
Abstract
Background
Ascaris roundworms are the parasitic nematodes responsible for causing human and porcine ascariasis. Whereas A. lumbricoides is the most common soil-transmitted helminth infecting humans in the world, A. suum causes important economic losses in the porcine industry. The latter has been proposed as a model for the study of A. lumbricoides since both species are closely related. The third larval stage of these parasites carries out an intriguing and complex hepatopulmonary route through the bloodstream of its hosts. This allows the interaction between larvae and the physiological mechanisms of the hosts circulatory system, such as the fibrinolytic system. Parasite migration has been widely linked to the activation of this system by pathogens that are able to bind plasminogen and enhance plasmin generation. Therefore, the aim of this study was to examine the interaction between the infective third larval stage of A. suum and the host fibrinolytic system as a model of the host-Ascaris spp. relationships.
Methods
Infective larvae were obtained after incubating and hatching fertile eggs of A. suum in order to extract their cuticle and excretory/secretory antigens. The ability of both extracts to bind and activate plasminogen, as well as promote plasmin generation were assayed by ELISA and western blot. The location of plasminogen binding on the larval surface was revealed by immunofluorescence. The plasminogen-binding proteins from both antigenic extracts were revealed by two-dimensional electrophoresis and plasminogen-ligand blotting, and identified by mass spectrometry.
Results
Cuticle and excretory/secretory antigens from infective larvae of A. suum were able to bind plasminogen and promote plasmin generation in the presence of plasminogen activators. Plasminogen binding was located on the larval surface. Twelve plasminogen-binding proteins were identified in both antigenic extracts.
Conclusions
To the best of our knowledge, the present results showed for the first time, the pro-fibrinolytic potential of infective larvae of Ascaris spp., which suggests a novel parasite survival mechanism by facilitating the migration through host tissues.

Similar content being viewed by others


Background
Ascaris suum, generally considered as the most prevalent intestinal parasite in domestic pigs, is the nematode responsible for causing porcine ascariasis. This soil-transmitted infection presents high prevalence rates around the world and entails significant economic losses in the swine industry due to the reduction of production efficiency and organ condemnations [1]. In addition, A. suum has been postulated as a model for the study of A. lumbricoides infection in humans, which involves a parasitosis affecting an estimated 804 million people, most commonly children and adolescents [2, 3]. One of the most striking characteristics of the life-cycle of these parasites is the complex migratory route carried out by their third-stage larvae (L3) before establishing in the intestine. After being released from the eggs in the small intestine, larvae head for the caecum and proximal colon to undertake a hepatopulmonary migration through the bloodstream. Once L3 reach the alveoli, they ascend the trachea in order to be swallowed through the oesophagus and return to the small intestine, where they eventually reach the adult stage [4]. Despite being a process with high adaptive cost for the parasite, it could confer evolutionary benefits in terms of establishment in the host, as it has been proposed for nematode parasites whose larvae undergo migrations that begin and end in the same location [5]. However, the molecular mechanisms governing L3 migration processes in ascariasis remain unclear [6, 7]. For this reason, the knowledge of these key aspects of the life-cycle of the parasite could contribute to develop new intervention strategies in human and porcine ascariasis by studying the molecular basis of the host-parasite relationships [7, 8]. Thereby, the interaction between different species of pathogens and the fibrinolytic system of their corresponding host has already been studied [9, 10].
The fibrinolytic system is the main mechanism responsible for degrading blood clots in mammals [11]. Its activity is based on the conversion of a circulating zymogen in plasma (plasminogen) into its proteolytically active enzyme (plasmin). Plasminogen possesses lysine-binding sites called kringle domains, which interact with the lysine residues of different proteins and cellular receptors. Plasminogen is transformed into plasmin by cleavage of a peptide bond by the action of two proteases, the tissue plasminogen activator (tPA) and the urokinase-type plasminogen activator (uPA) [12]. The serine protease plasmin generated exerts its proteolytic activity against a broad range of substrates, including fibrin of blood clots and different components of the extracellular matrix [13]. Consequently, plasminogen recruitment and activation to plasmin by secreted and surface proteins of many groups of bacteria, fungi and parasites have been described and related to their invasion and migration processes, among others, facilitating their establishment in the host [9, 10, 14, 15].
In order to contribute to the knowledge of the host-Ascaris spp. relationships, especially during the parasitic intraorganic migration, the aim of this study was to examine the interaction between the cuticle and excretory/secretory antigenic extracts of the L3 of A. suum (AsL3C and AsL3ES) and the host fibrinolytic system.
Methods
Collection of third-stage larvae of A. suum
AsL3 were obtained following the protocol described by Vlaminck et al. [16] with minor modifications. Adult female worms of A. suum were collected from the intestines of naturally infected pigs from a local abattoir and dissected in order to extract the uterus and obtain the eggs. All the parasite material was washed with phosphate-buffered saline (PBS), pH 7.2. The eggs were suspended in a 2% potassium dichromate (K2Cr2O7) solution and placed in culture plates for their incubation at 27 °C for approximately 40 days in a place restricted from light. The incubation process was monitored by light microscopy observation.
Once most of the eggs were embryonated, they were collected from culture plates and transferred to test tubes to be treated with 5–6% sodium hypochlorite (NaClO) (commercial bleach) at 37 °C for 15 min. The eggs were washed at least 5 times with PBS by centrifugation at 200× g for 5 min. The suspension was transferred into an Erlenmeyer flask containing glass beads and a magnetic stir bar and maintained for approximately 1 h shaking slowly (60× rpm). Egg hatching was monitored by observing the solution under an optical microscope. After most of the larvae were outside the egg, the suspension was poured into a cotton gauze layer (2.5 g) on a Baermann apparatus filled with PBS and maintained at 37 °C and 5% CO2 overnight in a place restricted from light following the method described by Urban et al. [17]. Larvae were then collected from the neck of the funnel and washed with PBS. Some of them were used to obtain the antigenic extracts subsequently described, while others were fixed in a solution of 10% formaldehyde until use.
