
Abstract
Background
Several members of genus Babesia are important pathogens causing babesiosis in dogs. In China, at least five Babesia species have been described in dogs or ticks. This study sought to determine the prevalence and molecular characteristics of various Babesia spp. in dogs in cities in Shaanxi Province in China, including Xi’an and Hanzhong.
Methods
A total of 371 blood samples were collected from pet dogs presenting to veterinary clinics in the cities of Xi’an and Hanzhong in Shaanxi, China. Babesia spp. DNA was detected via amplification of partial 18S rRNA genes by semi-nested PCR. Almost full-length 18S rRNA, ITS, partial TRAP and complete cytb genes were recovered for analysis of the genetic characteristics and relationships with known isolates.
Results
A single species, Babesia gibsoni, was identified in dogs in Xi’an and Hanzhong. Consistently, B. gibsoni was also detected in 14 ticks collected from positive dogs. Sequence similarities and phylogenetic analysis suggested that the isolates identified herein showed a closer genetic relationship with isolates from East Asian countries rather than India, Bangladesh, or the USA. Sequence analysis based on tandem repeat analysis of the TRAP gene further revealed that specific haplotypes were circulating in both Xi’an and Hanzhong, with no specific regionality. In addition, 10.9% of all isolates with atovaquone (ATV)-resistance were identified because of M121I mutation in the deduced cytb protein.
Conclusions
This study revealed a high prevalence rate of Babesia infection. Babesia gibsoni was the only Babesia species identified in cases of canine babesiosis in the cities of Xi’an and Hanzhong cities in Shaanxi, China. In addition, the TRAP gene presented high genetic diversity across isolates. Such information is useful for elucidating the epidemiological characteristics of canine babesiosis, as well as the overall genetic diversity of Babesia spp. circulating in dog populations in Shaanxi Province.

Similar content being viewed by others

Background
Babesiosis refers to a serious, worldwide, tick-borne hemoprotozoan disease caused by various Babesia spp. These Babesia spp. are classified as either large (3–5 μm) or small (1.5–2.5 μm) species based on their morphological size in erythrocytes [1]. Several members of the genus Babesia have gained increased attention due to their clinical significance in both veterinary and human medicine [2]. Canine babesiosis, a significant disease in dogs, is caused by at least seven validated Babesia species, including B. gibsoni, B. conradae, B. vulpes, B. vogeli, B. canis, B. rossi and B. caballi [3]. In addition, several unclassified Babesia spp. have also been detected in dogs [4,5,6]. Of these, B. vogeli and B. gibsoni present the most widespread distribution. Babesia spp. mainly spread between dogs through tick bites, although other ways of transmission, including dog bites, blood transfusions, and transplacental transmission is possible [7,8,9].
The clinical signs of canine babesiosis are similar for infection with both small and large Babesia spp. and include pyrexia, anemia, jaundice, hemoglobinuria, splenomegaly and weakness [3, 10,11,12,13]. The severity of canine babesiosis varies from mild to serious illness, largely depending on the specific species of infecting parasite, and conditions of the affected dog, such as age, nutritional and immune status [10,11,12]. For example, B. rossi, a highly virulent species, is associated with higher mortality in infected dogs compared to other Babesia species due to various complications of infection, such as disseminated intravascular coagulation and multi-organ dysfunction [14].
In China, serological investigation and molecular surveys of Babesia spp. infection have shown the geographical circulation of B. gibsoni, B. caballi, B. canis, B. rossi and B. vogeli in dog populations. As the most prevalent Babesia species, B. gibsoni is responsible for canine babesiosis in the central, southern and eastern regions of China, as well as Taiwan [15,16,17,18,19,20]. Babesia vogeli has been identified in dogs in Gansu, Jiangxi, Guangdong and Hunan provinces [21, 22] and in ticks in Henan [23]. Babesia canis has been reported in dogs in Henan [24] and various 18S rRNA gene sequences of B. canis have been identified in dogs and ticks in Hunan and Qinghai provinces, respectively. Babesia rossi isolated identified in dogs in Hunan are further available in the GenBank database. Babesia caballi has been detected in ticks in the Xinjiang Uygur autonomous region and Jilin Province, as well as in horses (rather than dogs) in Gansu Province [25, 26]. Importantly, as a zoonotic agent, B. caballi has also been detected in one human blood sample (GenBank: KJ715182).
To date, no information is available regarding the prevalence of babesiosis in dogs in Shaanxi Province in China. To better understand the prevalence of various Babesia spp. in dogs, blood samples were collected in the cities of Xi’an and Hanzhong to characterize the prevalence and genetic diversity of Babesia spp. infections in dogs in Shaanxi Province.
