
Abstract
Background
Jackals are medium-sized canids from the wolf-like clade, exhibiting a unique combination of ancestral morphotypes, broad trophic niches, and close phylogenetic relationships with the wolf and dog. Thus, they represent a potential host of several pathogens with diverse transmission routes. Recently, populations of the Eurasian golden jackal Canis aureus have expanded into the Western Palaearctic, including most of Europe. The aim of our study was to examine Eurasian golden jackals from Romania, Czech Republic and Austria for a wide spectrum of vector-borne protists and to evaluate the role of this species as a reservoir of disease for domestic dogs and/or humans.
Results
Diagnostic polymerase chain reaction (PCR) DNA amplifications revealed 70% of jackals to be positive for Hepatozoon, 12.5% positive for piroplasms, and one individual positive for Leishmania infantum. Phylogenetic analyses of partial 18S rDNA sequences invariably placed sequenced isolates of Hepatozoon into the H. canis clade. For piroplasms, both the 18S and cox1 sequences obtained confirmed the presence of Babesia canis and “Theileria annae” in 5 and 2 individuals, respectively, providing the first records of these two piroplasmids in Eurasian golden jackals. A single animal from Dolj County (Romania) was PCR-positive for L. infantum, as confirmed also by sequencing of ITS1-5.8S.
Conclusions
Apparently, expanding populations of jackals can play a significant role in spreading and maintaining new Babesia canis foci in Central Europe. The role of jackals in the epidemiology of “Theileria annae” and H. canis is probably similar to that of red foxes and should be taken into account in further research on these parasites. Also the presence of L. infantum deserves attention. Our study confirms that once established, the populations of Eurasian golden jackals constitute natural reservoirs for many canine vector-borne diseases, analogous to the role of the coyotes in North America.
Similar content being viewed by others

Background
Wildlife reservoirs for diseases of companion animals and humans merit serious attention (e.g., [1–4]), due to recent dynamics in their populations and distribution ranges [5] and the resulting changes in disease epidemiology. High population densities and ubiquitous presence make red foxes (Vulpes vulpes) the most commonly discussed wild carnivore in Europe. However, the current dramatic expansion of Eurasian golden jackal [6–8] is likely to change the situation. In contrast to the red fox, the Eurasian golden jackal is closely related to and may hybridize with the domestic dog [9]. A preference for lowland habitats, ability to establish vital populations near human settlements and close phylogenetic relationships with dogs increase the probability of interactions between these two canid species. Considering these facts together, Eurasian golden jackals might become the most significant reservoir of diseases of zoonotic and veterinary importance among the European canids. The Eurasian golden jackal has been reported as a host of pathogens of zoonotic and/or veterinary importance, including a range of vector-borne pathogens such as Ehrlichia canis, Anaplasma phagocytophilum, the spotted fever group rickettsiae [10], Dirofilaria spp. [11], and recently also Thelazia callipaeda [12]. Among tick-borne protists, Hepatozoon canis is the only pathogen reported in golden jackals in Europe [13, 14]. There is a near absence of published data regarding piroplasms in Eurasian golden jackals. Several studies have reported the presence of Leishmania infantum in Eurasian golden jackals [15, 16] and suggested their importance as a potential reservoir of zoonotic L. infantum.
The aim of our study was to examine tissues of Eurasian golden jackals from Romania, Czech Republic and Austria for a spectrum of vector-borne protists and to evaluate the role of this species as a potential reservoir of diseases for domestic dogs and/or humans. Using phylogenetic analyses of nuclear and mitochondrial markers, we provide more detailed insight into parasites’ intraspecific variability, ruling out possible existence of jackal-specific lineages of the parasites investigated.
Methods
Fifty-four Eurasian golden jackals were legally hunted between October 2013 and May 2015 in 11 counties of Romania with established jackal populations. To sample animals from the edge of distribution range, we also included a single available animal from the Czech Republic, shot in the Moravia-Silesia Region near Nový Jičín (July 2014), and a single Austrian road-killed Eurasian golden jackal from Wiener Neudorf in Lower Austria (January 2012). All carcasses were frozen and transported to the laboratory for necropsy, wherein blood, blood clot and spleen samples were collected and frozen at -20 °C. Bone marrow used for Leishmania detection was collected and frozen from a subset of 36 Romanian golden jackals only.