Collection of cuticle and excretory/secretory antigenic extracts from A. suum third-stage larvae
AsL3C and AsL3ES extracts were obtained following the methodology described by Wedrychowicz et al. [18] and González-Miguel et al. [19] with some modifications. In order to isolate the cuticle antigens from the AsL3, these larvae were incubated in a saline solution containing 0.25% cetyl trimethyl ammonium bromide (CTAB) at 37 °C for 4 h shaking. After removing larvae by centrifugation at 200× g for 5 min, the supernatant was filtered through a filter of 0.22 µm. Proteins were precipitated with a solution of 0.002 M sodium acetate and 9 volumes of 96% ethanol at − 20 °C for 48 h. The suspension was centrifuged at 10.000× g for 10 min and the resulting pellet was re-suspended in PBS. The same quantity of AsL3 was cultured in RPMI-1640 medium (Sigma-Aldrich, St. Louis, USA) supplemented with an antibiotic-antimycotic solution (100×) (Sigma-Aldrich) at 37 °C and 5% CO2 for 24 h to obtain the excretory/secretory products. Then, the suspension was centrifuged at 200× g for 5 min to remove larvae, and the resulting medium was filtered through a filter of 0.22 µm. After dialyzing against water at 4 °C for 48 h shaking, proteins were concentrated using Amicon Ultra-15 centrifugal filter devices (Millipore, Burlington, USA). Finally, both extracts were mixed with a cocktail of proteases inhibitors [20], their protein concentrations measured by BCA protein assay reagent kit (Thermo Fisher Scientific, Waltham, USA) and stored at − 80 °C until their use.
Plasminogen binding assays
The ability of AsL3C and AsL3ES to bind plasminogen was studied by an enzyme-linked immunosorbent assay (ELISA) according to the methods by González-Miguel et al. [19] with minor modifications. In brief, multi-well microplates (Corning, New York, USA) were coated with 1 µg/well of AsL3C or AsL3ES diluted in carbonate buffer, pH 9.6, at 4 °C overnight and blocked with 1% bovine serum albumin (BSA) diluted in PBS at 37 °C for 30 min. Wells were successively incubated with increasing amounts (0–2 µg/well) of human plasminogen (Acris Antibodies, Herford, Germany), an anti-plasminogen IgG developed in sheep (Acris Antibodies) at a 1:2000 dilution and a peroxidase-conjugated anti-sheep IgG (Sigma-Aldrich) at a 1:4000 dilution. These incubations were carried out in blocking solution at 37 °C for 1 h. Wells were washed three times with PBS between each step. Reactions were revealed with ortho-phenylene-diamine. A competition assay was performed in parallel including 25 mM of ε-aminocaproic acid (ε-ACA), a lysine analogue, during plasminogen incubation. Some wells were coated with BSA as a negative control. Optical densities (OD) were measured at 492 nm in a Microplate Absorbance Reader iMark (Bio-Rad, Hercules, USA). Each sample was analysed in triplicate.
In order to confirm plasminogen binding and characterize the parasitic protein bands responsible for this process, a western blot was employed [21]. To carry out the SDS-PAGE electrophoresis, 15 µg/well of AsL3C or AsL3ES were loaded into 12% polyacrylamide gels. Two µg of plasminogen were loaded as a positive control and the presence of the antigenic extracts was omitted in some wells to serve as a negative control. Once proteins were separated by molecular weight, some gels were developed with silver stain, while others were transferred to nitrocellulose membranes by semi-dry transfer to perform the western blot. Membranes were blocked with 2% BSA in wash buffer (PBS 0.05% Tween 20) at room temperature for 1 h and incubated with 10 µg/ml of human plasminogen (Acris Antibodies) in blocking solution at 4 °C overnight. Membranes were then successively incubated with an anti-plasminogen IgG developed in sheep (Acris Antibodies) at a 1:1000 dilution and a peroxidase-conjugated donkey anti-sheep IgG (Sigma-Aldrich) at a 1:2000 dilution in blocking solution at 37 °C for 1 h 30 min. Membranes were washed three times with wash buffer between each step. The reactions were revealed with 4-cloronaftol with peroxidase substrate. Gels and membranes were scanned in GS-800 Densitometer (Bio-Rad) and analysed with the Quantity One software v.4.6.5 (Bio-Rad). All assays were performed in triplicate.
Immunofluorescence plasminogen binding assay
To locate plasminogen binding on the larval surface, an immunofluorescence assay was performed and analysed by confocal microscopy following the method described by Ramajo-Hernández et al. [21] with minor modifications. AsL3 previously fixed in 10% formaldehyde were successively incubated with 10 µg/ml of human plasminogen (Acris Antibodies) diluted in 1% BSA in PBS at room temperature for 2 h shaking and 10 µg/ml of a fluorescein isothiocyanate (FITC)-labelled anti-plasminogen antibody (Acris Antibodies) diluted in PBS at 4 °C shaking overnight in the dark. AsL3 were washed four times with PBS between each step. The presence of plasminogen was omitted in some assays as a negative control. In parallel, a competition assay with 40 mM of ε-ACA was carried out during plasminogen incubation. AsL3 were then observed under the 63× water immersion objective of a TCS SP2 DM IRB microscope (Leica, Wetzlar, Germany), using an Argon 488 laser and 500 nm filter at the microscopy facility of the Instituto de Recursos Naturales y Agrobiología de Salamanca (IRNASA-CSIC), Spain. All assays were performed in triplicate.
Plasminogen activation assay
In order to evaluate the ability of both antigenic extracts to activate plasminogen and promote plasmin generation, a chromogenic assay was carried out according to the methodology described by González-Miguel et al. [19] and Fernandes et al. [22] with some modifications. One µg of AsL3C or AsL3ES was incubated in the presence of 1 µg of human plasminogen (Acris Antibodies), 3 µg of the plasmin chromogenic substrate S-2251 (Sigma-Aldrich) and a plasminogen activator [15 ng of tPA (Sigma-Aldrich) or 10 ng of uPA (Sigma-Aldrich)] in a final volume of 100 µl of PBS in multi-well microplates (Corning). In some wells the antigenic extract was replaced by BSA as negative control and in others the presence of plasminogen activators was omitted to study the ability of antigenic extracts to generate plasmin on their own. All the mixtures were maintained at 37 °C for 2 h 30 min and the hydrolysis of the substrate was analysed at 415 nm in a Microplate Absorbance Reader iMark (Bio-Rad). Each sample was analysed in triplicate.