Methods
Collection of canine blood samples and ticks
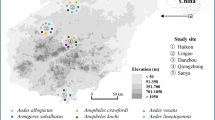
Xi’an city is located in the middle of the Guanzhong Plain with the Qinling Mountains to the south, and its average elevation is 400 meters above sea level. Xi’an is characterized by a semi-moist monsoon climate with a clear distinction between the four seasons, and the annual average temperature is approximately 13 °C. Hanzhong city is located in the Hanjiang River basin with the Qinling Mountains to the north, and its average elevation is 550 meters above sea level. Hanzhong is characterized by a subtropical moist monsoon climate, with the annual average temperature of approximately 14 °C. From 2017 to 2018, EDTA-anticoagulated whole blood samples were aseptically collected from 371 pet dogs at four veterinary clinics located in urban areas, including two in Xi’an city and two in Hanzhong city in Shaanxi Province, China (Fig. 1). Among these, 260 (144 from Hanzhong and 116 from Xi’an) whole blood samples were collected from dogs suspected of having babesiosis that presented with at least two of the following clinical signs: fever; jaundice; anemia; thrombocytopenia; or hemoglobinuria. Moreover, all babesiosis-suspected dogs had a history of outdoor activities in the field and tick bites based on their owners’ statements. Additionally, 111 apparently healthy dogs (64 from Xi’an and 47 from Hanzhong) attended for prophylactic procedures were randomly selected, none of which presented with any of the above-mentioned clinical signs based on their owners’ statements. Furthermore, 15 adult and 2 nymphal ticks were collected from 9 babesiosis-suspected dogs. The tick species were first identified morphologically [27], and then further confirmed by analysis of the 710 bp cox1 gene [28].

Geographic distribution of canine blood samples used for Babesia spp. detection in Xi’an and Hanzhong cities in the Shaanxi Province of China
DNA extraction and detection of Babesia
Total DNA was extracted from canine blood samples using an E.Z.N.A.® Blood DNA Kit (Omega, Norcross, GA, USA) according to the manufacturer’s recommendations. Meanwhile, total DNA was also extracted individually from all ticks using the E.Z.N.A.® Tissue DNA Kit (Omega) according to the manufacturer’s instructions.
Babesia spp. DNA was detected using primers targeting the 18S ribosomal RNA (rRNA) gene using a semi-nested polymerase chain reaction (PCR) [29]. Primary primer sets BS1 and PiroC, and secondary PiroA and PiroC, were used to amplify a 352-bp 18S rRNA gene fragment (Table 1).
Amplification of nearly complete 18S rRNA gene, ITS region, partial TRAP gene and complete cytb gene
A nearly complete 18S rRNA gene, ITS region, partial thrombospondin-related adhesive protein (TRAP) gene and complete cytb gene were recovered from positive samples to investigate the genetic relationships among Babesia species. All the primer sequences were shown in Table 1 in detail. The nearly full length 18S rRNA gene and the ITS region were amplified using the primer pairs P1/P2 and ITSF/ITS2, respectively, as previously described [15]. The 1100 bp TRAP gene was amplified by nested PCR using external primer set BgTRAPtF1 and BgTRAPtR1 and internal primer set BgTRAPtF2 and BgTRAPtR2 [30].
The complete cytb gene was amplified with primer set Bgcytb-F and Bgcytb-R designed in-house based on the conserved regions of cytb gene sequences of the genus Babesia available in the GenBank database. PCR was performed in a 50 μl volume including 25 μl Premix Taq (Takara), 3μl extracted total DNA as a template, 2 μl of each primer (10 pmol) and 18 μl water. The PCR cycling condition comprised an initial denaturation at 94 °C for 5 min, followed by 35 cycles denaturation at 94 °C for 40 s, annealing at 50 °C for 40 s, and elongation at 72 °C for 1 min, with a final extension step at 72 °C for 7 min.
Cloning and sequencing of PCR products
All PCR amplicons were electrophoresed in 1.0% agarose gels, and those with expected size were purified using a Gel Extraction kit (TaKaRa). Partial 18S rRNA gene sequences were subjected to sequencing in both directions using an Applied Biosystems 3130 Genetic Analyzer with a BigDye v3.1 Terminator cycle sequencing kit (Applied Biosystems Inc., Carlsbad, CA, USA). The purified PCR products were cloned into pMD19-T vector (TaKaRa, Dalian, China), and then the recombinant plasmid was transformed into Escherichia coli DH5α competent cells. Positive clones were identified by PCR and at least 3 positive clones were sequenced for each PCR product to determine a consensus sequence.
Sequence and phylogenetic analyses
DNA sequences of the 18S rRNA, ITS, TRAP and cytb genes recovered from positive samples were assembled and edited using the Lasergene program (DNASTAR, Inc., Madison, WI, USA). The obtained sequences were subjected to BLAST analysis against the GenBank database. The nucleotide identity was calculated using the ClustalW method implemented in Lasergene. The alignment among the new sequences obtained here and the reference sequences retrieved from GenBank was conducted using MAFFT (version 7) [31]. The optimal nucleotide substitution model, GTR+Γ+I, was determined using jModeltest 0.1 [32]. Phylogenetic trees were reconstructed using the Maximum Likelihood (ML) method based on the four above-mentioned 4 gene sequences using MEGA7 [33], with 1000 replications for bootstrap test.
