only, is required to validate these findings. Although the erroneous increase in IL-17 levels may have been due to the heterophilic binding by serum RF [51, 52], there were no significant differences in RF positivity or titers between responders and non-responders in the present study.
Conclusions
Our results show that anti-TNF-α therapy not only neutralizes the effects of TNF-α, but it also down-regulates the IL-6/Th17 axis in EULAR responders. In contrast, anti-TNF-α therapy causes a significant increase in Th17 cells in RA patients with an inadequate response to anti-TNF-α therapy. A high baseline level of IL-17 may have a predictive value for poor therapeutic response to TNF-α inhibitors. Although the small sample size of this study did not allow for a definitive conclusion, our findings at least suggest that Th17-dependent inflammation is implicated in the pathogenesis of a subset of RA patients with poor response to TNF-α inhibitors, and that treatment targeting IL-17 may be beneficial for these patients [21, 22].
Abbreviations
- Anti-CCP:
-
anti-cyclic citrullinated peptide
- CIA:
-
collagen-induced arthritis
- DAS28:
-
28-joint disease activity score
- DMARDs:
-
disease-modifying anti-rheumatic drugs
- ELISA:
-
enzyme-linked immunosorbent assay
- ESR:
-
erythrocyte sedimentation rate
- EULAR:
-
European League Against Rheumatism
- IL:
-
interleukin
- MTX:
-
methotrexate
- NSAIDs:
-
non-steroid anti-inflammatory drugs
- PBMCs:
-
peripheral blood mononuclear cells
- PC5:
-
Phycoerythrin-Cyanin 5
- PE:
-
phycoerythrin
- RA:
-
rheumatoid arthritis
- RF:
-
rheumatoid factor
- RT:
-
room temperature
- Th17 cells:
-
T helper-type 17 cells
- TNF-α:
-
tumor necrosis factor-α.
References
Christodoulou C, Choy EH: Joint inflammation and cytokine inhibition in rheumatoid arthritis. Clin Exp Med. 2006, 6: 13-19. 10.1007/s10238-006-0088-5.
Choy EH, Panayi GS: Cytokine pathways and joint inflammation in rheumatoid arthritis. N Engl J Med. 2001, 344: 907-916. 10.1056/NEJM200103223441207.
Feldmann M, Maini RN: TNF defined as a therapeutic target for rheumatoid arthritis and other autoimmune diseases. Nat Med. 2003, 9: 1245-1250. 10.1038/nm939.
van der Heijde D, Klareskog L, Rodriguez-Valverde V, Codreanu C, Bolosiu H, Melo-Gomes J, Tornero-Molina J, Wajdula J, Pedersen R, Fatenejad S, TEMPO Study Investigators: Comparison of etanercept and methotrexate, alone and combined, in the treatment of rheumatoid arthritis: Two-year clinical and radiographic results from the TEMPO study, a double-blind, randomized trial. Arthritis Rheum. 2006, 54: 1063-1074. 10.1002/art.21655.
Breedveld FC, Weisman MH, Kavanaugh AF, Cohen SB, Pavelka K, van Vollenhoven R, Sharp J, Perez JL, Spencer-Green GT: The PREMIER study: a multicenter, randomized, double-blind clinical trial of combination therapy with adalimumab plus methotrexate versus methotrexate alone or adalimumab alone in patients with early, aggressive rheumatoid arthritis who had not had previous methotrexate treatment. Arthritis Rheum. 2006, 54: 26-37. 10.1002/art.21519.
Kievit W, Fransen J, Adang EMM, den Broeder AA, Moens HJB, Visser H, van de Laar MA, van Riel PL: Long-term effectiveness and safety of TNF-blocking agents in daily clinical practive: results from the Dutch rheumatoid arthritis monitoring register. Rheumatology. 2011, 50: 196-203. 10.1093/rheumatology/keq325.