DNA isolation and polymerase chain reaction
DNA from Romanian and Czech golden jackal blood samples was isolated using the Genomic DNA Mini Kit (Geneaid Biotech, Taipei city, Taiwan) from 200 μl of blood or blood clot equivalent in size. In one Romanian animal in which blood was not available, 20 mg of spleen tissue were used for DNA isolation. In a subset of 36 golden jackals, DNA was isolated from 20 mg of bone marrow using a High Pure PCR Template Preparation Kit (Roche Life Science, Mennheim, Germany). The processing of the Austrian sample is described elsewhere [13].
Hepatozoon and Piroplasmida PCR
All 56 samples were screened using two different diagnostic PCRs amplifying fragments of Hepatozoon spp. (~670 bp) or piroplasms (~560 bp) small ribosomal subunit (18S rDNA) to identify the positive ones.
Genus specific primers were used for Hepatozoon spp. detection [17]. For positive samples, further PCR was performed to obtain ~1700 bp fragments of 18S rDNA [18].
For piroplasms detection nested PCR was used [19]. For positive samples, modified nested PCR amplifying ~1700 bp fragments of 18S rDNA using the combination of BT1 F, BT outer R primers in first run and BT Inner R [20] and Piro0F2 [21] primers in second run was used. To confirm the determination of piroplasmids and to obtain further phylogenetic characterization, two modified nested PCR protocols amplifying fragments of cytochrome c oxidase subunit 1 (cox1) gene were performed [21, 22]. Table 1 provides details on the primers used, PCR conditions and obtained fragments for all PCRs.
All PCRs were performed using commercial master mix following the manufacturer’s instructions. A total volume of 25 μl was prepared for each reaction containing 12.5 μl of master mix, 10 pmol of each primer, 2 μl of template DNA or 1 μl of PCR product from the first run in the case of nested PCR, plus PCR water. DNA samples from a fox naturally infected by Hepatozoon and from a dog naturally infected with B. gibsoni, were used as positive controls; in both cases, the infection was confirmed microscopically.
Products from selected PCRs (~1700 bp of 18S rDNA of Hepatozoon PCR and products from second runs of all piroplasmida PCRs) were purified using a Gel/PCR DNA Fragments Extraction Kit (Geneaid Biotech Ltd., Taipei city, Taiwan) and directly sequenced using the amplification primers. For sequencing, a ~1700 bp fragment of Babesia 18S rDNA, the additional sequencing primers BTH-1 F, BTH-1R, 600 F, 1200 F [20] were used and one more sequencing primer 18S-R4 (5’-CTT GCG CAT ACT AGG CAT TCC TCG-3’) was designed to cover the whole length of 18S amplified fragment during the sequencing. Because of low quality of obtained sequences of “T. annae” cox1 fragments, PCR products from second run of nested PCR were cloned using a TOPO®-TA Cloning Kit (Thermo Fisher Scientific, Carlsbad, USA). Plasmids from cloning reactions were isolated using a GenElute™ Plasmid Miniprep Kit (Sigma-Aldrich, St. Louis, USA) and sequenced according to the manufacturer’s instructions. All sequencing reactions were done by Macrogen capillary sequencing services (Macrogen Europe, Amserdam, the Netherlands).
Leishmania PCR
Based on our previous experiences [23, 24] and the extensive review focused on molecular diagnosis of Leishmania infections [25] we have decided to use two different PCR protocols (amplifying the same target locus) for the Leishmania DNA screening of the subset of 36 samples. Quantitative PCR (iQSYBER Green Supermix, BioRad, Hercules, Foster City, CA, USA) was performed using the primers ITS-219 F and ITS-219R to amplify a ~280 bp product of the internal transcribed spacer (ITS1) region of the Leishmania rRNA operon [26]. To confirm the results of qPCR obtained, the same 36 DNA samples were also analysed by conventional PCR to amplify a ~300 bp ITS1-5.8S fragment using primers LITSR and L5.8S [27]. The DNA of Leishmania major was used as a positive control. The conventional PCR products were visualized on 2% agarose gels, purified with a High Pure PCR Product Purification Kit (Roche) and submitted for direct DNA sequencing (ABI 3730 automated sequencer, Applied Biosystems) to the Center for Genomic Technologies at the Faculty of Science, Charles University in Prague.