Tandem repeat analysis of TRAP gene fragments
Tandem Repeats Finder (version 4.09; http://tandem.bu.edu/trf/trf.html), a program for the detection of tandem repeats (TR) in a DNA sequence, was used to find out the location and to display TR DNA sequences in TRAP gene fragments, as previously described [34, 35].
Results
Detection and identification of Babesia spp
Of the 260 babesiosis-suspected dogs, PCR products with expected size were successfully amplified from 73 and 94 samples collected in Xi’an and Hanzhong, respectively. Sequencing and further BLAST analysis showed that all sequences had the highest nucleotide identity with sequences of B. gibsoni (more than 99.0%), suggesting B. gibsoni infection in all babesiosis-suspected dogs. The overall prevalence of B. gibsoni in babesiosis-suspected dogs was 64.2% (62.9% in Xi’an and 65.3% in Hanzhong, respectively; Table 2). Meanwhile, B. gibsoni was also detected in 4.7% and 10.6% of healthy dog samples collected in Xi’an (3/64) and Hanzhong (5/47), respectively, with an overall infection rate of 7.2% in healthy dogs. The positive rates of B. gibsoni infection in relation to season, age, sex, and breed of dogs are shown in Table 3.
All the infested ticks were identified as Haemaphysalis longicornis based on their morphological characters and the position of their cox1 gene sequences in the phylogenetic tree (Additional file 1: Figure S1). Consistent with the finding that 8 out of 9 dogs infested with ticks tested positive for B. gibsoni, 14 ticks collected from these 8 dogs also tested positive. In contrast, 3 other ticks sampled from a single B. gibsoni-negative dog also tested negative.
Genetic variation and phylogenetic analysis of the 18S rRNA gene and ITS region
Nearly full-length 18S rRNA gene sequences of B. gibsoni obtained from all positive samples (MN928814-MN928833) shared 99.3–100% nucleotide identity with each other, as well as a high identity of 99.0–100% with known 18S rRNA gene sequences. They specifically showed 100% nucleotide identity with Shikoku 1 (GenBank: AB478329), Aomori (GenBank: AB118032), and Okinawa (GenBank: AB478328) from Japan, Jeju 4 (GenBank: AB478323) from South Korea and TWN5 (GenBank: JQ710685) from Taiwan. Partial 18S rRNA gene sequences recovered from tick samples were identical to those obtained from the positive canine blood samples. The 1100 bp ITS sequences recovered from all the positive samples (MN928834-MN928851) were also closely related, with less than 0.2% divergence from each other, and less than 1% divergence from known ITS sequences.
Phylogenetic trees were reconstructed based on representative 18S rRNA and ITS sequences (only 1 of the sequences with 100% identity was included from each city, except XABg43 and XABg35) using the ML method. Both 18S rRNA (> 1200 bp) and ITS (> 900 bp) sequences deposited on GenBank and those determined in the present study were used to perform phylogenetic analysis. The 18S rRNA tree was low-resolution since it was highly conserved among different variants of B. gibsoni. Generally, all 18S rRNA sequences were divided into two clades, albeit not well supported (Fig. 2). Three isolates recovered in this study were located in the first clade, including two (XABg43 and XABg35) clustering with those from Nanjing, Taiwan, Japan and the USA, and one (XABg8) clustered with those from Taiwan and Japan. All others were located in another clade and had a close phylogenetic relationship with canine isolates detected in Wuhan and Japan. In addition, the representative ITS sequences (only one of the sequences with 100% identity was included) in this study clustered together and had a closer phylogenetic relationship with sequences isolated from dogs in Wuhan and Taiwan, rather than India and the USA (Fig. 3).

Phylogenetic tree based on 18S rRNA gene sequences of B. gibsoni indicating genetic relationships between the new sequences obtained in this study and known sequences. Numbers at each node indicate bootstrap values (only numbers > 70 are shown). The tree was mid-point rooted for clarity, and the scale-bar represents the number of nucleotide substitutions per site. Representative strains were used to reconstruct the tree and marked by circles

Phylogenetic tree based on ITS sequences of B. gibsoni indicating genetic relationship between the new sequences obtained in this study and known sequences. Numbers at each node indicate bootstrap values (only numbers > 70 are shown). The tree was mid-point rooted for clarity, and the scale-bar represents the number of nucleotide substitutions per site. Representative strains were used to reconstruct the tree and marked by circles
Genetic variation and phylogenetic analysis of the TRAP gene
The TRAP gene (1100 bp) fragment was recovered from all B. gibsoni isolates; all sequences (MN928852-MN928879) presented 98.4–100% nucleotide identity and 95.6–100% amino acid identity with each other. When compared with known TRAP sequences of B. gibsoni, they showed 86.8–99.8% nucleotide identity and 73.3–100% amino acid identity. Sequence comparison of TRAP sequences between those obtained here and other known relevant complete sequences showed that amino acid residues between 265 and 431 of the TRAP protein, including both partial von Willebrand Factor-like A (vWF-like A) and thrombospondin type I repeat (TSR) domains, were well-conserved (Additional file 2: Figure S2). Amino acid mutations primarily occurred after the TSR domain, and a large fragment deletion was observed at position 449–495, similar to isolates Kansai 59 (GenBank: KR013043), BgTRAP-Shikoku 2 (GenBank: AB478349), BgTRAP-Houshu 1 (GenBank: AB478341), BgTRAP-Houshu 2 (GenBank: AB478342), BgTRAP-Jeju 1 (GenBank: AB478343) and TWN6 (GenBank: JN247443). Amino acid mutations occurred in the transmembrane region of 7 sequences, mainly from Val (V) to Glu (E) and Tyr (Y) to Asp (D).
