
Abstract
Background
Culicoides biting midges are potential vectors of different pathogens. However, especially for eastern Europe, there is a lack of knowledge on the host-feeding patterns of this vector group. Therefore, this study aimed to identify Culicoides spp. and their vertebrate hosts collected in a wetland ecosystem.
Methods
Culicoides spp. were collected weekly from May to August 2017, using Biogents traps with UV light at four sites in the Danube Delta Biosphere Reserve, Romania. Vectors and hosts were identified with a DNA barcoding approach. The mitochondrial cytochrome c oxidase subunit 1 was used to identify Culicoides spp., while vertebrate hosts were determined targeting cytochrome b or 16S rRNA gene fragments. A maximum likelihood phylogenetic tree was constructed to verify the biting midge identity against other conspecific Palaearctic Culicoides species. A set of unfed midges was used for morphological confirmation of species identification using slide-mounted wings.
Results
Barcoding allowed the species identification and detection of corresponding hosts for 1040 (82.3%) of the 1264 analysed specimens. Eight Culicoides spp. were identified with Culicoides griseidorsum, Culicoides puncticollis and Culicoides submaritimus as new species records for Romania. For 39 specimens no similar sequences were found in GenBank. This group of unknown Culicoides showed a divergence of 15.6–16.3% from the closest identified species and clustered in a monophyletic clade, i.e. a novel species or a species without reference sequences in molecular libraries. For all Culicoides spp., nine mammalian and 24 avian species were detected as hosts. With the exception of C. riethi (n = 12), at least one avian host was detected for all Culicoides spp., but this host group only dominated for Culicoides kibunensis and the unknown Culicoides sp.. The most common host group were mammals (n = 993, 87.6% of all identified blood sources) dominated by cattle (n = 817, 70.6%).
Conclusions
Most Culicoides spp. showed a broad host-feeding pattern making them potential bridge vectors. At the same time, new records of biting midge species for Romania, as well as a potentially unknown Culicoides species, highlight the lack of knowledge regarding the biting midge species and their genetic diversity in eastern Europe.

Similar content being viewed by others


Background
Biting midges of the genus Culicoides Latreille, 1809 (Diptera: Ceratopogonidae) are vectors of a variety of pathogens. These include protozoans [1,2,3], filarial worms [4] and numerous viruses [5]. Their relevance as vectors is primarily related to veterinary health, though outbreaks of the Culicoides-borne Oropouche virus in humans regularly occur in the Neotropics [6]. In Europe, several biting midge species are able to transmit bluetongue virus (BTV), African horse sickness virus and Schmallenberg virus (SBV) [7]. These viruses are responsible for outbreaks of non-contagious diseases in ruminants, causing huge economic losses, e.g. due to restrictions on animal trade [8].
The expansion of BTV from the Mediterranean basin to central Europe up to Scandinavia [9,10,11] prompted studies on Culicoides taxonomy [12,13,14], ecology [15,16,17] and vector competence [18,19,20]. In contrast, only few studies focused on the Culicoides fauna in southeastern Europe. Severe BTV outbreaks were observed between 2014 and 2015 in the Balkan Peninsula [21, 22]. In Romania, BTV was confirmed for the first time in 2014 [23]. The most comprehensive studies on the Culicoides fauna conducted in Romania date back to the end of the 20th century [24, 25]. More recent studies of the Culicoides fauna in Romania only focused on the known vectors of BTV. Thus, with the exception of C. imicola Kieffer, 1913 or C. nubeculosus (Meigen 1830) [26, 27], biting midges were recorded as species groups considered the most important vectors of BTV/SBV, i.e. C. obsoletus group and C. pulicaris group, or as “other Culicoides” [28, 29]. Currently, species-specific information on the distribution of other Culicoides taxa in Romania is missing.
The identification of blood sources from engorged vectors is a useful method to understand vector-host interactions and the ecology of associated pathogens [30, 31]. The host-feeding patterns of Culicoides have received much less attention compared to other vector groups (e.g. mosquitoes and ticks) [32, 33]. In Europe, most of the vertebrate hosts identified from engorged biting midges are ruminants [34,35,36]. However, other mammalian species such as humans and pigs can also be frequent [37,38,39]. In comparison, avian hosts are generally a more diverse, but less frequent group compared to mammals [34, 37, 38, 40]. Information about hosts of Culicoides species from eastern Europe was obtained by recent efforts undertaken in natural areas of Bulgaria [41] and Serbia [42]. In Serbia, blood-meal analysis predominantly detected ruminant hosts, whereas in Bulgaria, a large diversity of avian hosts was recorded for ornithophilic biting midges. To the best of our knowledge, such studies do not exist for Romania. Therefore, the aim of this study was to investigate the host-feeding patterns of Culicoides species collected from four sampling sites in the Danube Delta Biosphere Reserve (DDBR).
Methods
Trapping methods and study sites
Biting midges were collected at four sites in the DDBR as part of a pilot longitudinal arbovirus surveillance programme [43] (Fig. 1, Additional file 1: Text S1). The trapping site Letea is characterized by a semi-open enclosure for cattle and goats built of wood, reeds and rushes, located a short distance from a small canal and almost 1 km from a deciduous forest. In Sulina, the sampling site was a covered cow stable with two or three animals kept at night with a stagnant water body (canal) and a large dung heap in close proximity. The local host communities of both anthropogenic sites (Letea and Sulina) are predominantly characterized by cattle, horse, cat, poultry and humans accompanied by dogs. In contrast, the site at Dunărea Veche lays at the confluence of two branches of the Danube and adjacent small canals; a large crop field is bordered by these waters. The site Lake Roșuleț is an old fishery surrounded by a shallow, stagnant canal and rows of trees isolating the area from the surrounding marshland. Only few humans (farmers and fishermen) with dogs and cats are present in Dunărea Veche and Lake Roșuleț. The host community of both sites is predominantly characterized by a high diversity of wild mammals and birds.