Sequence analysis
All obtained sequences were edited and analysed using Geneious® 9.1.2 [28] and compared with those available in the GenBank database by BLASTn analysis (http://blast.ncbi.nlm.nih.gov/Blast.cgi). Alignments of non-coding (18S rDNA) sequences were generated using the ClustalW algorithm [29]. For cox1 sequences, the nucleotide dataset was translated into amino acids and aligned in BioEdit 7.0.5.3 [30] using the ClustalW algorithm and aligned sequences were translated back to nucleotides. Nucleotide sequences in the edited dataset were then used to infer phylogeny. Evolution models were determined by a likelihood ratio test using R software (R Core Team, 2012). Phylogenetic analyses were performed using the maximum likelihood method in PhyML 3.0 software [31]. Phylogenetic trees were visualized and edited in FigTree v1.4.1 (http://tree.bio.ed.ac.uk/software/figtree/).
Results
Diagnostic PCR revealed that 39/56 (70%) of the golden jackals were positive for Hepatozoon, including the specimens from the Czech Republic and Austria. All seven (12.5%) piroplasm-positive animals originated from Romania and were co-infected with Hepatozoon. Furthermore, one sample from Dolj County (Romania) was PCR-positive for L. infantum in both protocols used, as confirmed by the sequence of the partial ITS1-5.8S fragment. Table 2 and Fig. 1 provide data for the distribution of the positive animals in Romania and Table 3 provides details of the nearest hits in a BLAST search.

Sampling localities in Romania with positive (red and/or green dots) or negative (black dot) results. a Hepatozooncanis; occurrence of Rhipicephalus sanguineus (s.l.) is marked with an × (Mihalca et al. [39]. b Piroplasmida (Babesia canis, red, “Theileria annae”, green); occurrence of Dermacentor reticulatus is marked with an × (Mihalca et al. [39]). c Leishmania infantum; occurrence of Phlebotomus neglectus and Phlebotomus perfiliewi is marked with an × (Dumitrache et al. [49])
The BLAST analysis of partial 18S rDNA sequences retrieved from piroplasm diagnostic PCR identified 5 positive samples as Babesia canis (99–100% identity) and 2 as “Theileria annae” (100% nucleotide identity) (Table 3). Additionally, the ~1700 bp fragments were amplified from two B. canis-positive golden jackals, both confirming the B. canis genotype B [32, 33] in BLAST. Phylogenetic analysis of 504 bp fragment of 18S rDNA of Babesia (sensu stricto) (s.s.) [34] species infecting dogs placed all B. canis sequences retrieved from golden jackals into “group B” (Fig. 2), together with sequences from domestic dogs. Phylogenetic analysis of 506 bp of 18S rDNA sequences of piroplasmid clade I according to Schnittger et al. [34] placed both “T. annae” sequences from our samples into a common clade with sequences from foxes and dogs, forming a sister clade to Babesia microti sequences, being more distantly related to B. leo and B. rodhaini (Fig. 3).

Maximum likelihood tree based on 504 nt-long alignment of 18S rDNA sequences of dogs infected by Babesia spp.; sequences of B. caballi (AY309955, EU888901, EU642514) used as the outgroup are not shown; bootstrap values from 1000 replicates shown only above 60%

Maximum likelihood tree based on 506 nt-long alignment of 18S rDNA sequences of “clade I” (according to Schnittger et al. [34]); sequences of B. conradae (AF158702) and B. lengau (GQ411415, AF158700) used as the outgroup are not shown; bootstrap values from 1000 replicates shown only above 60%
Furthermore, a ~900–1000 bp cox1 fragment was obtained from all piroplasm-positive samples. BLAST determination of these sequences confirmed the results of 18S fragment analysis (Table 3). Phylogenetic analysis of 768 bp of all available piroplasm cox1 sequences was performed. All five B. canis sequences were placed within the B. canis clade (Fig. 4), whereas six unique clones of “Theileria annae” cox1 obtained from two different animals formed a sister clade with those of B. microti.