Rubbert-Roth A, Finckh A: Treatment options in patients with rheumatoid arthritis failing initial TNF inhibitor therapy: a critical review. Arthritis Res Ther. 2009, 11: S1-10.1186/ar2666.
Bettelli E, Oukka M, Kuchroo VK: Th-17 cells in the circle of immunity and autoimmunity. Nat Immunol. 2007, 8: 345-350.
Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver CT: Interleukin17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineage. Nat Immunol. 2005, 6: 1123-1132. 10.1038/ni1254.
Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q, Dong C: A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005, 6: 1133-1141. 10.1038/ni1261.
Jovanovic DV, Di Battista JA, Martel-Pelletier J, Jolicoeur FC, He Y, Zhang M, Mineau F, Pelletier JP: IL-17 stimulates the production of proinflammatory cytokines, IL-1β and TNF-α, by human macrophages. J Immunol. 1998, 160: 3513-3521.
Agarwal S, Misra R, Aggarwal A: Interleukin 17 levels are increased in juvenile idiopathic arthritis synovial fluid and induce synovial fibroblasts to produce proinflammatory cytokines and matrix metalloproteinases. J Rheumatol. 2008, 35: 515-519.
Shen H, Goodall JC, Gaston JSH: Frequency and phenotype of peripheral blood Th17 cells in ankylosing spondylitis and rheumatoid arthritis. Arthritis Rheum. 2009, 60: 1647-1656. 10.1002/art.24568.
Colin EM, Asmawidjaja PS, van Hamburg JP, Mus AMC, van Driel M, Hazes JMW, van Leeuwen JP, Lubberts E: 1,25-Dihydroxyvitamn D3 modulates Th17 polarization and interleukin-22 expression by memory T cells from patients with early rheumatoid arthritis. Arthritis Rheum. 2010, 62: 132-42. 10.1002/art.25043.
Chabaud M, Lubberts E, Joosten L, van den Berg W, Miossec P: IL-17 derived from juxta-articular bone and synovium contributes to joint degradation in rheumatoid arthritis. Arthritis Res Ther. 2001, 3: 168-177.
Raza K, Falciani F, Curnow SJ, Ross EJ, Lee CY, Akbar AN, Lord JM, Gordon C, Buckley CD, Salmon M: Early rheumatoid arthritis is characterized by a distinct and transient synovial fluid cytokine profile of T cells and stromal cell origin. Arthritis Res Ther. 2005, 7: R784-795. 10.1186/ar1733.
Katz Y, Nadiv O, Beer Y: Interleukin-17 enhances tumor necrosis factor alpha-induced synthesis of interleukins1, 6, and 8 in skin and synovial fibroblasts: a possible role as a "fine-tuning cytokine" in inflammation processes. Arthritis Rheum. 2001, 44: 2176-2184. 10.1002/1529-0131(200109)44:9<2176::AID-ART371>3.0.CO;2-4.
Van Bezooijen RL, Van Der Wee-Pals L, Papapoulos SE, Lowik CW: Interleukin 17 synergies with tumor necrosis factor alpha to induce cartilage destruction in vitro. Ann Rheum Dis. 2002, 61: 870-876. 10.1136/ard.61.10.870.
Van Hamburg JP, Asmawidjaja PS, Davelaar N, Mus AMC, Colin EM, Hazes JMW, Dolhain RJ, Lubberts E: Th17 cells, but not Th1 cells, from patients with early rheumatoid arthritis are potent inducers of matrix metalloproteinases and proinflammatory cytokines upon synovial fibroblast interaction, including autocrine interleukin-17A production. Arthritis Rheum. 2011, 63: 73-83. 10.1002/art.30093.
Lubberts E, Koenders MI, Oppers-Walgreen B, van den Bersselaar L, Coenen-de Roo CJ, Joosten LA, van den Berg WB: Treatment with a neutralizing anti-murine interleukin-17 antibody after the onset of collagen-induced arthritis reduces joint inflammation, cartilage destruction, and bone erosion. Arthritis Rheum. 2004, 50: 650-659. 10.1002/art.20001.