Comparative analysis of the deduced amino acid sequences revealed the presence of 9 different haplotypes of TRAP based on tandem repeat analysis. More specifically, haplotypes 1 and 2, 3 and 4, 5 and 6, and 7, 8, and 9 had the same insertion/deletion pattern after the TSR region, respectively (Fig. 4). In both cities, all of these haplotypes were identified, with haplotypes 3 and 5 being the predominant ones. In addition to 59 isolates identified in this study, haplotype 5 also included BgTRAP-Jeju 1 (GenBank: AB478343) from South Korea and TWN6 (GenBank: JN247443) from Taiwan (Fig. 4). Although no tandem repeats were found in the consensus patterns, haplotype 1 identified in this study presented different insertion/deletion patterns compared to several other East Asian isolates from Japan, Korea and Taiwan (Fig. 4). TR analysis of the partial TRAP gene revealed 0 to 5 repeats in Xi’an and Hanzhong isolates (Table 4). Four consensus patterns (GGA GGA, GAG GAA GAG GAA, GGA GGA GGA GGA AGA GGA AGA and GCG GAG GAA GAG GAA GAG GAA GAG GAG) were common to most isolates found in Shaanxi Province, China.

Alignment of representative TRAP amino acid sequences after the TSR region in each haplotype recovered in this study alongside samples in the GenBank database. A large fragment deletion was identified (box). Haplotype 1 identified in this study showed different insertion/deletion patterns compared to several other East Asian isolates from Japan, Korea and Taiwan (indicated in green). The transmembrane region is indicated in gray
The representatives (only one of the sequences with 100% identity was included in each city) of partial TRAP gene fragments were used to reconstruct a phylogenetic tree using the ML method. The results revealed that all B. gibsoni isolates were classified into two well-supported clusters (Fig. 5). The isolates identified in this study, as well as those from Taiwan, South Korea, and Japan formed the first cluster, and those identified in India and Bangladesh formed the second cluster. In the first cluster, the Shaanxi isolates had the closest relationship with BgTRAP-Jeju 1, BgTRAP-Houshu 1, BgTRAP-Houshu 2, BgTRAP-Shikoku 2, Kansai 59 and TWN6, followed by other Asian B. gibsoni isolates, including Taiwan, South Korea and Japan; they were more distantly related to isolates from India and Bangladesh.

Phylogenetic tree based on TRAP gene sequences of B. gibsoni indicating genetic relationship between the new sequences obtained in this study and known sequences. Numbers at each node indicate bootstrap values (only numbers > 70 are shown). The tree was mid-point rooted for clarity, and the scale-bar represents the number of nucleotide substitutions per site. Representative strains were used to reconstruct the tree and marked by circles
Genetic variation and phylogenetic analysis of the cytb gene
One hundred and seventy-five full length cytb gene sequences were obtained from B. gibsoni-positive samples (MN928880 to MN928897), which presented 99.5–100% nucleotide identity and 99.2–100% amino acid identity with each other. In addition, these sequences also shared 99.0–100% nucleotide and 99.2–100% amino acid identity with known cytb gene sequences of B. gibsoni, especially WH58 (GenBank: KP666169) from the city of Wuhan in China. Comparative analysis of the deduced amino acid sequences revealed that amino acid residue Ile at position 121 was present in 19 isolates (19/175, 10.9%), which is responsible for the atovaquone (ATV)-resistant phenotype confirmed in previous studies [36, 37]. However, Ile and Val were not identified at positions 220 and 303, respectively, which have also been associated with ATV-resistance.
Consistent with the nucleotide identity analysis, the representative cytb gene sequences (only one of the sequences with 100% identity was included in each city) of B. gibsoni, including these determined in the present study, demonstrated a close phylogenetic relationship with each other due to shorter genetic distances shared among them (Fig. 6). Generally, the cytb gene sequences recovered in this study had the closest relationship with isolate WH58 (GenBank: KP666169) from Wuhan, China, rather than other isolates from Japan.