Maximum likelihood tree based on 768 nt-long alignment of cox1 sequences of all piroplasms; sequences of Plasmodium sp. (AY791691, AB550280, KJ569502, AB379667) used as the outgroup are not shown; bootstrap values from 1000 replicates shown only above 60%
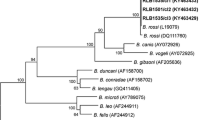
A fragment of ~1700 bp of Hepatozoon 18S rDNA was obtained from eight Romanian jackals and from both individuals originated from the Czech Republic and Austria. Sequence analysis by BLAST confirmed 99–100% identity of H. canis (Table 3). In phylogenetic analysis, all 10 sequences clustered within the H. canis clade from dogs and other wild canids (Fig. 5). Only limited number of ~1700 bp of Hepatozoon 18S rDNA sequences is available in GenBank; thus shorter fragment was used for phylogenetic analysis to cover the broad geographical and host range of this parasite.

Maximum likelihood tree based on 722 nt-long alignment of 18S rDNA sequences of Hepatozoon spp; sequences of Hemolivia spp. (KF992713, KF992698, KF992702) used as the outgroup are not shown; bootstrap values from 1000 replicates shown only above 60%
The obtained sequences of 18S rDNA (KX712122–KX712130), cox1 (KX712131- KX712138) and ITS1-5.8S (KX712139) fragments were deposited in the GenBank database. Identical sequences were deposited under the same accession number.
Discussion
Free-ranging canids are key hosts for a broad range of parasites including those transmitted by arthropods. The distribution and abundance of the large European carnivores (e.g. wolves, lynxes, bears) is surprisingly dynamic, showing fluctuation not only in abundance within particular areas but also in geographical distribution [5]. The gradual spread of Eurasian golden jackals is remarkable from an ecological perspective insofar as this mesocarnivore species is a predator historically largely new in central European ecosystems [6–8, 35]. The role of jackals is interesting also from an epidemiological perspective, given that they can act as reservoir hosts for several pathogens transmitted by arthropod vectors [10–16].
Our study revealed a significant proportion (70%) of examined jackals testing positive for H. canis. Although typically found in dogs in the Mediterranean region within the range of its only known vector, R. sanguineus (s.l.) ticks the parasite is common in red foxes but rare in dogs in areas where R. sanguineus (s.l.) is absent [4, 36]. We detected H. canis in golden jackals both inside and outside the R. sanguineus (s.l.) range in Romania (as illustrated in Fig. 1), and in both animals from Austria and the Czech Republic. Previously, the pathogen has been reported in golden jackals from Austria and Hungary [13, 14]. All sequences of 18S rDNA obtained from jackals in our study cluster intermixed with sequences from dogs and foxes available in GenBank. Despite the observed heterogeneity of the obtained sequences, the analysis did not reveal any host- or geography-related pattern in the sequences’ clustering suggesting ongoing transmission between involved carnivore species.
Although piroplasms have been reported in other jackal species/lineages from Africa and Asia [37, 38], surprisingly no records exist of piroplasms in Eurasian golden jackals in Europe. To overcome the low sequence variability of the 18S gene of piroplasmids, the amplification of partial mitochondrial cox1 gene in all piroplasm-positive samples was performed. The cox1 sequences and resulting phylogenetic analyses indicated a significantly higher degree of sequence heterogeneity and confirmed the presence of B. canis and “T. annae” in samples examined in this study.
We detected B. canis only in jackals from Romania, where the origin of positive animals corresponds with the known distribution of D. reticulatus [39] as well as with reported clinical cases of canine babesiosis [40–42] (Fig. 1). Phylogenetic analysis separated the clade of B. canis into two lineages corresponding with the previously described genotypes “A” and “B”. All five sequences from golden jackals from our study cluster within the genotype B carrying the AG nucleotides at positions 609–610 (according to AY072926) [32, 33]. The amplification of ~1700 bp 18S rDNA fragments was successful in two B. canis-positive animals only, probably due to co-infection and unspecific parallel amplification of H. canis 18S rDNA and despite our attempts to optimize PCR conditions.
Our study has suggests the Eurasian golden jackal to be a novel host for “Theileria annae”, however, the final proof of their reservoir role requires also other than DNA-based data. This piroplasmid is widespread all over Europe in red foxes and occasionally causes clinical piroplasmosis in dogs [20, 43]. The species belongs to a basal piroplasmid clade referred to either as clade VIII sensu Lack et al. [44] or clade I sensu Schnittger et al. [34], distant from “typical” Babesia and Theileria. Generic placement of the taxon described as T. annae [43] is unresolved and the clade rather represents a separate genus [44]. The nomenclature of this species is recently under the discussion [45, 46] and to prevent further confusion, we refer this species to as “Theileria annae” throughout our paper. The importance of Eurasian golden jackals in this piroplasmid’s transmission cycle remains questionable, it is well possible that they, like dogs, are only accidental hosts while red foxes remain its major natural host and reservoir [20, 43].