Hueber W, Patel DD, Dryja T, Wright AM, Koroleva I, Bruin G, Antoni C, Draelos Z, Gold MH, Psoriasis Study Group, Durez P, Tak PP, Gomez-Reino JJ, Rheumatoid Arthritis Study Group, Foster CS, Kim RY, Samson CM, Falk NS, Chu DS, Callanan D, Nguyen QD, Uveitis Study Group, Rose K, Haider A, Di Padova F: Effects of AIN457, a fully human antibody to interleukin-17A, on psoriasis, rheumatoid arthritis and uveitis. Sci Transl Med. 2010, 2: 52ra72-10.1126/scitranslmed.3001107.
Genovese MC, Van den Bosch F, Roberson SA, Bojin S, Biagini IM, Ryan P, Sloan-Lancaster J: LY2439821, A humanized anti-IL-17 monoclonal antibody, in the treatment of patients with rheumatoid arthritis: a phase 1 randomized double-blind, placebo-controlled, proof of concept study. Arthritis Rheum. 2010, 62: 929-939. 10.1002/art.27334.
Iwamoto S, Iwai S, Tsujiyama K, Kurahashi C, Takeshita K, Naoe M, Masunaga A, Ogawa Y, Oguchi K, Miyazaki A: TNF-alpha drives human CD14+ monocytes to differentiate into CD70+ dendritic cells evolving Th1 and Th17 responses. J Immunol. 2007, 179: 1449-1457.
Ma HL, Napierata L, Stedman N, Benoit S, Collins M, Nickerson-Nutter C, Young DA: Tumor necrosis factor α blockade exacerbates murine psoriasis-like disease by enhancing Th17 function and decreasing expansion of Treg cells. Arthritis Rheum. 2010, 62: 430-440.
Notley CA, Inglis JJ, Alzabin S, McCann FE, McNamee KE, Williams RO: Blockade of tumor necrosis factor in collagen-induced arthritis reveals a novel immunoregulatory pathway for Th1 and Th17 cells. J Exp Med. 2008, 205: 2491-2497. 10.1084/jem.20072707.
Gottlieb AB, Chamian F, Masud S, Cardinale I, Abello MV, Lowes MA, Chen F, Magliocco M, Krueger JG: TNF inhibition rapidly down-regulates multiple proinflammatory pathways in psoriasis plaques. J Immunol. 2005, 175: 2721-2729.
Kageyama Y, Ichikawa T, Nagafusa T, Torikai E, Shimazu M, Nagano A: Etanercept reduces the serum levels of interleukin-23 and macrophage inflammatory protein-3 alpha in patients with rheumatoid arthritis. Rheumatol Int. 2007, 28: 137-143. 10.1007/s00296-007-0388-4.
Yue C, You X, Zhao L, Wang H, Tang F, Zhang F, Zhang X, He W: The effects of adalimumab and methotrexate treatment on peripheral Th17 cells and IL-17/IL-6 secretion in the rheumatoid arthritis patients. Rheumatol Int. 2009, 30: 1553-1557.
Aerts NE, De Knop KJ, Leysen J, Ebo DG, Bridts CH, Weyler JJ, Stevens WJ, De Clerck LS: Increased IL-17 production by peripheral T helper cells after tumor necrosis factor blockade in rheumatoid arthritis is accompanied by inhibition of migration-associated chemokine receptor expression. Rheumatology. 2010, 49: 2264-2272. 10.1093/rheumatology/keq224.
Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, Healey LA, Kaplan SR, Liang MH, Luthra HS, Medsger TA, Mitchell DM, Neustadt DH, Pinals RS, Schaller JG, Sharp JT, Wilder RL, Hunder GG: The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988, 31: 315-324. 10.1002/art.1780310302.