Two-dimensional (2-D) electrophoresis of AsL3C and AsL3ES extracts and immunoblot assays
For the characterisation of plasminogen-binding proteins from AsL3C and AsL3ES extracts, 2-D electrophoresis and plasminogen-ligand blotting were performed as previously described [19]. For that purpose, both antigenic extracts were purified using the ReadyPrep 2-D Cleanup Kit (Bio-Rad) and assayed in a 2-D electrophoresis. After re-suspending 60 µg of protein from each extract in 125 µl of a rehydration buffer [7 M urea, 2 M thiourea, 4% 3-((3-cholamidopropyl) dimethylammonio)-1-propanesulfonate (CHAPS)], the mixtures were incubated in the presence of ampholytes and dithiothreitol (DTT). Samples were applied to 7 cm IPG strips (Bio-Rad) with a linear pH range of 3–10 and once they were absorbed by the gel strip, proteins were separated in a first dimension depending on their isoelectric point (pI) using a Protean IEF Cell (Bio-Rad). After that, strips were reduced and alkylated using successively dilutions of DTT and iodoacetamide in equilibrated buffer [6 M urea, 2% SDS, 0.05 M Tris/HCl (1.5 M, pH 8.8), 30% glycerol]. The second-dimension separation by molecular weight (MW) was carried out in 12% polyacrylamide gels and proteins were detected by a silver stain. 2-D gels were scanned with the GS-800 Densitometer (Bio-Rad) and the resulting images were analysed with the Quantity One software v.4.6.5 (Bio-Rad).
To perform western blots, 2-D gels were transferred to nitrocellulose membranes using a semi-dry transfer in a Trans-Blot SD Semi-Dry Transfer cell (Bio-Rad). Membranes were then blocked with 2% BSA in wash buffer (PBS 0.05% Tween 20) for 1 h at room temperature and incubated with 10 µg/ml of human plasminogen (Acris Antibodies) in blocking solution at 4 °C overnight. Then, membranes were successively incubated with a sheep anti-human plasminogen IgG (Acris Antibodies) at a 1:1000 dilution and a peroxidase-conjugated donkey anti-sheep IgG (Sigma-Aldrich) at a 1:2000 dilution in blocking solution at 37 °C for 1 h 30 min. Between each step, membranes were washed three times with wash buffer. Plasminogen-binding spots were revealed with 4-chloronaphthol and matched to their corresponding 2-D gels using the PDQuest software v.8.0.1 (Bio-Rad). Plasminogen incubation was omitted in some membranes as negative controls. Membranes were scanned with the GS-800 Densitometer (Bio-Rad) and analysed with the Quantity One software v.4.6.5 (Bio-Rad). All assays were performed in triplicate.
Mass spectrometry (MS), protein identification and bioinformatics analyses
Selected plasminogen-binding spots were manually excised from the silver stained 2-D gels and analysed by MS at the proteomics facility of the Servei Central de Suport a la Investigació Experimental (SCSIE) of the University of Valencia, Spain. Samples were digested with sequencing grade trypsin (Promega, Madison, USA) [23] and the resulting digestion mixture was dried in a vacuum centrifuge and resuspended in 7 µl of 2% acetonitrile (ACN), 0.1% trifluoroacetic acid (TFA), spotting 1 µl onto the target plate. After the droplets were air-dried at room temperature, 1 µl of matrix [10 mg/ml α-Cyano-4-hydroxycinnamic acid - α-Cyano-2,4-difluorocinnamic acid - α-Cyano-2,3,4,5,6-pentafluorocinnamic acid mixture (CHCA) (Sigma-Aldrich) in 0.1% TFA-ACN/H2O (1:1, v/v)] was added and allowed to air-dry at room temperature. The resulting mixtures were analysed in a 5800 Matrix-Assisted Laser Desorption/Ionization Time-Of-Flight (MALDI TOF/TOF) (AB Sciex, Framingham, USA) in positive reflectron mode. Five of the most intense precursors were selected for every position for the MS/MS analysis. MS/MS data were acquired using the default 1 kV MS/MS method with collision-induced dissociation (CID) active. The MS and MS/MS information was sent to MASCOT via the Protein Pilot (AB Sciex). The samples that did not have significant identification were analysed by liquid chromatography and tandem mass spectrometry (LC-MS/MS). Five µl of each sample were loaded onto a trap column (NanoLC Column, 3 µ C18-CL, 350 µm × 0.5 mm, Eksigent) and desalted with 0.1% TFA at 3 µl/min for 5 min. The peptides were then loaded onto an analytical column (LC Column, 3 µ C18-CL, 75 µm × 12 cm, Nikkyo) equilibrated in 5% ACN, 0.1% formic acid (FA). Elution was carried out with a linear gradient of 5 to 40% B in A (A: 0.1% FA; B: ACN, 0.1% FA) at a flow rate of 300 nl/min for 15 min. Peptides were analysed in a mass spectrometer nanoESIqQTOF (5600 TripleTOF, AB Sciex). Samples were ionized applying 2.8 kV to the spray emitter and analysed in a data-dependent mode. Survey MS1 scans were acquired from 350–1250 m/z for 250 ms. The quadrupole resolution was set to UNIT for MS2 experiments, which were acquired 100–1500 m/z for 50 ms in ‘high sensitivity’ mode. Up to 50 ions were selected for fragmentation after each survey scan. Dynamic exclusion was set to 15 s. The system sensitivity was controlled with 2 fmol of 6 proteins (LC Packings). Database searches were performed on the National Center for Biotechnology Information (NCBI) database with tryptic specificity allowing one missed cleavage and a tolerance on the mass measurement of 100 ppm in MS mode and 0.8 Da for MS/MS ions. Carbamidomethylation of Cys was used as a fixed modification and oxidation of Met and deamidation of Asn and Gln as variable modifications.