From the epidemiological perspective, jackals in the studied area host the range of tick species typical for other carnivores including domestic dogs. The same set of individuals of Romanian Eurasian golden jackals was surveyed recently, reporting Ixodes ricinus, I. hexagonus, Dermacentor reticulatus, Haemaphysalis punctata, H. concinna and Rhipicephalus sanguineus (s.l.) [47]. However, the vectorial competence of individual species of ixodids parasitizing jackals deserves further attention, especially related to transmission of Hepatozoon canis in non-Rhipicephalus areas.
The only jackal, which tested positive for L. infantum, originated from southern Romania, close to the Bulgarian border (Fig. 1). Although Romania has not been regarded in the last 80 years as endemic for canine leishmaniasis, two recent reports of this parasite in dogs brought the disease to our attention [48, 49]. The prevalence of L. infantum in our study is relatively low (1/36), and it remains to be clarified whether the apparent recent re-emergence of canine leishmaniasis in Romania might be related to jackal expansion and its role as a known reservoir [15, 16].
Conclusion
As a close relative of the domestic dog, the Eurasian golden jackal apparently hosts a wide spectrum of vector-borne pathogenic protists that affect dogs in Europe. Expanding populations of jackals rapidly colonize new localities in central Europe, and the vagrant animals may play a significant role in spreading B. canis into new localities in which D. reticulatus is present. The jackals’ role in the epidemiology of “T. annae” and H. canis is probably similar to that of red foxes and should be taken into account in further research on these parasites. Despite the low prevalence of L. infantum in our study, migrating jackals could represent a significant risk for spreading canine leishmaniasis into new areas.
References
Duscher GG, Leschnik M, Fuehrer HP, Joachim A. Wildlife reservoirs for vector-borne canine, feline and zoonotic infections in Austria. Int J Parasitol Parasites Wildl. 2014;4:88–96.
Mackenstedt U, Jenkins D, Romig T. The role of wildlife in the transmission of parasitic zoonoses in peri-urban and urban areas. Int J Parasitol Parasites Wildl. 2014;4:71–9.
Otranto D, Cantacessi C, Dantas-Torres F, Brianti E, Pfeffer M, Genchi C, et al. The role of wild canids and felids in spreading parasites to dogs and cats in Europe. Part II: Helminths and arthropods. Vet Parasitol. 2015a;213:24–37.
Otranto D, Cantacessi C, Pfeffer M, Dantas-Torres F, Brianti E, Deplazes P, et al. The role of wild canids and felids in spreading parasites to dogs and cats in Europe. Part I: Protozoa and tick-borne agents. Vet Parasitol. 2015b;213:24–37.
Chapron G, Kaczensky P, Linnell JDC, von Arx M, Huber D, Andrén H, et al. Recovery of large carnivores in Europe’s modern human-dominated landscapes. Science. 2014;346:1517–9.
Arnold J, Humer A, Heltai M, Murariu D, Spassov N, Hackländer K. Current status and distribution of golden jackals Canis aureus in Europe. Mamm Rev. 2012;42:1–11.
Trouwborst A, Krofel M, Linnell JD. Legal implications of range expansions in a terrestrial carnivore: the case of the golden jackal (Canis aureus) in Europe. Biodivers Conserv. 2015;24:2593–610.
Jann G. Die Chorologie des Goldschakals (Canis aureus Linnaeus, 1758) in Europa. The distribution of the golden jackal (Canis aureus Linnaeus, 1758) in Europe. 2016. doi:10.13140/RG.2.1.4869.2080. Accessed 8 Dec 2016.
Galov A, Fabbri E, Caniglia R, Arbansic H, Lapalombella S, Florijančic T, et al. First evidence of hybridization between golden jackal (Canis aureus) and domestic dog (Canis familiaris) as revealed by genetic markers. R Soc Open Sci. 2015;2:150450.