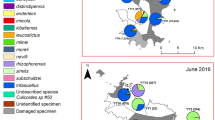
Sampling sites (1: Letea; 2: Dunărea Veche; 3: Sulina; 4: Lake Roșuleț) of Culicoides in the Danube Delta Biosphere Reserve (Romania) during the sampling period in 2017. Landcover variables are aggregated land cover data (Corine Land Cover (CLC) 2018 raster data, https://land.copernicus.eu/). CLC codes: water bodies, 511–523; natural, 311–423; rural, 211–244; urban, 111–142
Between May and August 2017, one Biogents Sentinel trap (BG trap; Biogents, Regensburg, Germany (http://www.biogents.com/)) equipped with an ultraviolet lamp was operated at each site for one night per week resulting in a total of 60 trap nights. The climate of the study area is continental with an annual mean temperature of 11 ℃ (− 1 ℃ in January and 22 ℃ in July) and around 350 mm of mean precipitation per year. Sampling in the present study was conducted during a hot and dry summer. A mean temperature of 21 ℃ and mean precipitation under 30 mm was recorded in the Danube Delta between May and August 2017 (http://www.meteoromania.ro/clima/monitorizare-climatica/).
Sample processing
Insects were frozen, shipped on dry ice and stored at − 80 ℃ in the laboratory. Due to the large amount of non-engorged and engorged Culicoides, only a random subsample of 1264 engorged specimens from all four sampling sites and every month of collection were selected. During the progress of sequencing, a dominance of cattle was observed for the sites Sulina and Letea. Therefore, we focused specifically on the engorged Culicoides from the sites Dunărea Veche and Lake Roșuleț, where a wider range of wildlife host blood meals were likely to be detected. Dry, frozen storage was preferred over ethanol storage to allow virus isolation and characterization at a later time. Biting midges were separated by engorged status and wing patterns under a stereomicroscope (Olympus ZSX12, Tokyo, Japan). In addition, a small set of unfed specimens (n = 37) from each sampling site (Sulina, n = 10; Letea, n = 9; Dunărea Veche, n = 10; Lake Roșuleț, n = 8) were used for morphological identification, which were selected as morphologically representative for the different Culicoides species in the samples. Wings were mounted on slides in Euparal (Carl Roth, Karlsruhe, Germany) and species identified by morphology using the key of Mathieu et al. [14].
For DNA extraction, each specimen was placed into an individual sterile 2 ml tube (Eppendorf, Hamburg, Germany) with 5–9 zirconium beads (1 mm, Carl Roth) and 200 μl of Dulbecco’s modified Eagle’s medium (Sigma-Aldrich, St. Louis, MO, USA) with 100 μg/ml streptomycin (PAN-Biotech, Aidenbach, Germany) and 2.5 μg/ml amphotericin B (PAN-Biotech). The samples were homogenised with a TissueLyser II (Qiagen, Hilden, Germany) twice for 3 min at 30 Hertz. The suspension was clarified by centrifugation at 8000× rpm for 2 min at 4 ℃. Total nucleic acid was extracted from 100 μl of supernatant, using the MagMAX™RNA/DNA Pathogen Kit with a KingFisher™ Flex Magnetic Particle Processor (Thermo Fisher Scientific, Waltham, MA, USA).
Molecular identification of biting midges
A 658-bp fragment of the mitochondrial cytochrome c oxidase subunit 1 gene (cox1) was amplified PCR, using the primers HCO2198 and LCO1490 [44]. One microliter template was added to a 10 μl reaction mix, containing 6.6 μl of Hotstar Taq Master Mix (Qiagen), 2.2 μl of molecular grade water (included in the Master Mix kit) and 0.6 μl of each 10 μM primer. The following cycling program was used: initial denaturation at 95 ℃ for 15 min, followed by 40 cycles of 30 s denaturation at 94 ℃, 45 s annealing at 40 ℃ and 1 min extension at 72 ℃, and final extension step for 10 min at 72 ℃. Each PCR run included DNA of Culex quinquefasciatus Say, 1823 (positive control) and ultrapure water (negative control). All amplicons were visualised on 2% agarose gels and PCR products sequenced with LGC Genomics (Berlin, Germany).
Molecular identification of Culicoides hosts
Hosts were identified using two PCR protocols targeting the cytochrome b (cytb) and 16S rRNA gene fragment [45,46,47]. Both protocols were described in detail in a previous study by Börstler et al. [32]. If the amplification with the first pair of primers failed [45, 46], another PCR was applied using the second pair of primers [47]. The same applied to potential mixed blood meals as indicated by double peaks at different positions in the sequence electropherograms. These samples were also analysed with both PCRs. As observed in our previous studies [32, 33], the PCR targeting the cytb gene fragment generally has a higher amplification rate for mammals, and the PCR targeting the 16S rRNA gene fragment a higher amplification rate for birds. We used the DNA of a mammal (African green monkey, Chlorocebus sabaeus (L.)) and a bird (European blackbird, Turdus merula L.) as positive controls. The negative control was ultrapure water, which was included in each PCR run. These amplicons were also visualised and sequenced as described above.
Data analysis
Sequences were visualised and edited with Geneious version 9.1.7 (Biomatters, Auckland, New Zealand). The resulting sequences were submitted for species identification using the basic alignment search tool (BLAST) in the GenBank DNA sequence database (https://blast.ncbi.nlm.nih.gov/) and the Barcode of Life Database [48]. In order to rule out potential contamination, samples indicating human host DNA were repeated separately in an individual PCR reaction. Identity values for the Culicoides and host species generally ranged between 98 and 100%. Sequences with lower identity values were repeated. One exception was the newly described haplotype of C. punctatus (Meigen, 1804), which showed identity values between 96 and 97%. In addition, information on the fauna of the DDBR were used to interpret the sequences. For example, domestic pig has become quite a rarity in the study area (Additional file 1: Text S1). Therefore, these sequences were classified as wild boar, which is a common wild mammal in the DDBR.