The molecular function and biological processes of the identified proteins were assigned according to the gene ontology database (http://www.geneontology.org) and the Swiss-Prot/UniProt database (http://beta.uniprot.org). Prediction of the secondary structures and three-dimensional modelling of the resulting sequences was conducted with the Swiss Model server [24] (http://swissmodel.expasy.org/). The 3-D models were visualized with the RasMol software v. 2.7.5.2.
Statistical analysis
Plasminogen binding and plasminogen activation assays results were analysed with the Student’s t-test. Data are expressed as the mean ± standard deviation (SD) of three independent experiments. Significant differences were defined as a P-value of < 0.05 for a confidence level of 95%.
Results
AsL3C and AsL3ES bind plasminogen
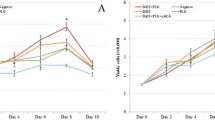
The ability of AsL3C and AsL3ES to bind plasminogen was assayed by ELISA. The results showed that both antigenic extracts were able to bind plasminogen since their optical densities were significantly higher than those obtained for the negative controls with BSA (AsL3C: t(4) = 27.4955, P = 0.0001; t(4) = 22.3828, P = 0.0001; t(4) = 19.4370, P = 0.0001; t(4) = 24.9594, P = 0.0001); AsL3ES: t(4) = 75.8832, P = 0.0001; t(4) = 193.3830, P = 0.0001; t(4) = 23.1134, P = 0.0001; t(4) = 37.6490, P = 0.0001). In addition, this binding was directly proportional to the amount of plasminogen (Fig. 1). The competition assay with ε-ACA showed similar values to the negative control both for AsL3C and AsL3ES, indicating the involvement of lysine residues in the plasminogen interaction (Fig. 1).

Plasminogen binding to AsL3C (a) and AsL3ES (b) by ELISA. Key: squares, 1 µg of AsL3C or AsL3ES incubated with increasing amounts of plasminogen (0–2 µg); circles, negative controls replacing the antigenic extracts by BSA; triangles, competition assays with 25 mM ε-ACA during plasminogen incubation. Each point represents the mean of three replicates ± SD. *P < 0.05
Plasminogen binding by AsL3C and AsL3ES was corroborated and characterized by western blot. Both antigenic extracts revealed plasminogen-binding bands that were absent in the negative control (Fig. 2). The SDS-PAGE showed approximately 30 bands for AsL3C and 20 for AsL3ES distributed in a broad MW range (between 10 and 100 kDa), of which 12 (10–75 kDa) and 5 (30–75 kDa) were revealed as plasminogen-binding bands in the immunoblots, respectively (Fig. 2).

Plasminogen binding to AsL3C and AsL3ES by western blot. Lanes 1–3: SDS-PAGE with 15 µg of AsL3C (Lane 1), 15 µg of AsL3ES (Lane 2) and 2 µg of plasminogen (Lane 3); Lanes 4–7: western blot incubated with plasminogen: AsL3C (Lane 4), AsL3ES (Lane 5) plasminogen (Lane 6) and the negative control (Lane 7). The reference of molecular weights is indicated on the left
Plasminogen is attached to the larval surface
The location of plasminogen binding on the larval surface was analysed by immunofluorescence and confocal microscopy. Plasminogen binding was observed along the larval surface, mainly in the anterior part of the body. No binding was revealed in the negative control. The assay performed in the presence of ε-ACA showed a much lower binding level than that carried out in its absence, suggesting the participation of lysine residues in the plasminogen binding (Fig. 3).

Plasminogen binding on the A. suum larval surface by immunofluorescence and confocal microscopy. a–c Fluorescence images from larvae surface. a Incubation with plasminogen and a (FITC)-labelled anti-plasminogen antibody. b The presence of plasminogen was omitted in the previous assay as a negative control. c Competition assay with 40 mM ε-ACA during plasminogen incubation. d–f Corresponding light microscopy images to a–c. Scale-bars: 25 µm
AsL3C and AsL3ES activate plasminogen and promote plasmin generation
The ability of AsL3C and AsL3ES to activate plasminogen and enhance plasmin generation was studied in a chromogenic assay. Both antigenic extracts activated plasminogen and promoted plasmin generation in the presence of both plasminogen activators, tPA and uPA, since the results obtained for these assays were significantly higher than those obtained by their corresponding negative controls [(AsL3C: (tPA) t(4) = 5.8712, P = 0.0042; (uPA) t(4) = 2.8378, P = 0.0470) (AsL3ES: (tPA) t(4) = 19.0919, P = 0.0001; (uPA) t(4) = 8.2024, P = 0.0012)] (Fig. 4). Comparing the results of both activators, the activation of plasminogen was higher by AsL3C and AsL3ES in the presence of tPA and uPA, respectively. Furthermore, AsL3C showed a slight ability to activate plasminogen in the absence of the activators since significant differences between the optical densities obtained by this experimental group and its negative control were found (t(4) = 10.3923, P = 0.0005) (Fig. 4).

Plasminogen activation and plasmin generation by AsL3C (a) and AsL3ES (b). Key: filled bars, 1 µg of AsL3C or AsL3ES incubated with 1 µg of plasminogen, 3 µg of S-2251 and a plasminogen activator (15 ng of tPA or 10 ng of uPA); empty bars, negative controls replacing the antigenic extracts by BSA. Each point represents the mean of three replicates ± SD. The asterisk (*) indicates significant differences (P < 0.05) with the control groups on its left
Identification of plasminogen-binding proteins in AsL3C and AsL3ES
In order to identify the proteins responsible for binding plasminogen, both antigenic extracts were analysed by 2-D electrophoresis and plasminogen-ligand blotting in a pH range of 3–10 and, subsequently, by MS. Silver stain of AsL3C 2-D gel revealed 560 spots with pIs between 3.3–9.7 and MWs between 12–176 kDa (Fig. 5a). The corresponding immunoblot revealed 54 plasminogen-binding spots with pIs between 5.1–9.5 and MWs between 13.7–176 kDa (Fig. 5b), representing a binding rate of 9.64% of total spots revealed. The AsL3ES 2-D gel showed a fewer number of spots (387) distributed in narrower ranges of pI (3.7–8.9) and MW (13.5–158 kDa) (Fig. 5c). In this case, only 16 spots were revealed as plasminogen-binding spots, with pIs between 5.1–7.1 and MWs between 48–109 kDa (Fig. 5d), representing a binding rate of 4.13%. In the case of the negative control, no spot was revealed in the membranes performed in the absence of plasminogen (not shown).