Phylogenetic tree based on cytb gene sequences of B. gibsoni indicating the genetic relationship between the new sequences obtained in this study and known sequences. Numbers at each node indicate bootstrap values (only numbers > 70 are shown). The tree was mid-point rooted for clarity, and the scale-bar represents the number of nucleotide substitutions per site. Representative strains were used to reconstruct the tree and shown in bold
Discussion
Tick-borne Babesia spp. infections in dogs continue to increase worldwide, primarily in many developing countries but also in several developed countries. Previous epidemiological studies regarding Babesia infection in dogs and ticks demonstrated that four species of Babesia were circulating in China, and B. gibsoni was the predominant pathogen causing babesiosis in the central, southern and eastern regions of China [18,19,20, 22, 23, 38]. However, little information concerning the epidemiology of Babesia spp. in dogs in Shaanxi has been available. The results of an investigation of the prevalence of tick-borne pathogens in ten provinces of China showed that no B. gibsoni DNA was detected in 56 canine blood samples collected from Yangling in Shaanxi Province [19]. Nevertheless, another study reported the presence of B. gibsoni by detecting antibodies specific against the TRAP protein, with a positive rate of 8.33% in Xi’an in Shaanxi Province [39]. In the present study, molecular epidemiological analysis of Babesia infection in dogs was performed in Xi’an and Hanzhong, two cities in Shaanxi Province. The results suggest that B. gibsoni is the only Babesia species infecting all positive samples, as well as ticks collected from dogs positive for B. gibsoni infection. Taken together, these data indicate that the B. gibsoni parasite is widely distributed in China, although further investigations are warranted.
In the present study, the prevalence rate of B. gibsoni infection in babesiosis-suspected dogs was 64.2% in Xi’an and Hanzhong. Interestingly, 7.2% of healthy dogs were also positive for B. gibsoni, consistent with a prior report of B. vogeli infection in healthy pet dogs in Shenzhen, China [40]. These results suggest that both Xi’an and Hanzhong are severely affected by B. gibsoni. In terms of season and age of dogs, our results were consistent with those reported from India [41] and Australia [42], although they are in contrast to those from Jiangxi Province in China [22]. In this study, babesiosis-suspected cases were confirmed by detecting the DNA of various Babesia spp., strengthening the reliability of the prevalence rate identified for B. gibsoni in different season, age, sex and breed of dogs, as cases initially presenting with babesiosis-like symptoms may be infected by other known pathogens, such as Rickettsiales, Hepatozoon spp., Theileria spp., Dirofilaria spp., and Leishmania spp. [43,44,45,46,47], as well as unknown pathogens, and even non-infectious factors.
Similarity and phylogenetic analyses revealed that 18S rRNA and ITS genes of B. gibsoni isolates identified in this study were highly conserved, and shared a closer relationship with those identified in Japan, Taiwan, Korea, and other parts of China, and were more distantly related to those in India, Bangladesh and the USA [30, 48], suggesting that geographic clustering pattern exists to a certain extent. Phylogenetic and similarity analyses based on the TRAP gene revealed a high genetic diversity of B. gibsoni isolates identified in this study, which may suggest that having a diverse TRAP protein may be favorable for pathogens to escape the host immune system [49]. All the isolates identified in this study were generally more closely related to each other and those detected in Japan, Taiwan, Korea, and other parts of China, while they were more distant to those in India, Bangladesh and the USA. Similarity and phylogenetic analyses based on the cytb gene suggest that B. gibsoni isolates determined in this study were closely related to those previously identified from Japan, although they formed two lineages based on regionality.
The TRAP gene is one of the more highly immunogenic proteins in B. gibsoni and possesses function in the parasite’s motility and invasion of red blood cells [50, 51]. This protein contains three domains: a vWF-like A domain; a TSR domain; and a short acidic cytoplasmic tail domain (CTD) [50, 51]. Consistent with previous studies, the vWF-like A and TSR domains were conserved in all isolates identified in this study, which have been used to develop a serological diagnostic assay and a vaccine candidate [52, 53]. However, regions between the TSR and the CTD present high genetic polymorphisms, including a large fragment deletion, which are suitable for genetic diversity and phylogenetic analysis of different B. gibsoni isolates [17, 54]. Moreover, amino acid mutations in the transmembrane region of 11 sequences were identified that have not been reported in previous studies, and the mechanism(s) by which these amino acid mutations may affect the function of TRAP protein require further investigation. TR analysis identified nine different haplotypes circulating in Xi’an and Hanzhong; all haplotypes were detected in both cities, suggesting no specific distribution pattern although Xi’an and Hanzhong are located on the north and south sides of the Qinling Mountains, respectively.
To our knowledge, few studies have focused on analyzing the cytb gene of Babesia. However, the cytb gene of B. gibsoni has gained increasing attention because amino acid residue Ile at position 121 has been associated with an ATV-resistant phenotype [36, 37]. To date, only one cytb sequence of B. gibsoni identified from China has been available in the GenBank database. To the best of our knowledge, our study is the first report concerning an AA substitution in the cytb gene of B. gibsoni related to ATV-resistance. Our results showed that M121I was observed in the deduced amino acid sequences recovered from canine blood samples in both Xi’an and Hanzhong, revealing that B. gibsoni with ATV-resistance has been naturally circulating in China. In the present study, its prevalence reached 10.9%, which is higher than it in Japan [55]. Interestingly, AA mutations V220I or I303V, which may also be linked to the ATV-resistance [37], were not identified in this study.