To assess the phylogenetic relationship of Culicoides identified in the DDBR with other previously reported species in the Palaearctic, a maximum likelihood tree was constructed with MEGAX [49] with additional conspecific and outgroup sequences (Forcipomyia spp. and Cx. quinquefasciatus) from GenBank (Additional file 2: Table 1). The HKY + G model was identified as the best-fit model of nucleotide substitution by Jmodeltest 2.1.10 [50] based on calculations of Bayesian and Akaikeʼs information criteria. Robustness of nodes was assessed by 1000 bootstrap replicates. The Culicoides spp. sequences generated in this study were deposited in the GenBank database under the accession numbers MN274523-274532 and MN340302-340312.
Results
Molecular identification of biting midges
Sequencing a fragment of the cox1 gene allowed the molecular identification of 1134 (89.7%) of the analysed 1264 engorged Culicoides (Table 1). Five species were identified for engorged biting midges: C. griseidorsum Kieffer, 1918; C. kibunensis Tokunaga, 1937; C. punctatus; C. riethi Kieffer 1914; and C. submaritimus Tokunaga & Murachi, 1959. Culicoides subfasciipennis Kieffer, 1919/C. pallidicornis Tokunaga & Murachi, 1959 were not differentiated to the species level. Furthermore, 39 sequences (3.1% of the analysed specimens) could not be identified to species level by comparison with other Culicoides sequences available on GenBank. The sequences of these specimens had a high similarity indicative of belonging to the same species and represent the seventh taxon hereafter referred to as “unknown Culicoides”. The eighth taxon detected was C. puncticollis (Becker, 1903), only present in the non-engorged fed biting midges selected for morphological identification. Four of the seven detected engorged species were confirmed by morphology: C. griseidorsum; C. kibunensis; C. riethi; and C. punctatus. In contrast, engorged C. submaritimus and C. subfasciipennis/C. pallidicornis were identified solely by barcoding and were not found in the small set of unfed specimens. Culicoides puncticollis was identified by morphology and cox1 barcoding, but only from the same subset of 37 unfed specimens (Additional file 3: Figure S1). As the cox1 sequences are not suitable to differentiate between C. subfasciipennis and C. pallidicornis [51, 52], these specimens were classified as C. subfasciipennis/C. pallidicornis. The unknown Culicoides species had similar wing patterns to C. kibunensis (Fig. 2).

Two wing pictures for the unknown Culicoides species collected in the Danube Delta Biosphere Reserve (Romania) during 2017
In order to perform a identity verification of the generated Culicoides cox1 sequences, we constructed a maximum likelihood phylogenetic tree including conspecific Culicoides and outgroup sequences (Fig. 3). A distinct haplotype of C. punctatus (designated as C. punctatus P) was identified in almost half (n = 207, 45.5%) of the 454 C. punctatus specimens analysed. These clustered within a separate monophyletic clade showing a genetic distance of approximately 4% to C. punctatus (Fig. 3). For the unknown Culicoides we could not find any similar sequences in the databases. This group of specimens showed a divergence of 15.6–16.3% from the closest identified Culicoides species (data not shown). The sequences of these specimens had a high similarity with each other and clustered with C. kibunensis in a monophyletic clade (Fig. 3).

Maximum likelihood phylogenetic tree of cox1 sequences for Culicoides species collected in the Danube Delta Biosphere Reserve (Romania) during 2017. Silhouettes indicate observed host-feeding patterns regarding the relative frequencies of mammalian and avian hosts. The tree was inferred using an HKY + G model (1000 bootstrap replicates) and rooted with Forcipomyia sp. and Culex quinquefasciatus. Branch support values of ≥ 50% are displayed and GenBank accession numbers of sequences shown on the branch tips
Culicoides punctatus (n = 455, 36.0% of all analysed specimens), C. griseidorsum (n = 276, 21.8%), C. subfasciipennis/C. pallidicornis (n = 242, 19.1%) and C. kibunensis (n = 102, 8.1%) were the most frequent taxa identified (Table 1, Additional file 4: Table S2). Culicoides riethi (n = 12, 0.95%) was found in the traps set near livestock in the sites Sulina and Letea, while Culicoides submaritimus (n = 8, 0.63%) was only found for Dunărea Veche and Lake Roșuleț, respectively.
Molecular identification of Culicoides hosts
Overlapping the two sets of sequences obtained for Culicoides identification and their hosts, information was available for 1040 (91.7%) of the 1134 molecular identified biting midges (Table 1). Blood-meal identification was not possible for 93 specimens due to failed PCR amplification. In addition, eight mixed blood meals were detected. With the exception of C. punctatus (n = 455) and C. riethi (n = 12), mixed blood meals where found for engorged specimens of all five Culicoides spp. Two Culicoides specimens contained blood from two mammalian hosts, while the other six specimens had mixed blood meals from a bird and a mammal.
A total of 33 vertebrate species were identified including nine species of mammals (27.3%) and 24 species of birds (72.7%) (Table 1). Mammals dominated the host spectrum (n = 1064, 92.0% of all 1156 identified blood sources). Cattle (Bos taurus) was the most abundant species (n = 817, 70.7%), followed by wild boar (n = 101, 8.7%). Other mammalian hosts were each found at a rate below 5%. Birds amounted to 8% of all the identified hosts with the Eurasian reed warbler (Acrocephalus scirpaceus; n = 13, 1.12%) and the carrion crow (Corvus corone; n = 16, 1.38%) as most frequent.