2-D images of AsL3C (a, b) and AsL3ES (c, d). a, c 2-D electrophoresis gels of 12% polyacrylamide and pH range of 3–10 revealed with silver stain. b, d Homologous western blots showing plasminogen-binding spots. The reference of molecular weights is indicated on the left. On the gel images, the reference of isoelectric point is indicated. Circled and numbered spots correspond to matched plasminogen-binding spots
The matching of spots revealed by immunoblots with their homologous in the silver-stained 2-D gels allowed us to select a total of 41 and 16 plasminogen-binding spots in the AsL3C and AsL3ES proteomes, respectively. Among them, 30 spots (20 from AsL3C and 10 from AsL3ES) were manually excised from the 2-D gels and submitted to analysis by MS. All of them were identified, corresponding to 12 different proteins deposited in databases as A. suum proteins. The proteins identified were maguk p55 subfamily member 6, phosphoenolpyruvate carboxykinase, disulphide isomerase, ATP synthase subunit, glucose-6-phosphate isomerase, actin-2, fructose-bisphosphate aldolase 1, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), 32 kDa beta-galactoside-binding lectin, 60s ribosomal protein l23, prion-like domain-bearing protein 25 and thachykinin-like peptides receptor 99d. Between 1 and 6 isoforms of each protein were identified. One of these proteins (glucose-6-phosphate isomerase) appeared in both antigenic extracts, 9 proteins derived from AsL3C and 2 proteins (prion-like domain-bearing protein 25 and thachykinin-like peptides receptor 99d) from AsL3ES. Table 1 shows the identity of these proteins with their corresponding NCBI accession code, their MWs and pIs, and the molecular function and biological process in which they are involved. Among these, 5 proteins were related to metabolic pathways, and 4 of them were linked with glycolytic processes.
Finally, in silico three-dimensional modelling of the identified plasminogen-binding proteins predicted their 3-D structures as it is shown in the Fig. 6. Lysine residues were highlighted and were visualized on the outside of the proteins as potential plasminogen-binding sites.

Molecular modelling of the plasminogen-binding proteins from AsL3 identified by MS. The secondary structure of the proteins was predicted with the Swiss-Model web server (http://swissmodel.expasy.org/) by analogy with the X-ray crystallography available models. The three-dimensional models of the molecules were visualized with the RasMol application v. 2.7.5.2. Lysine residues of proteins as possible plasminogen-binding sites are highlighted as red balls
Discussion
The plasmin broad substrate specificity makes this enzyme susceptible to be used by parasites to complement their own protease repertoire, which could facilitate their migration through the host tissues. In addition to parasites, other types of blood and/or tissue pathogens manipulate the host fibrinolytic system by means of the recruitment of plasminogen from the host plasma and its activation to plasmin [9, 10]. Despite A. suum being a parasite that spends most of its biological cycle in the intestinal tract of its host, its infective L3 stage carries out a wide migratory route through the host bloodstream [4]. For this reason, it was hypothesized that AsL3 interact with the host fibrinolytic system as a survival mechanism facilitating their intraorganic migration. In order to study this phenomenon, we decided to use cuticle and excretory/secretory antigenic extracts from the parasite larvae. Although the complete protein isolation of both extracts entails some limitations as a result of potential somatic products appearing in these compartments, we obtained two extracts enriched with cuticle and excretory/secretory proteins, respectively.
In the present study, we revealed the pro-fibrinolytic potential of the AsL3, due to the ability of their cuticle and excretory/secretory antigenic extracts to bind plasminogen in a lysine-dependent manner, which correlates with the results obtained for other helminth and protozoan parasites [10]. Plasminogen binding was located on the parasitic surface (Fig. 3), as previous studies have demonstrated for other parasites [21, 25]. This suggests that plasminogen-binding proteins are exposed to the larval surface, which could allow them to interact with plasminogen at the host-parasite interface. In addition, both antigenic extracts activated plasminogen and enhanced plasmin generation in the presence of host plasminogen activators (tPA and uPA), results that are consistent with those obtained for other parasites [21, 22, 26,27,28,29,30,31]. Plasmin generation by tPA was more effective in the presence of AsL3C, which could be due to the role of this molecule as the main activator responsible for the degradation of fibrin at a vascular level [11]. Hence AsL3 could use their surface associated plasminogen-binding proteins to promote plasmin generation in their immediate habitat while they are migrating through the bloodstream in order to avoid the clot formation. On the contrary, the generation of plasmin by uPA was higher in the presence of AsL3ES. uPA produces pericellular proteolysis since it binds to specific cellular receptors and activates cell-bound plasminogen [11]. Consequently, larvae could use their excretory/secretory plasminogen-binding proteins to enhance plasmin generation in specific cellular sites at a systemic level, as it has been postulated for other pathogens [30, 32]. Our results also showed that AsL3C induced plasmin generation by its own, in the absence of the plasminogen activators. Most of the studies carried out with parasitic extracts have revealed their dependence on the physiological plasminogen activators (tPA and/or uPA) in order to promote plasmin generation. This could indicate the presence of plasminogen activators in the AsL3 surface, as it has been already described for different genera of bacteria [14, 15], so that their identification could indicate a matter of future investigation. Regarding AsL3 pro-fibrinolytic potential, all these results suggest that there could be a combined action between both antigenic compartments at the host-parasite interface. Thus, AsL3 could use the proteolytic action of plasmin not only in their immediate environment, but also by directing it to those places where its activity is required at the pericellular level.