Conclusions
Our findings indicate that B. gibsoni is the causative agent responsible of canine babesiosis in Xi’an and Hanzhong cities in the Shaanxi Province of China. Its high prevalence rate reveals a possibly serious epidemic in local dog populations. In addition, substantial great genetic diversity was observed based on analysis of the partial TRAP gene, and an AA mutation associated with ATV-resistance was confirmed.
Availability of data and materials
Data supporting the conclusions of this article are included within the article and its additional files. The 18S rRNA, ITS, TRAP and cytb gene sequences generated in this study for phylogenetic analysis were submitted to the GenBank database under the accession numbers MN928814-MN928897.
Abbreviations
- rRNA:
-
ribosomal RNA
- PCR:
-
polymerase chain reaction
- TRAP :
-
thrombospondin-related adhesive protein
- ML:
-
Maximum Likelihood
- TR:
-
tandem repeat
- vWF-like A:
-
von Willebrand Factor-like A
- TSR:
-
a thrombospondin type I repeat
- CTD:
-
cytoplasmic tail domain
- ATV:
-
atovaquone
References
Solano-Gallego L, Baneth G. Babesiosis in dogs and cats—expanding parasitological and clinical spectra. Vet Parasitol. 2011;181:48–60.
Dantas-Torres F, Chomel BB, Otranto D. Ticks and tick-borne diseases: a one health perspective. Trends Parasitol. 2012;28:437–46.
Baneth G. Antiprotozoal treatment of canine babesiosis. Vet Parasitol. 2018;254:58–63.
Kubo S, Tateno M, Ichikawa Y, Endo Y. A molecular epidemiological survey of Babesia, Hepatozoon, Ehrlichia and Anaplasma infections of dogs in Japan. J Vet Med Sci. 2015;77:1275–9.
Lehtinen LE, Birkenheuer AJ, Droleskey RE, Holman PJ. In vitro cultivation of a newly recognized Babesia sp. in dogs in North Carolina. Vet Parasitol. 2008;151:150–7.
Ozubek S, Aktas M. Molecular evidence of a new Babesia sp. in goats. Vet Parasitol. 2017;233:1–8.
Fukumoto S, Suzuki H, Igarashi I, Xuan X. Fatal experimental transplacental Babesia gibsoni infections in dogs. Int J Parasitol. 2005;35:1031–5.
Jefferies R, Ryan UM, Jardine J, Broughton DK, Robertson ID, Irwin PJ. Blood, bull terriers and Babesiosis: further evidence for direct transmission of Babesia gibsoni in dogs. Aust Vet J. 2007;85:459–63.
Víchová B, Miterpáková M, Iglódyová A. Molecular detection of co-infections with Anaplasma phagocytophilum and/or Babesia canis canis in Dirofilaria-positive dogs from Slovakia. Vet Parasitol. 2014;203:167–72.
Irwin PJ. Canine babesiosis: from molecular taxonomy to control. Parasit Vectors. 2009;2(Suppl. 1):S4.
Schoeman JP. Canine babesiosis. Onderstepoort J Vet Res. 2009;76:59–66.
Schnittger L, Rodriguez AE, Florin-Christensen M, Morrison DA. Babesia: a world emerging. Infect Genet Evol. 2012;12:1788–809.
Jacobson LS. The South African form of severe and complicated canine babesiosis: clinical advances 1994–2004. Vet Parasitol. 2006;138:126–39.
Solano-Gallego L, Sainz Á, Roura X, Estrada-Peña A, Miró G. A review of canine babesiosis: the European perspective. Parasit Vectors. 2016;9:336.
He L, Miao X, Hu J, Huang Y, He P, He J, et al. First molecular detection of Babesia gibsoni in dogs from Wuhan, China. Front Microbiol. 2017;8:1577.
Lin HR, Mei XT, Hong YF, Zhao YB, Guo XN, Yang DJ, et al. Sequence analysis of the thrombospondin-related adhesive protein gene and heat shock protein 70 gene of Babesia gibsoni isolated from dogs in Nanjing, China. Infect Genet Evol. 2017;56:111–6.
Lee CC, Hsieh YC, Huang CC, Tsang CL, Chung YT. Sequence and phylogenetic analysis of the thrombospondin-related adhesive protein (TRAP) gene of Babesia gibsoni isolates from dogs in Taiwan. J Vet Med Sci. 2010;72:1329–35.
Wong SS, Teng JL, Poon RW, Choi GK, Chan KH, Yeung ML, et al. Comparative evaluation of a point-of-care immunochromatographic test SNAP 4Dx with molecular detection tests for vector-borne canine pathogens in Hong Kong. Vector Borne Zoonotic Dis. 2011;11:1269–77.