With the exception of C. riethi (n = 12), at least one avian host was detected for all Culicoides spp. Birds dominated the blood-meal sources of C. kibunensis and the unknown Culicoides sp. (68.8% and 72.2% of the detected hosts, respectively) (Table 1). Culicoides kibunensis had the highest diversity of hosts, with seven (77.8%) of the nine mammalian hosts and 18 (75%) of 24 species of avian hosts. Furthermore, humans were the most frequent mammalian host for this species (n = 10, 13.0% of all identified hosts). In contrast, the three most frequent Culicoides spp. (C. griseidorsum, C. punctatus and C. subfasciipennis/C. pallidicornis) showed high proportions of cattle (between 63.9 and 85.9% of all identified blood sources per taxon). The second most frequent hosts were goat (Capra hircus) for C. griseidorsum (17.3%) and wild boar for C. punctatus (6.6%), C. punctatus P (14.4%) and C. subfasciipennis/C. pallidicornis (9.7%) (Table 1). No differences were observed between C. punctatus and its distinct haplotype C. punctatus P. Furthermore, for C. submaritimus (n = 8) only blood meals from humans (n = 5), carrion crows (n = 3) and cattle (n = 1) were detected.
Discussion
The relevance of Culicoides spp. as important vectors of pathogens is well known. Thus, information about their diversity and host-feeding patterns is crucial to understand parasite-host interactions and the ecology of associated pathogens [30]. DNA barcoding is an important tool in biodiversity studies [53,54,55,56,57]. Thereby, barcoding also helped to identify cryptic and new Culicoides species [58,59,60]. In this study, successful sequencing of 1040 engorged insects demonstrated that barcoding is a useful tool for both, Culicoides and host identification. However, it must be considered that the different genetic markers can have pitfalls and do not necessarily reflect morphological differences [56, 61], i.e. using a single marker might be insufficient for an accurate identification of species.
A total of seven Culicoides species-level taxa were detected for the four sites in the DDBR. In the phylogenetic tree, specimens of the same taxon clustered in well-supported terminal clades. The only exception was C. subfasciipennis/C. pallidicornis. The separation between these two species is based on a variable light spot on the wing’s anal cell of C. subfasciipennis [14]. However, the analysis indicated no sequence differences of the cox1 gene. The discriminatory characters on the wing might be unreliable and further studies are required to clarify the status of both species [51, 52].
Culicoides griseidorsum, C. puncticollis and C. submaritimus were recorded for the first time in Romania, increasing the number of known Culicoides species for the country to 49 species [25]. Culicoides submaritimus has been considered a synonym of C. maritimus Tokunaga, 1940 by some authors [62, 63], while recent studies treated C. submaritimus as a distinct species [14, 64]. In the present study, C. submaritimus was identified by its similarity with cox1 sequences from Turkey, which are the only sequences available on GenBank for this species, while no cox1 sequences were available for C. maritimus. Neither C. submaritimus, nor C. maritimus are included in the inventory of Culicoides biting midges of Romania [25], although more recent studies include the country in the distribution of C. maritimus [14, 65].
The observed genetic variation for the analysed C. punctatus in two distinct clades is within intraspecific boundaries [59]. Such sibling species may vary in their vectorial capacity [66], e.g. vector competence or host-feeding patterns of members in the Anopheles gambiae complex. However, we did not detect differences in the host-feeding patterns between either taxa. Furthermore, the specimens clustering within the clade designated as “unknown Culicoides” showed genetic distances of 15.6–16.3% from the closest described species. These distances are similar to those observed between the other Culicoides species in our study. Comparable distances were found in other Culicoides spp. [67, 68] or mosquitoes [69], indicating that these specimens belong to a separate new species or a species without reference sequences in molecular libraries.
The overall host spectrum covered species expected for the DDBR, including livestock species like buffalo (Bubalis bubalis). Therefore, most of the analysed Culicoides spp. had a broad host-feeding range. Only mammalian hosts were detected for C. riethi, but the small sample size of only 12 engorged specimens does not allow an accurate conclusion on the species’ host-feeding pattern. Both, mammalian and avian hosts were detected for all other biting midge taxa to various extents. The broad host choice matches previous studies, which find similar results for different Culicoides spp. [70, 71]. Humans and carrion crow were the only hosts of C. submaritimus (n = 8). Cattle, wild boar or goat dominated the hosts of the three most frequent Culicoides taxa (C. punctatus, C. subfasciipennis/C. pallidicornis and C. griseidorsum). The high frequency of cattle probably relates to the large number of free-roaming cattle available in the DDBR and their large body mass [72]. However, as observed before [41, 67, 73, 74], despite this distinct dominance of mammalian hosts, different avian hosts were detected for the three Culicoides taxa.
Culicoides kibunensis is considered predominantly ornithophilic [37, 38, 75, 76]. With 18 species of birds and seven species of mammals, this vector of avian malaria [37, 38] showed the highest overall host diversity. The wide range of bird species is not surprising, considering the diversity of this vertebrate group in the DDBR. Nevertheless, the observed generalist host-feeding pattern including humans match previous studies [34, 37, 38]. Interestingly, the unknown Culicoides species showed a similar host-feeding pattern as C. kibunensis, with which it formed a monophyletic clade in the phylogenetic tree. These observations support the hypothesis of a positive correlation between biting midge phylogenetic relatedness and their feeding behaviour [40, 77]. In contrast, other studies speculated that such similarities in host-feeding patterns are not necessarily driven by phylogenetic relatedness, but might be the result of other factors (e.g. body size-driven host choice due to larger emissions of CO2 or volatile compounds) [71].
Host availability probably has a significant impact on the observed host-feeding patterns of Culicoides spp. Although no quantitative information on the host community is available, the prevalence of humans and domestic animals at Dunărea Veche and Lake Roșuleț is known. Humans, dogs and cats had relative low abundance at both sites compared to birds or free-ranging cattle and horses. Nevertheless, humans, dogs or cats were detected as hosts for all analysed Culicoides species. Thus, caution regarding the distribution of biting midges and the potential host has to be considered when interpreting host-feeding patterns of Culicoides. For example, a high proportion of C. griseidorsum were found to have fed on goats, but this host was widely available at Letea, where most of this species were collected (Additional file 4: Table S2, Additional file 5: Table S3).