Plasmin produced by plasminogen activation can degrade fibrin networks and different proteins of the extracellular matrix [13], elements that could entail a barrier in the migration of parasites through the host tissues. The ability of AsL3 to enhance plasmin generation could be used by the parasite as a survival mechanism allowing it to degrade these components in order to facilitate its migration through the host tissues, as it has been postulated for other pathogens [27, 31, 33,34,35,36]. Regarding other survival-related functions, the degradation of proteins for nutrition, or of immunoglobulins and complement components for immune evasion mechanisms have also been suggested to be the result of the interaction between pathogens and the fibrinolytic system of their hosts [10]. Consequently, the plasmin generated by AsL3 could also contribute to the larval feeding or to modulate the host immune response. The latter could indicate that AsL3 can reach their definitive location at the small intestine of the host without being eliminated by the immune system during their migration through the bloodstream.
Apart from the effects related to the establishment and survival of the parasite in the host, the overproduction of plasmin by parasites has been linked with the appearance of pathological processes in the host, such as cell proliferation and migration, or inflammation [10, 37]. Intriguingly, one of the most distinctive lesions of ascariasis is the emergence of fibrotic, necrotic and hemorrhagic focus in the liver known as milk/white spots [38], which are accompanied by migratory trajectories of the parasitic larvae [6]. Even though it seems that these lesions are produced as a result of mechanical injury and inflammatory response induced by the larval migration in the liver [39], the overproduction of plasmin by the parasite could also contribute to this inflammatory phenomenon. These lesions not only cause damages on the health of the infected animals, but also comprise a significant problem for the porcine industry, since they produce less suitable livers for human consumption, producing economic losses for abattoirs [1]. Similar spots appear in the lungs, which are described as chronic reactions due to the larval migration [40], and in the intestinal mucosa, as a result of the larval penetration in the caecum and proximal colon [6].
In this study, 12 different proteins were identified as potential plasminogen receptors. Among them, GAPDH, fructose-bisphosphate aldolase 1 and actin-2 have been widely studied for their ability to interact with the fibrinolytic system of the host, as well as with the possible linkage of this interaction with the pathogen invasion processes [10]. Their roles as potential plasminogen receptors in a wide variety of unrelated biological groups show their high evolutionary conservation and the importance of plasminogen recruitment for pathogens in general and parasites in particular [10]. In addition, these proteins have been previously described as “moonlighting proteins” [41], which means that they are able to perform distinct functions depending on whether they are located inside or outside the cell, or in different compartments of the cell [42]. Following this idea, GAPDH and fructose-bisphosphate aldolase can function in the cytosol as glycolytic enzymes, while they could bind plasminogen outside the cell as part of the excretory/secretory system [43]. In this case, this secondary function would be possible due to the interaction between the so-called plasminogen kringle domains with the lysine residues of their receptors, which determines the phenomenon of plasminogen binding [44]. According to the 3-D models developed in this work, our results showed that lysine residues seem to be located externally in all the identified plasminogen-binding proteins, which would facilitate the accessibility of plasminogen (Fig. 6). Other proteins identified in this study that have been previously described as potential plasminogen receptors in other parasites are beta-galactoside-binding lectin and disulphide isomerase, which were found in the host-parasite interface of Dirofilaria immitis and Fasciola hepatica, respectively [29, 30]. The remaining proteins (maguk p55 subfamily member 6, phosphoenolpyruvate carboxykinase, ATP synthase subunit, glucose-6-phosphate isomerase, 60s ribosomal protein l23, prion-like domain-bearing protein 25 and thachykinin-like peptides receptor 99d) have been related to the plasminogen recruitment for the first time in this study. Interestingly, the ATP synthase subunit has a previous connection with plasminogen binding since different subunits of this protein bind angiostatin, a proteolytic fragment of plasminogen, in human cells [45].
Conclusions
To our knowledge, we demonstrate for the first time the interaction between AsL3 and the fibrinolytic system of the host. Both cuticle and excretory/secretory antigenic extracts of the parasite bind plasminogen and enhance plasmin generation, which could facilitate the intraorganic migration of the parasite. As previously mentioned, A. suum is currently considered as a model to study A. lumbricoides. This is possible because both species have the same life-cycle and there are cross infections and gene flow between them, since they are able to produce hybrids [46,47,48]. This fact allows to extrapolate our results to the knowledge of the molecular mechanisms governing a human health problem of increasing importance that affects about a billion people in the world, such as human ascariasis. Future studies aimed at unravelling the molecular relationships between Ascaris roundworms and their hosts in an early phase of infection could be of paramount importance in order to design new tools that allow us to control these parasitosis before the adult worms are established in their definitive location.
Availability of data and materials
Data supporting the conclusions of this article are included within the article. The datasets used and/or analysed during the present study are available from the corresponding author upon reasonable request.
Abbreviations
- L3:
-
third-stage larvae
- tPA:
-
tissue plasminogen activator
- uPA:
-
urokinase-type plasminogen activator
- AsL3C:
-
cuticle antigenic extract of the L3 of A. suum
- AsL3ES:
-
excretory/secretory antigenic extract of the L3 of A. suum
- AsL3:
-
third-stage larvae of A. suum
- PBS:
-
phosphate-buffered saline
- CTAB:
-
cetyl trimethyl ammonium bromide
- ELISA:
-
enzyme-linked immunosorbent assay
- BSA:
-
bovine serum albumin
- ε-ACA:
-
ε-aminocaproic acid
- OD:
-
optical densities
- FITC:
-
fluorescin isothiocyanate
- 2-D:
-
two-dimensional
- CHAPS:
-
3-((3-cholamidopropyl) dimethylammonio)-1-propanesulfonate
- DTT:
-
dithiothreitol
- pI:
-
isoelectric pint
- MW:
-
molecular weight
- MS:
-
mass spectrometry
- ACN:
-
acetonitrile
- TFA:
-
trifluoroacetic acid
- CHCA:
-
α-Cyano-4-hydroxycinnamic acid - α-Cyano-2,4-difluorocinnamic acid - α-Cyano-2,3,4,5,6-pentafluorocinnamic acid mixture
- MALDI TOF/TOF:
-
Matrix-Assisted Laser Desorption/Ionization Time-Of-Flight
- CID:
-
collision-induced dissociation
- LC-MS/MS:
-
liquid chromatography and tandem mass spectrometry
- FA:
-
formic acid
- NCBI:
-
National Center for Biotechnology Information
- SD:
-
standard deviation
- GAPDH:
-
glyceraldehyde-3-phosphate dehydrogenase
References
Thamsborg SM, Nejsum P, Mejer H. Impact of Ascaris suum in livestock. In: Holland C, editor. Ascaris: the neglected parasite. Cambridge: Academic Press, Elsevier; 2013. p. 363–81.