Ledingham J, Deighton C, British Society for Rheumatology Standards, Guidelines and Audit Working Group: Update on the British Society for Rheumatology guidelines for prescribing TNFalpha blockers in adults with rheumatoid arthritis (update of previous guidelines of April 2001). Rheumatology (Oxford). 2005, 44: 157-163.
Prevoo ML, van't Hof MA, Kuper HH, van Leeuwen MA, van de Putte LBA, van Riel PL: Modified disease activity scores that include twenty-eight-joint counts: development and validation in a prospective longitudinal study of patients with rheumatoid arthritis. Arthritis Rheum. 1995, 38: 44-48. 10.1002/art.1780380107.
Van Gestel AM, Prevoo ML, van't Hof MA, van Rijswijk MH, van de Putte LB, van Riel PL: Development and validation of the European League Against Rheumatism response criteria for rheumatoid arthritis. Comparison with the preliminary American College of Rheumatology and the World Health Organization/International League Against Rheumatism Criteria. Arthritis Rheum. 1996, 39: 34-40. 10.1002/art.1780390105.
Yamada H, Nakashima Y, Okazaki K, Mawatari T, Fukushi JI, Kaibara N, Hori A, Iwamoto Y, Yoshikai Y: Th1 but not Th17 cells predominate in the joints of patients with rheumatoid arthritis. Ann Rheum Dis. 2008, 67: 1299-1304.
Chen DY, Lan JL, Chen YM, Lin CC, Chen HH, Hsieh CW: Potential role of Th17 cells in the pathogenesis of adult-onset Still's disease. Rheumatology. 2010, 49: 2305-2312. 10.1093/rheumatology/keq284.
Atzeni F, Antivalle M, Pallavicini FB, Caporali R, Bazzani C, Gorla R, Favalli EG, Marchesoni A, Sarzi-Puttini P: Predicting response to anti-TNF treatment in rheumatoid arthritis patients. Autoimmun Rev. 2009, 8: 431-437. 10.1016/j.autrev.2009.01.005.
Kameda H, Ueki Y, Saito K, Nagaoka S, Hidaka T, Atsumi T, Tsukano M, Kasama T, Shiozawa S, Tanaka Y, Takeuchi T, Japan Biological Agent Study Integrated Consortium: Etanercept (ETN) with methotrexate (MTX) is better than ETN monotherapy in patients with active rheumatoid arthritis despite MTX therapy: a randomized trial. Mod Rheumatol. 2010, 20: 531-538. 10.1007/s10165-010-0324-4.
Zaba LC, Cardinale I, Gilleaudeau P, Sullivan-Whalen M, Suárez-Fariñas M, Fuentes-Duculan J, Novitskaya I, Khatcherian A, Bluth MJ, Lowes MA, Krueger JG: Amelioration of epidermal hyperplasia by TNF inhibition is associated with reduced Th17 responses. J Exp Med. 2007, 204: 3183-3194. 10.1084/jem.20071094.
Nomura T, Abe Y, Kamada H, Shibata H, Kayamuro H, Inoue M, Kawara T, Arita S, Furuya T, Yamashita T, Nagano K, Yoshikawa T, Yoshioka Y, Mukai Y, Nakagawa S, Taniai M, Ohta T, Serada S, Naka T, Tsunoda S, Tsutsumi Y: Therapeutic effect of PEGylated TNFR1-selective antagonistic mutant TNFF in experimental autoimmune encephalomyelitis mice. J Control Release. 2011, 149: 8-14. 10.1016/j.jconrel.2009.12.015.
Sato K, Suematsu A, Okamoto K, Yamaguchi A, Morishita Y, Kadono Y, Tanaka S, Kodama T, Akira S, Iwakura Y, Cua DJ, Takayanagi H: Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med. 2006, 203: 2673-2682. 10.1084/jem.20061775.
Kim KW, Cho ML, Park MK, Yoon CH, Park SH, Lee SH, Kim HY: Increased interleukin-17 production via a phosphoinositide 3-kinase/Akt and nuclear factor kappaB-dependent pathway in patients with rheumatoid arthritis. Arthritis Res Ther. 2005, 7: R139-148. 10.1186/ar1470.
Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, Levy DE, Leonard WJ, Littman DR: IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. 2007, 8: 967-974. 10.1038/ni1488.
Romagnani S: Human Th17 cells. Arthritis Res Ther. 2008, 10: 10-16. 10.1186/ar2360.
Chen Z, Tato CM, Muul L, Laurence A, ÓShea JJ: Distinct regulation of interleukn-17 in human T helper lymphocytes. Arthritis Rheum. 2007, 56: 2936-2946. 10.1002/art.22866.
Emery P, Keystone E, Tony HP, Cantagrel A, van Vollenhoven R, Sanchez A, Alecock E, Lee J, Kremer J: IL-6 receptor inhibition with tocilizumab improves treatment outcomes in patients with rheumatoid arthritis refractory to anti-tumor necrosis factor biologicals: results from a 24-week multicentre randomized placebo-controlled trial. Ann Rheum Dis. 2008, 67: 1516-1523.
Korn T, Bettelli E, Gao W, Awasthi A, Jäger A, Strom TB, Oukka M, Kuchroo VK: IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature. 2007, 448: 484-487. 10.1038/nature05970.
Stritesky GL, Yeh N, Kaplan MH: IL-23 promotes maintenance but not commitment to the Th17 lineage. J Immunol. 2008, 181: 5948-5955.
Liu FL, Chen CH, Chu SJ, Chen JH, Lai JH, Sytwu HK, Chang DM: Interleukin (IL)-23 p19 expression induced by IL-1β in human fibroblast-like synoviocytes with rheumatoid arthritis via active nuclear factor-κB and AP-1 dependent pathway. Rheumatology. 2007, 46: 1266-1273. 10.1093/rheumatology/kem055.
Lundy SK, Sarkar S, Tesmer LA, Fox DA: Cells of the synovium in rheumatoid arthritis. T lymphocytes. Arthritis Res Ther. 2007, 9: 202-212. 10.1186/ar2107.
Zhang J, Wang X, Fahmi H, Wojcik S, Fikes J, Yu Y, Wu J, Luo H: Role of TL1A in the pathogenesis of rheumatoid arthritis. J Immunol. 2009, 183: 5350-5357. 10.4049/jimmunol.0802645.
DeForge LE, Loyet KM, Delarosa D, Chinn J, Zamanian F, Chuntharapai A, Lee J, Hass P, Wei N, Townsend MJ, Wang J, Wong WL: Evaluation of heterophilic antibody blocking agents in reducing false positive interference in immunoassays for IL-17AA, IL-17FF, and IL-17AF. J Immunol Methods. 2010, 362: 70-81. 10.1016/j.jim.2010.09.004.
Todd DJ, Knowlton N, Amato M, Frank MB, Schur PH, Izmailova ES, Roubenoff R, Shadick NA, Weinblatt ME, Centola M, Lee DM: Erroneous augmentation of multiplex assay measurements in patients with rheumatoid arthritis due to heterophilic binding by serum rheumatoid factor. Arthritis Rheum. 2011, 63: 894-903. 10.1002/art.30213.
Acknowledgements
This study was supported by grant TCVGH-993803C from Taichung Veterans General Hospital, Taiwan.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
DYC and JLL participated in the study design, acquisition of data, interpretation of results, and manuscript preparation. YMC participated in the study design, acquisition of data, interpretation of results, and assisted in drafting the manuscript. HHC, CWH and CCL contributed to analysis and interpretation of data, and assisted in drafting the manuscript. All authors approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Chen, DY., Chen, YM., Chen, HH. et al. Increasing levels of circulating Th17 cells and interleukin-17 in rheumatoid arthritis patients with an inadequate response to anti-TNF-α therapy. Arthritis Res Ther 13, R126 (2011). https://doi.org/10.1186/ar3431
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/ar3431