Information on the host-feeding patterns can be also used to estimate dispersal distances of Culicoides spp. [77]. Biting midges from the sampling site Dunărea Veche were engorged with blood from buffalo and goat. These hosts are only available in the nearest village more than 4 km from the trapping site, which is in the range of a previous study on Culicoides [78]. Maximum dispersal distances of more than 3 km over one night were recorded regularly. Winds over the delta’s flat landscape might favour passive dispersal [79,80,81,82]. Thereby, besides active midge movement, wind dispersal is considered an important mode of long-distance dispersal for Culicoides-borne pathogens [83,84,85].
Conclusions
The broad host range of different mammalian and avian species indicates that most of the analysed Culicoides species in the DDBR are potential bridge vectors. However, the actual vector competence of these species is largely unknown. Of the dominant Culicoides species analysed, C. punctatus was previously indicated as a potential vector of BTV and SBV [86, 87]. Free roaming cattle, the most abundant and most frequently detected hosts in the region, could have an important role in amplification and spread of pathogens between wild ruminants and livestock [88]. At the same time, the new records of biting midge taxa for the country presented here and the detection of a potentially unknown Culicoides taxon highlight the lack of knowledge regarding the biting midge species and their genetic diversity in Europe.
Availability of data and materials
The data supporting the conclusions of this article are included within the article and its additional files.
Abbreviations
- DDBR:
-
Danube delta biosphere reserve
- DNA:
-
Deoxyribonucleic acid
- BTV:
-
Bluetongue virus
- SBV:
-
Schmallenberg virus
- BG trap:
-
Biogents sentinel trap
- PCR:
-
Polymerase chain reaction
References
Linley JR. Biting midges as vectors of nonviral animal pathogens. J Med Entomol. 1985;22:589–99.
Chagas CRF, Bukauskaite D, Ilgunas M, Iezhova T, Valkiunas G. A new blood parasite of leaf warblers: molecular characterization, phylogenetic relationships, description and identification of vectors. Parasit Vectors. 2018;11:538.
Svobodová M, Dolnik OV, Ivan Č, Rádrová J. Biting midges (Ceratopogonidae) as vectors of avian trypanosomes. Parasit Vectors. 2017;10:224.
Debrah LB, Nausch N, Opoku VS, Owusu W, Mubarik Y, Berko DA, et al. Epidemiology of Mansonella perstans in the middle belt of Ghana. Parasit Vectors. 2017;10:15.
Borkent A. The biting midges, the Ceratopogonidae (Diptera). In: Marquardt WC, editor. Biology of disease vectors. 2nd ed. Burlington MA: Elsevier Academic Press; 2005. p. 113–26.
da Rosa JF, de Souza WM, de Paula Pinheiro F, Figueiredo ML, Cardoso JF, Acrani GO, et al. Oropouche virus: clinical, epidemiological, and molecular aspects of a neglected orthobunyavirus. Am J Trop Med Hyg. 2017;96:1019–30.
Sick F, Beer M, Kampen H, Wernike K. Culicoides biting midges—underestimated vectors for arboviruses of public health and veterinary importance. Viruses. 2019;11:376.
Pinior B, Firth CL, Loitsch A, Stockreiter S, Hutter S, Richter V, et al. Cost distribution of bluetongue surveillance and vaccination programmes in Austria and Switzerland (2007–2016). Vet Rec. 2018;182:257.
Maan S, Maan NS, Ross-smith N, Batten CA, Shaw AE, Anthony SJ, et al. Sequence analysis of bluetongue virus serotype 8 from the Netherlands 2006 and comparison to other European strains. Virology. 2008;377:308–18.
Foxi C, Delrio G, Falchi G, Marche MG, Satta G, Ruiu L. Role of different Culicoides vectors (Diptera: Ceratopogonidae) in bluetongue virus transmission and overwintering in Sardinia (Italy). Parasit Vectors. 2016;9:440.
Purse BV, Mellor PS, Rogers DJ, Samuel AR, Mertens PPC, Baylis M. Climate change and the recent emergence of bluetongue in Europe. Nat Rev Microbiol. 2005;3:171–81.
Nolan DV, Carpenter S, Barber J, Mellor PS, Dallas JF, Mordue AJ, et al. Rapid diagnostic PCR assays for members of the Culicoides obsoletus and Culicoides pulicaris species complexes, implicated vectors of bluetongue virus in Europe. Vet Microbiol. 2007;124:82–94.
Augot D, Sauvage F, Jouet D, Simphal E, Veuille M, Couloux A, et al. Discrimination of Culicoides obsoletus and Culicoides scoticus, potential bluetongue vectors, by morphometrical and mitochondrial cytochrome oxidase subunit I analysis. Infect Genet Evol. 2010;10:629–37.
Mathieu B, Cêtre-Sossah C, Garros C, Chavernac D, Balenghien T, Carpenter S, et al. Development and validation of IIKC: an interactive identification key for Culicoides (Diptera: Ceratopogonidae) females from the Western Palaearctic region. Parasit Vectors. 2012;5:137.
Cuéllar AC, Kjær LJ, Kirkeby C, Skovgard H, Nielsen SA, Stockmarr A, et al. Spatial and temporal variation in the abundance of Culicoides biting midges (Diptera: Ceratopogonidae) in nine European countries. Parasit Vectors. 2018;11:112.
Lühken R, Kiel E, Steinke S, Fladung R. Topsoil conditions correlate with the emergence rates of Culicoides chiopterus and Culicoides dewulfi (Diptera: Ceratopogonidae) from cowpats. Parasitol Res. 2015;114:1113–7.
Garros C, Viennet E, Rossi S, Balenghien T. Adaptation of a species-specific multiplex PCR assay for the identification of blood meal source in Culicoides (Ceratopogonidae: Diptera): applications on Palaearctic biting midge species, vectors of orbiviruses. Infect Genet Evol. 2011;11:1103–10.
Carpenter S, Veronesi E, Mullens B, Venter G. Vector competence of Culicoides for arboviruses: three major periods of research, their influence on current studies and future directions. Rev Sci Tech. 2015;34:97–112.