Peng W, Criscione CD. Ascariasis in people and pigs: new inferences from DNA analysis of worm populations. Infect Genet Evol. 2012;12:227–35.
Jourdan PM, Lamberton PHL, Fenwick A, Addiss DG. Soil-transmitted helminth infections. Lancet. 2018;391:252–65.
Dold C, Holland CV. Ascaris and ascariasis. Microbes Infect. 2011;13:632–7.
Read AF, Skorping A. The evolution of tissue migration by parasitic nematode larvae. Parasitology. 1995;111:359–71.
Holland CV, Behnke JM, Dold C. Larval ascariasis: impact, significance, and model organisms. In: Holland C, editor. Ascaris: the neglected parasite. Cambridge: Academic Press, Elsevier; 2013. p. 107–25.
Ebner F, Kuhring M, Radonić A, Midha A, Renard BY, Hartmann S. Silent witness: dual-species transcriptomics reveals epithelial immunological quiescence to helminth larval encounter and fostered larval development. Front Immunol. 2018;9:1868.
Jex AR, Liu S, Li B, Young ND, Hall RS, Li Y, et al. Ascaris suum draft genome. Nature. 2011;479:529–33.
Figuera L, Gómez-Arreaza A, Avilán L. Parasitism in optima forma: exploiting the host fibrinolytic system for invasion. Acta Trop. 2013;128:116–23.
González-Miguel J, Siles-Lucas M, Kartashev V, Morchón R, Simón F. Plasmin in parasitic chronic infections: friend or foe? Trends Parasitol. 2016;32:325–35.
Rijken DC, Lijnen HR. New insights into the molecular mechanisms of the fibrinolytic system. J Thromb Haemost. 2009;7:4–13.
Cesarman-Maus G, Hajjar KA. Molecular mechanisms of fibrinolysis. Br J Haematol. 2005;129:307–21.
Syrovets T, Simmet T. Novel aspects and new roles for the serine protease plasmin. Cell Mol Life Sci. 2004;61:873–85.
Lähteenmäki K, Kuusela P, Korhonen TK. Bacterial plasminogen activators and receptors. FEMS Microbiol Rev. 2001;25:531–52.
Bhattacharya S, Ploplis VA, Castellino FJ. Bacterial plasminogen receptors utilize host plasminogen system for effective invasion and dissemination. J Biomed Biotechnol. 2012;2012:482096.
Vlaminck J, Masure D, Wang T, Nejsum P, Hokke CH, Geldhof P. A phosphorylcholine-containing glycolipid-like antigen present on the surface of infective stage larvae of Ascaris spp. is a major antibody target in infected pigs and humans. PLoS Negl Trop Dis. 2016;10:e0005166.
Urban JF Jr, Douvres FW, Tromba FG. A rapid method for hatching Ascaris suum eggs in vitro. Proc Helminthol Soc Wash. 1981;48:241–3.
Wedrychowicz H, Holmes PH, Bairden K, Tait A. Surface and excretory/secretory antigens of fourth-stage larvae and adult Ostertagia circumcincta. Vet Parasitol. 1994;53:117–32.
González-Miguel J, Morchón R, Mellado I, Carretón E, Montoya-Alonso JA, Simón F. Excretory/secretory antigens from Dirofilaria immitis adult worms interact with the host fibrinolytic system involving the vascular endothelium. Mol Biochem Parasitol. 2012;181:134–40.
Maizels RM, Blaxter ML, Robertson BD, Selkirk ME. Parasite antigens, parasite genes: a laboratory manual for molecular parasitology. Cambridge: Cambridge University Press; 1991.
Ramajo-Hernández A, Pérez-Sánchez R, Ramajo-Martín V, Oleaga A. Schistosoma bovis: plasminogen binding in adults and the identification of plasminogen-binding proteins from the worm tegument. Exp Parasitol. 2007;115:83–91.
Fernandes RS, Fernandes LGV, de Godoy AS, Miyasato PA, Nakano E, Farias LP, et al. Schistosoma mansoni venom allergen-like protein 18 (SmVAL18) is a plasminogen-binding protein secreted during the early stages of mammalian-host infection. Mol Biochem Parasitol. 2018;221:23–31.
Shevchenko A, Jensen ON, Podtelejnikov AV, Sagliocco F, Wilm M, Vorm O, et al. Linking genome and proteome by mass spectrometry: large-scale identification of yeast proteins from two dimensional gels. Proc Natl Acad Sci USA. 1996;93:14440–5.
Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201.
Acosta H, Rondón-Mercado R, Avilán L, Concepción JL. Interaction of Trypanosoma evansi with the plasminogen-plasmin system. Vet Parasitol. 2016;226:189–97.
Avilan L, Calcagno M, Figuera M, Lemus L, Puig J, Rodriguez AM. Interaction of Leishmania mexicana promastigotes with the plasminogen-plasmin system. Mol Biochem Parasitol. 2000;110:183–93.
Almeida L, Vanegas G, Calcagno M, Concepción JL, Avilan L. Plasminogen interaction with Trypanosoma cruzi. Mem Inst Oswaldo Cruz. 2004;99:63–7.