Xu D, Zhang J, Shi Z, Song C, Zheng X, Zhang Y, et al. Molecular detection of vector-borne agents in dogs from ten provinces of China. Parasit Vectors. 2015;8:501.
Yao DW, Jiang JY, Yu ZZ, Yao DQ, Yang DJ, Zhao YB. Canine babesiosis in China caused by Babesia gibsoni: a molecular approach. Iran J Parasitol. 2014;9:163–8.
Niu Q, Yang J, Liu Z, Gao S, Pan Y, Guan G, et al. First molecular detection of piroplasm infection in pet dogs from Gansu, China. Front Microbiol. 2017;8:1029.
Zheng W, Liu M, Moumouni PF, Liu X, Efstratiou A, Liu Z, et al. First molecular detection of tick-borne pathogens in dogs from Jiangxi, China. J Vet Med Sci. 2017;79:248–54.
Chen Z, Liu Q, Liu JQ, Xu BL, Lv S, Xia S, et al. Tick-borne pathogens and associated co-infections in ticks collected from domestic animals in central China. Parasit Vectors. 2014;7:237.
Wang J, Liu J, Yang J, Liu Z, Wang X, Li Y, et al. Molecular detection and genetic diversity of Babesia canis canis in pet dogs in Henan Province, China. Parasitol Int. 2019;71:37–40.
Song R, Wang Q, Guo F, Liu X, Song S, Chen C, et al. Detection of Babesia spp., Theileria spp. and Anaplasma ovis in Border Regions, northwestern China. Transbound Emerg Dis. 2018;65:1537–44.
Wang J, Liu J, Yang J, Wang X, Li Z, Jianlin X, et al. The first molecular detection and genetic diversity of Babesia caballi and Theileria equi in horses of Gansu province, China. Ticks Tick Borne Dis. 2019;10:528–32.
Sun Y, Xu RM, We CC. Haemaphysalis ticks (Ixodoidea, Ixodidae) in China-systematic and key to subgenera. Acta Parasitology Et Medica Entomologica Sinica. 2011;18:251–8.
Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotechnol. 1994;3:294–9.
Rar VA, Fomenko NV, Dobrotvorsky AK, Livanova NN, Rudakova SA, Fedorov EG, et al. Tickborne pathogen detection, western Siberia, Russia. Emerg Infect Dis. 2005;11:1708.
Terao M, Akter S, Yasin MG, Nakao R, Kato H, Alam MZ, et al. Molecular detection and genetic diversity of Babesia gibsoni in dogs in Bangladesh. Infect Genet Evol. 2015;31:53–60.
Katoh K, Frith MC. Adding unaligned sequences into an existing alignment using MAFFT and LAST. Bioinformatics. 2012;28:3144–6.
Posada D. Selection of models of DNA evolution with jModelTest. Methods Mol Biol. 2009;537:93–112.
Kumar S, Stecher G, Tamura K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–4.
Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–80.
Gelfand Y, Rodriguez A, Benson G. TRDB—the Tandem Repeats Database. Nucleic Acids Res. 2007;35:D80–7.
Iguchi A, Matsuu A, Ikadai H, Hasanuzzaman Talukder M, Hikasa Y. Development of in vitro atovaquone-resistant Babesia gibsoni with a single-nucleotide polymorphism in cytb. Vet. Parasitol. 2012;2012(185):145–50.
Matsuu A, Miyamoto K, Ikadai H, Okano S, Higuchi S. Cloning of the Babesia gibsoni cytochrome b gene and isolation of three single nucleotide polymorphisms from parasites present after atovaquone treatment. Am J Trop Med Hyg. 2006;74:593–7.
Cao J, Yang Q, Zhang J, Zhou Y, Zhang H, Gong H, et al. Seroprevalence survey of Babesia gibsoni infection and tick species in dogs in East China. Vet Parasitol. 2015;214:12–25.
Zhang J, Liu Q, Wang D, Li W, Beugnet F, Zhou J. Epidemiological survey of ticks and tick-borne pathogens in pet dogs in south-eastern China. Parasite. 2017;24:35.
Li XW, Zhang XL, Huang HL, Li WJ, Wang SJ, Huang SJ, et al. Prevalence and molecular characterization of Babesia in pet dogs in Shenzhen, China. Comp Immunol Microbiol Infect Dis. 2020;70:101452.
Singh A, Singh H, Singh NK, Singh ND, Rath SS. Canine babesiosis in northwestern India: molecular detection and assessment of risk factors. Biomed Res Int. 2014. https://doi.org/10.1155/2014/741785.
Hii SF, Traub RJ, Thompson MF, Henning J, O’Leary CA, Burleigh A, et al. Canine tick-borne pathogens and associated risk factors in dogs presenting with and without clinical signs consistent with tick-borne diseases in northern Australia. Aust Vet J. 2015;93:58–66.