Barber J, Harrup LE, Silk R, Veronesi E, Gubbins S, Bachanek-Bankowska K, et al. Blood-feeding, susceptibility to infection with Schmallenberg virus and phylogenetics of Culicoides (Diptera: Ceratopogonidae) from the United Kingdom. Parasit Vectors. 2018;11:116.
Pagès N, Talavera S, Verdún M, Pujol N, Valle M, Bensaid A, et al. Schmallenberg virus detection in Culicoides biting midges in Spain: first laboratory evidence for highly efficient infection of Culicoides of the obsoletus complex and Culicoides imicola. Transbound Emerg Dis. 2018;65:e1–6.
Kyriakis CS, Billinis C, Papadopoulos E, Vasileiou NGC, Athanasiou LV, Fthenakis GC. Bluetongue in small ruminants: an opinionated review, with a brief appraisal of the 2014 outbreak of the disease in Greece and the south-east Europe. Vet Microbiol. 2015;181:66–74.
Stojmanovski Z, Tabakovski B. Spatio-temporal characteristics of the bluetongue epizooty in the Balkan Peninsula from 2014 to february 2015. Maced Vet Rev. 2018;41:65–72.
Tilibașa EM, Popescu D, Badea C, Hora FȘ, Dărăbuș G. A report regarding the first occurrence of bluetongue in Romania, 2014. Scientific Works. Series C. Vet Med. 2015;61:277–80.
Damian-Georgescu A. New species of Ceratopogonidae (Diptera) for the Romanian fauna. Studii si Cercetari de Biologie, Seria Zoologie. 1972;24:423–32.
Damian-Georgescu A. Familia Ceratopogonidae, Genul Culicoides. Fauna româniei, insecta, Fasc 14 diptera, vol. 11. Bukuresti: Editura Academiei Romane; 2000.
Tilibașa EM, Dărăbuș G. Preliminary studies on dynamics of Culicoides spp. in western Romania in conjunction with some environmental factors. Parasit Vectors. 2014;7(Suppl. 1):O7.
Dărăbuș G, Tilibașa E, Oprescu I, Morariu S, Mederle N, Ilie M, Sujic T, Imre M. The abundance of Culicoides (Diptera: Ceratopogonidae) in Timiș county. Scient Works Med Vet. 2017;50:1689–99.
Ioniță M, Mitrea IL, Buzatu MC, Dascălu L, Ionescu A. Seasonal dynamics of haematophagous arthropod populations (ticks and Culicoides spp.)—vectors of pathogens in animals and humans, in different areas of Romania. Scient Works Med Vet. 2009;52:629–36.
Ilie A, Șerban C, Imre M, Sorescu D, Ilie M, Imre K, et al. A survey (or presences, dynamics, prevalences) of Culicoides (Diptera: Ceratopogonidae) in Gorj county, Romania preliminary results of entomological surveillance for bluetongue. Scient Works Med Vet. 2013;46:5–9.
Kent RJ. Molecular methods for arthropod bloodmeal identification and applications to ecological and vector-borne disease studies. Mol Ecol Resour. 2009;9:4–18.
Bitome-Essono PY, Ollomo B, Arnathau C, Durand P, Mokoudoum ND, Yacka-Mouele L, et al. Tracking zoonotic pathogens using blood-sucking flies as ’flying syringes’. Elife. 2017;6:e22069.
Börstler J, Jöst H, Garms R, Krüger A, Tannich E, Becker N, et al. Host-feeding patterns of mosquito species in Germany. Parasit Vectors. 2016;9:318.
Shahhosseini N, Friedrich J, Moosa-Kazemi SH, Sedaghat MM, Kayedi MH, Tannich E, et al. Host-feeding patterns of Culex mosquitoes in Iran. Parasit Vectors. 2018;11:669.
Lassen SB, Nielsen SA, Kristensen M. Identity and diversity of blood meal hosts of biting midges in Denmark. Parasit Vectors. 2012;5:143.
Hadj-Henni L, De Meulemeester T, Depaquit J, Noël P, Germain A, Helder R, et al. Comparison of vertebrate cytochrome b and prepronociceptin for blood meal analyses in Culicoides. Front Vet Sci. 2015;2:15.
Talavera S, Muñoz-Muñoz F, Verdun M, Pujol N, Pagès N. Revealing potential bridge vectors for BTV and SBV: a study on Culicoides blood feeding preferences in natural ecosystems in Spain. Med Vet Entomol. 2017;32:35–40.
Santiago-Alarcon D, Havelka P, Schaefer HM, Segelbacher G. Bloodmeal analysis reveals avian Plasmodium infections and broad host preferences of Culicoides (Diptera: Ceratopogonidae) Vectors. PLoS ONE. 2012. https://doi.org/10.1371/journal.pone.0031098.
Santiago-Alarcon D, Havelka P, Pineda E, Segelbacher G, Schaefer HM. Urban forests as hubs for novel zoonosis: blood meal analysis, seasonal variation in Culicoides (Diptera: Ceratopogonidae) vectors, and avian haemosporidians. Parasitology. 2013;140:1799–810.
Ninio C, Augot D, Delecolle JC, Dufour B, Depaquit J. Contribution to the knowledge of Culicoides (Diptera: Ceratopogonidae) host preferences in France. Parasitol Res. 2011;108:657–63.
Martínez-de la Puente J, Figuerola J, Soriguer R. Fur or feather? Feeding preferences of species of Culicoides biting midges in Europe. Trends Parasitol. 2015;31:16–22.
Bobeva A, Zehtindjiev P, Ilieva M, Dimitrov D, Mathis A, Bensch S. Host preferences of ornithophilic biting midges of the genus Culicoides in the eastern Balkans. Med Vet Entomol. 2015;29:290–6.
Vasic A, Zdravkovi N, Bojkovski J, Marinov M, Mathis A, Niculaua M, et al. Species diversity, host preference and arbovirus detection of Culicoides (Diptera: Ceratopogonidae) in south-eastern Serbia. Parasit Vectors. 2019;12:61.