Rojas M, Labrador I, Concepción JL, Aldana E, Avilan L. Characteristics of plasminogen binding to Trypanosoma cruzi epimastigotes. Acta Trop. 2008;107:54–8.
González-Miguel J, Morchón R, Carretón E, Montoya-Alonso JA, Simón F. Surface associated antigens of Dirofilaria immitis adult worms activate the host fibrinolytic system. Vet Parasitol. 2013;196:235–40.
González-Miguel J, Valero MA, Reguera-Gomez M, Mas-Bargues C, Bargues MD, Simón F, et al. Numerous Fasciola plasminogen-binding proteins may underlie blood-brain barrier leakage and explain neurological disorder complexity and heterogeneity in the acute and chronic phases of human fascioliasis. Parasitology. 2019;146:284–98.
Zhang S, Guo A, Zhu X, You Y, Hou J, Wang Q, et al. Identification and functional characterization of alpha-enolase from Taenia pisiformis metacestode. Acta Trop. 2015;144:31–40.
Bergmann S, Hammerschmidt S. Fibrinolysis and host response in bacterial infections. Thromb Haemost. 2007;98:512–20.
Jolodar A, Fischer P, Bergmann S, Büttner DW, Hammerschmidt S, Brattig NW. Molecular cloning of an alpha-enolase from the human filarial parasite Onchocerca volvulus that binds human plasminogen. Biochim Biophys Acta. 2003;1627:111–20.
Yang J, Qiu C, Xia Y, Yao L, Fu Z, Yuan C, et al. Molecular cloning and functional characterization of Schistosoma japonicum enolase which is highly expressed at the schistosomulum stage. Parasitol Res. 2010;107:667–77.
Sanderson-Smith ML, De Oliveira DM, Ranson M, McArthur JD. Bacterial plasminogen receptors: mediators of a multifaceted relationship. J Biomed Biotechnol. 2012;2012:272148.
Figuera L, Acosta H, Gómez-Arreaza A, Dávila-Vera D, Balza-Quintero A, Quiñones W, et al. Plasminogen binding proteins in secreted membrane vesicles of Leishmania mexicana. Mol Biochem Parasitol. 2013;187:14–20.
González-Miguel J, Morchón R, Carretón E, Montoya-Alonso JA, Simón F. Can the activation of plasminogen/plasmin system of the host by metabolic products of Dirofilaria immitis participate in heartworm disease endarteritis? Parasit Vectors. 2015;8:194.
Pérez J, García PM, Mozos E, Bautista MJ, Carrasco L. Immunohistochemical characterization of hepatic lesions associated with migrating larvae of Ascaris suum in pigs. J Comp Pathol. 2001;124:200–6.
Ronéus O. Studies on the aetiology and pathogenesis of white spots in the liver of pigs. Acta Vet Scand. 1966;16:1–112.
Liljegren CH, Aalbaek B, Nielsen OL, Jensen HE. Some new aspects of the pathology, pathogenesis, and aetiology of disseminated lung lesions in slaughter pigs. APMIS. 2003;111:531–8.
Karkowska-Kuleta J, Kozik A. Moonlighting proteins as virulence factors of pathogenic fungi, parasitic protozoa and multicellular parasites. Mol Oral Microbiol. 2014;29:270–83.
Jeffery CJ. Moonlighting proteins. Trends Biochem Sci. 1999;24:8–11.
Gómez-Arreaza A, Acosta H, Quiñones W, Concepción JL, Michels PA, Avilán L. Extracellular functions of glycolytic enzymes of parasites: unpredicted use of ancient proteins. Mol Biochem Parasitol. 2014;193:75–81.
Plow EF, Herren T, Redlitz A, Miles LA, Hoover-Plow JL. The cell biology of the plasminogen system. FASEB J. 1995;9:939–45.
Moser TL, Stack MS, Asplin I, Enghild JJ, Højrup P, Everitt L, et al. Angiostatin binds ATP synthase on the surface of human endothelial cells. Proc Natl Acad Sci USA. 1999;96:2811–6.
Criscione CD, Anderson JD, Sudimack D, Peng W, Jha B, Williams-Blangero S, et al. Disentangling hybridization and host colonization in parasitic roundworms of humans and pigs. Proc Biol Sci. 2007;274:2669–77.
Betson M, Nejsum P, Bendall RP, Deb RM, Stothard JR. Molecular epidemiology of ascariasis: a global perspective on the transmission dynamics of Ascaris in people and pigs. J Infect Dis. 2014;210:932–41.
Nejsum P, Hawash MB, Betson M, Stothard JR, Gasser RB, Andersen LO. Ascaris phylogeny based on multiple whole mtDNA genomes. Infect Genet Evol. 2017;48:4–9.
Acknowledgements
We would like to thank the cooperative society “Chacinera Albercana” (Salamanca, Spain) for providing us the adult female worms of A. suum, as well as Paula J. Gómez-González for improving the English version of the manuscript. The proteomic analysis was performed in the proteomics facility of SCSIE University of Valencia that belongs to ProteoRed, PRB2-ISCIII, supported by Grant PT13/0001.
Funding
This study was supported by the Centro para el Desarrollo Tecnológico Industrial (CDTI) (Grant number IDI-2016414). AD is supported by a doctoral fellowship from the University of Salamanca, co-funded by Banco Santander. JGM is supported by the JIN Grant “ULYSSES” (RTI2018-093463-J-100) funded by Ministerio de Ciencia, Innovación y Universidades (MCIU), Agencia Estatal de Investigación (AEI) and Fondo Europeo de Desarrollo Regional (FEDER, UE).
Author information
Authors and Affiliations
Contributions
JGM conceived and designed the study. AD and RM obtained the biological material. AD performed the experiments. AD, FS and JGM analysed and interpreted the data. AD, JGM and FS wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Diosdado, A., Simón, F., Morchón, R. et al. Pro-fibrinolytic potential of the third larval stage of Ascaris suum as a possible mechanism facilitating its migration through the host tissues. Parasites Vectors 13, 203 (2020). https://doi.org/10.1186/s13071-020-04067-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-020-04067-5