Baneth G. Perspectives on canine and feline hepatozoonosis. Vet Parasitol. 2011;181:3–11.
Nayyar Ghauri H, Ijaz M, Farooqi SH, Ali A, Ghaffar A, Saleem S, et al. A comprehensive review on past, present and future aspects of canine theileriosis. Microb Pathog. 2019;126:116–22.
Sainz Á, Roura X, Miró G, Estrada-Peña A, Kohn B, Harrus S, et al. Guideline for veterinary practitioners on canine ehrlichiosis and anaplasmosis in Europe. Parasit Vectors. 2015;8:75.
Schuller S, Francey T, Hartmann K, Hugonnard M, Kohn B, Nally JE, et al. European consensus statement on leptospirosis in dogs and cats. J Small Anim Pract. 2015;56:159–79.
Simón F, Siles-Lucas M, Morchón R, González-Miguel J, Mellado I, Carretón E, et al. Human and animal dirofilariasis: the emergence of a zoonotic mosaic. Clin Microbiol Rev. 2012;25:507–44.
Singh MN, Raina OK, Sankar M, Rialch A, Tigga MN, Kumar GR, et al. Molecular detection and genetic diversity of Babesia gibsoni in dogs in India. Infect Genet Evol. 2016;41:100–6.
Schmid-Hempel P. Immune defence, parasite evasion strategies and their relevance for ‘macroscopic phenomena’ such as virulence. Philos Trans R Soc Lond B Biol Sci. 2009;364:85–98.
Naitza S, Spano F, Robson KJ, Crisanti A. The thrombospondin-related protein family of apicomplexan parasites: the gears of the cell invasion machinery. Parasitol Today. 1998;14:479–84.
Morahan BJ, Wang L, Coppel RL. No TRAP, no invasion. Trends Parasitol. 2009;25:77–84.
Zhou J, Fukumoto S, Jia H, Yokoyama N, Zhang G, Fujisaki K, et al. Characterization of the Babesia gibsoni P18 as a homologue of thrombospondin related adhesive protein. Mol Biochem Parasitol. 2006;148:190–8.
Fukumoto S, Xuan X, Nishikawa Y, Inoue N, Igarashi I, Nagasawa H, et al. Identification and expression of a 50-kilodalton surface antigen of Babesia gibsoni and evaluation of its diagnostic potential in an enzyme-linked immunosorbent assay. J Clin Microbiol. 2001;39:2603–9.
Jia H, Aboge GO, Terkawi MA, Goo YK, Nishikawa Y, Kuriki K, et al. Genetic diversity of two selected antigen loci in Babesia gibsoni Asian genotype obtained from Japan and Jeju island of South Korea. Vet Parasitol. 2009;162:142–6.
Sakuma M, Fukuda K, Takayama K, Kobayashi Y, Shimokawa Miyama T, Setoguchi A, et al. Molecular epidemiological survey of the Babesia gibsoni cytochrome b gene in western Japan. J Vet Med Sci. 2012;74:1341–4.
Zheng WQ, Xuan XN, Fu RL, Tao HY, Liu YQ, Liu XQ, et al. Tick-borne pathogens in Ixodid ticks from Poyang Lake Region, southeastern China. Korean J Parasitol. 2018;56:589–96.
Acknowledgements
Not applicable.
Funding
This study was funded by the National Natural Science Foundation of China (No. 31700159 and 81702008), Scientific Research Foundation for High-level Talents of Chengde Medical University (202001) and Key discipline construction of colleges and universities in Hebei Province (Ji Jiao Gao No. [2013]4).
Author information
Authors and Affiliations
Contributions
WPG and LYD conceived and designed the study and critically revised the manuscript. WPG, GCX, DL, MS and RJ performed sample collection. WPG, GCX, DL, MS and RJ conducted the laboratory experiments. All the authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All blood samples were collected under the permission from the owners of the pet dogs, and performed according to Guidance for Experimental Animal Welfare and Ethical Treatment by the Ministry of Science and Technology of China (http://www.most.gov.cn/fggw/zfwj/zfwj2006/200609/t20060930_54389.htm). This study was approved by the Scientific Ethic Committee of Chengde Medical University (number 2017020).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Figure S1.
Phylogenetic tree based on cox1 gene sequences of ticks. Numbers at each node indicate bootstrap values (only numbers > 70 are shown). The tree was mid-point rooted for clarity and the scale-bar represents the number of nucleotide substitutions per site. Representative strains herein were used to reconstruct the tree and marked by circles.
Additional file 2: Figure S2.
Alignment of the representative TRAP amino acid sequences between partial vWF-like A and TSR regions in each haplotype identified in this study and others in the GenBank database. Partial vWF-like A and TSR regions were indicated by red and blue color, respectively.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Guo, WP., Xie, GC., Li, D. et al. Molecular detection and genetic characteristics of Babesia gibsoni in dogs in Shaanxi Province, China. Parasites Vectors 13, 366 (2020). https://doi.org/10.1186/s13071-020-04232-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-020-04232-w