Török E, Tomazatos A, Cadar D, Horváth C, Keresztes L, Jansen S, et al. Pilot longitudinal mosquito surveillance study in the Danube Delta Biosphere Reserve and the first reports of Anopheles algeriensis theobald, 1903 and Aedes hungaricus Mihályi, 1955 for Romania. Parasit Vectors. 2016;9:196.
Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotechnol. 1994;3:294–9.
Burkett-Cadena ND, Graham SP, Hassan HK, Guyer C, Eubanks MD, Katholi CR, et al. Blood feeding patterns of potential arbovirus vectors of the genus Culex targeting ectothermic hosts. Am J Trop Med Hyg. 2008;79:809–15.
Kitano T, Umetsu K, Tian W, Osawa M. Two universal primer sets for species identification among vertebrates. Int J Legal Med. 2007;121:423–7.
Kocher TD, Thomas WK, Meyer A, Edwards SV, Paabo S, Villablanca FX, et al. Dynamics of mitochondrial DNA evolution in animals: amplification and sequencing with conserved primers. Proc Natl Acad Sci. 1989;86:6196–200.
Ratnasingham S, Hebert PDN. The barcode of life data system (www.barcodinglife.org). Mol Ecol Notes. 2007; 7: 355–64.
Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35:1547–9.
Darriba D, Taboada GL, Doallo R, Posada D. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 2012;9:772.
Sarvašová A, Delecolle JC, Halan M, Mathieu B, Kočišová A. Morphological and molecular analysis of the genus Culicoides (Diptera: Ceratopogonidae) in Slovakia with five new records. Zootaxa. 2014;3872:541–60.
Filatov S, Szadziewski R. Annotated checklist and distribution of Culicoides biting midges of Ukraine (Diptera: Ceratopogonidae). J Nat Hist. 2017;51:487–511.
Pires AC, Marinoni L. DNA barcoding and traditional taxonomy unified through integrative taxonomy: a view that challenges the debate questioning both methodologies. Biota Neotrop. 2010;10:339–46.
Hebert PDN, Cywinska A, Ball SL, DeWaard JR. Biological identifications through DNA barcodes. Proc R Soc B Biol Sci. 2003;270:313–21.
Hajibabaei M, Singer GAC, Hebert PDN, Hickey DA. DNA barcoding: how it complements taxonomy, molecular phylogenetics and population genetics. Trends Genet. 2007;23:167–72.
Hernández-Triana LM, Brugman VA, Nikolova NI, Ruiz-Arrondo I, Barrero E, Thorne L, et al. DNA barcoding of British mosquitoes (Diptera, Culicidae) to support species identification, discovery of cryptic genetic diversity and monitoring invasive species. ZooKeys. 2019;832:57–76.
Whitworth TL, Dawson RD, Magalon H, Baudry E. DNA barcoding cannot reliably identify species of the blowfly genus Protocalliphora (Diptera: Calliphoridae). Proc R Soc B Biol Sci. 2007;274:1731–9.
Pagès N, Muñoz-Muñoz F, Talavera S, Sarto V, Lorca C, Núñez JI. Identification of cryptic species of Culicoides (Diptera: Ceratopogonidae) in the subgenus Culicoides and development of species-specific PCR assays based on barcode regions. Vet Parasitol. 2009;165:298–310.
Nielsen SA, Kristensen M. Delineation of Culicoides species by morphology and barcode exemplified by three new species of the subgenus Culicoides (Diptera: Ceratopogonidae) from Scandinavia. Parasit Vectors. 2015;8:151.
Sarvašová A, Kočišová A, Candolfi E, Mathieu B. Description of Culicoides (Culicoides) bysta n. sp., a new member of the Pulicaris group (Diptera: Ceratopogonidae) from Slovakia. Parasit Vectors. 2017;10:279.
Beebe NW. DNA barcoding mosquitoes: advice for potential prospectors. Parasitology. 2018;145:622–33.
Dik B, Yaǧci Ş, Linton YM. A review of species diversity and distribution of Culicoides Latreille, 1809 (Diptera: Ceratopogonidae) in Turkey. J Nat Hist. 2006;40:1947–67.
Rawlings P. A key based on wing patterns of biting midges (genus Culicoides Latreille—Diptera: Ceratopogonidae) in the Iberian Peninsula, for use in epidemiological studies. Graellsia. 1996;52:57–71.
Talavera S, Muñoz-Muñoz F, Pagès N. New insights on diversity, morphology and distribution of Culicoides Latreille 1809 (Diptera: Ceratopogonidae) from northeast Spain. Ann Soc Entomol Fr. 2011;47:214–31.
Szadziewski R, Borkent A, Dominiak P. Fauna Europaea: Ceratopogonidae. In: Beuk P, Pape T, ed. Fauna Europaea: Diptera, Nematocera. Fauna Europaea version 2.6; 2013. https://fauna-eu.org/cdm_dataportal/taxon/7161829a-94f3-4c36-943d-0d15f8fa1f77. Accessed 15 Aug 2019.
White BJ, Collins FH, Besansky NJ. Evolution of Anopheles gambiae in relation to humans and malaria. Annu Rev Ecol Evol S. 2011;42:111–32.
Pettersson E, Bensch S, Ander M, Chirico J, Sigvald R, Ignell R. Molecular identification of bloodmeals and species composition in Culicoides biting midges. Med Vet Entomol. 2013;27:104–12.
Ander M, Troell K, Chirico J. Barcoding of biting midges in the genus Culicoides: a tool for species determination. Med Vet Entomol. 2013;27:323–31.
Chan A, Chiang LP, Hapuarachchi HC, Tan CH, Pang SC, Lee R, et al. DNA barcoding: complementing morphological identification of mosquito species in Singapore. Parasit Vectors. 2014;7:569.
Tempelis CH, Nelson RL. Blood-feeding patterns of midges of the Culicoides variipennis complex in Kern county, California. J Med Entomol. 1971;8:532–4.
Hopken MW, Ryan BM, Huyvaert KP, Piaggio AJ. Picky eaters are rare: DNA-based blood meal analysis of Culicoides (Diptera: Ceratopogonidae) species from the United States. Parasit Vectors. 2017;10:169.
Viennet E, Garros C, Gardès L, Rakotoarivony I, Allène X, Lancelot R, et al. Host preferences of palaearctic Culicoides biting midges: implications for transmission of orbiviruses. Med Vet Entomol. 2013;27:255–66.
Lassen SB, Nielsen SA, Skovgård H, Kristensen M. Molecular identification of bloodmeals from biting midges (Diptera: Ceratopogonidae: Culicoides Latreille) in Denmark. Parasitol Res. 2011;108:823–9.
Calvo JH, Berzal B, Calvete C, Miranda MA, Estrada R, Lucientes J. Host feeding patterns of Culicoides species (Diptera: Ceratopogonidae) within the Picos de Europa National Park in northern Spain. Bull Entomol Res. 2012;102:692–7.
Černý O, Votýpka J, Svobodová M. Spatial feeding preferences of ornithophilic mosquitoes, blackflies and biting midges. Med Vet Entomol. 2011;25:104–8.
Martinez-de La Puente J, Merino S, Toms G, Moreno J, Morales J, Lobato E, et al. Factors affecting Culicoides species composition and abundance in avian nests. Parasitology. 2009;136:1033–41.
Augot D, Hadj-Henni L, Strutz SE, Slama D, Millot C, Depaquit J, et al. Association between host species choice and morphological characters of main sensory structures of Culicoides in the Palaeartic region. PeerJ. 2017;5:e3478.
Sanders CJ, Harrup LE, Tugwell LA, Brugman VA, England M, Carpenter S. Quantification of within- and between-farm dispersal of Culicoides biting midges using an immunomarking technique. J Appl Ecol. 2017;54:1429–39.
Greenberg JA, DiMenna MA, Hanelt B, Hofkin BV. Analysis of post-blood meal flight distances in mosquitoes utilizing zoo animal blood meals. J Vector Ecol. 2012;37:83–9.
Sanders CJ, Selby R, Carpenter S, Reynolds DR. High-altitude flight of Culicoides biting midges. Vet Rec. 2011;169:208.
Johansen CA, Farrow RA, Morrisen A, Foley P, Bellis G, Van Den Hurk AF, et al. Collection of wind-borne haematophagous insects in the Torres Strait, Australia. Med Vet Entomol. 2003;17:102–9.
Kluiters G, Swales H, Baylis M. Local dispersal of palaearctic Culicoides biting midges estimated by mark-release-recapture. Parasit Vectors. 2015;8:86.
Ducheyne E, De Deken R, Bécu S, Codina B, Nomikou K, Mangana-Vougiaki O, et al. Quantifying the wind dispersal of Culicoides species in Greece and Bulgaria. Geospat Health. 2007;1:177.
Sedda L, Brown HE, Purse BV, Burgin L, Gloster J, Rogers DJ. A new algorithm quantifies the roles of wind and midge flight activity in the bluetongue epizootic in northwest Europe. Proc R Soc B. 2012;279:2354–62.
Burgin LE, Gloster J, Sanders C, Mellor PS, Gubbins S, Carpenter S. Investigating incursions of bluetongue virus using a model of long-distance Culicoides biting midge dispersal: investigating bluetongue virus incursions. Transbound Emerg Dis. 2013;60:263–72.
Goffredo M, Catalani M, Federici V, Portanti O, Marini V, Mancini G, et al. Specie di Culicoides coinvolte nell’epidemia di Bluetongue 2012–2014 in Italia. Vet Ital. 2015;51:131–8.
Larska M, Lechowski L, Grochowska M, Zmudziński JF. Detection of the Schmallenberg virus in nulliparous Culicoides obsoletus/scoticus complex and C punctatus-the possibility of transovarial virus transmission in the midge population and of a new vector. Vet Microbiol. 2013;166:467–73.
Conraths FJ, Gethmann JM, Staubach C, Mettenleiter TC, Beer M, Hoffmann B. Epidemiology of bluetongue virus serotype 8, Germany. Emerg Infect Dis. 2009;15:433–5.
Acknowledgements
We would like to thank to Vasile Suhov and the residents of Sulina and Letea, who granted access to their properties. In addition, we are very grateful to Dr Andreas Krüger for his support and fruitful discussion. In addition, we thank Patricia Iftene for help during the field work, Daniel Truchado for his comments on the manuscript and to Daniel Ciobanu for help with literature research.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
AT, HJ, MS, JSC, DC and RL conceived and designed the study. AT, HJ and JS collected the data. AT, HJ, JSC, DC and RL analyzed the data. AT, DC and RL drafted the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The DDBR administration approved all research activities for trapping at specific study sites (9/19.04.2017, 5627/ARBDD/13.04.2017).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Text S1.
Description of the sampling sites with information on vegetation, surrounding environment and available hosts.
Additional file 2: Table S1.
Accession numbers of Culicoides spp., Forcipomyia spp. and Culex quinquefasciatus used for phylogenetic analysis.
Additional file 3: Figure S1.
Wing patterns for C. punctatus, C. punctatus P, C. kibunensis, C. puncticollis, C. riethi and C. griseidorsum collected in this study.
Additional file 4: Table S2.
Overview of the Culicoides species per sampling site.
Additional file 5: Table S3.
Overview of the frequency of each molecularly identified Culicoides host species per sampling site.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Tomazatos, A., Jöst, H., Schulze, J. et al. Blood-meal analysis of Culicoides (Diptera: Ceratopogonidae) reveals a broad host range and new species records for Romania. Parasites Vectors 13, 79 (2020). https://doi.org/10.1186/s13071-020-3938-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-020-3938-1