Waner T, Baneth G, Strenger C, Keysary A, King R, Harrus S. Antibodies reactive with Ehrlichia canis, Ehrlichia phagocytophila genogroup antigens and the spotted fever group rickettsial antigens, in free-ranging jackals (Canis aureus syriacus) from Israel. Vet Parasitol. 1999;82:121–8.
Ionică AM, Matei IA, D’Amico G, Daskalaki AA, Juránková J, Ionescu DT, et al. Role of golden jackals (Canis aureus) as natural reservoirs of Dirofilaria spp. in Romania. Parasit Vectors. 2016;9:240.
Mihalca AD, Ionică AM, D’Amico G, Daskalaki AA, Deak G, Matei IA, et al. Thelazia callipaeda in wild carnivores from Romania: new host and geographical records. Parasit Vectors. 2016;9:350.
Duscher GG, Kübber-Heiss A, Richter B, Suchentrunk F. A golden jackal (Canis aureus) from Austria bearing Hepatozoon canis - import due to immigration into a non-endemic area? Ticks Tick Borne Dis. 2013;4:133–7.
Farkas R, Solymosi N, Takács N, Hornyák Á, Hornok S, Nachum-Biala Y, Baneth G. First molecular evidence of Hepatozoon canis infection in red foxes and golden jackals from Hungary. Parasit Vectors. 2014;7:303.
Hervás J, Méndez A, Carrasco L, Gómez-Villamandos JC. Pathological study of visceral leishmaniasis in a jackal (Canis aureus). Vet Rec. 1996;139(12):293–5.
Cirović D, Chochlakis D, Tomanović S, Sukara R, Penezić A, Tselentis Y, Psaroulaki A. Presence of Leishmania and Brucella species in the golden jackal Canis aureus in Serbia. Biomed Res Int. 2014; doi:10.1155/2014/728516.
Inokuma H, Okuda M, Ohno K, Shimoda K, Onishi T. Analysis of the 18S rRNA gene sequence of a Hepatozoon detected in two Japanese dogs. Vet Parasitol. 2002;106:265–71.
Criado-Fornelio A, Rey-Valeiron C, Buling A, Barba-Carretero JC, Jefferies R, Irwin P. New advances in molecular epizootiology of canine hematic protozoa from Venezuela, Thailand and Spain. Vet Parasitol. 2007;144:261–9.
Zintl A, Finnerty EJ, Murphy TM, De Waal T, Gray JS. Babesias of red deer (Cervus elaphus) in Ireland. Vet Res. 2011;42:7.
Bartley PM, Hamilton C, Wilson C, Innes EA, Katzer F. Detection of Babesia annae DNA in lung exudate samples from red foxes (Vulpes vulpes) in Great Britain. Parasit Vectors. 2016; doi:10.1186/s13071-016-1364-1.
Tuvshintulga B, Sivakumar T, Battsetseg B, Narantsatsaral SO, Enkhtaivan B, Battur B, et al. The PCR detection and phylogenetic characterization of Babesia microti in questing ticks in Mongolia. Parasitol Int. 2016;64:527–32.
Gou H, Guan G, Liu A, Ma M, Xu Z, Liu Z, et al. A DNA barcode for Piroplasmea. Acta Trop. 2012;124:92–7.
Kassahun A, Sadlova J, Benda P, Kostalova T, Warburg A, Hailu A, Baneth G, Volf P, Votypka J. Natural infection of bats with Leishmania in Ethiopia. Acta Trop. 2015a;150:166–170.
Kassahun A, Sadlova J, Dvorak V, Kostalova T, Rohousova I, Frynta D, et al. Detection of Leishmania donovani and L. tropica in ethiopian wild rodents. Acta Trop. 2015b;145:39–44.
Akhoundi M, Downing T, Votypka J, Kuhls K, Lukeš J, Cannet A, et al. Leishmania infections: Molecular targets and diagnosis. Molecular Aspects of Medicine. 2017 (in press).
Talmi-Frank D, Kedem-Vaanunu N, King R, Bar-Gal GK, Edery N, Jaffe CL, Baneth G. Leishmania tropica infection in golden jackals and red foxes, Israel. Emerg Infect Dis. 2010;16:1973–5.
el Tai N, Osman OF, el Fari M, Presber W, Schönian G. Genetic heterogeneity of ribosomal internal transcribed spacer in clinical samples of Leishmania donovani spotted on filter paper as revealed by single-strand conformation polymorphisms and sequencing. Trans R Soc Trop Med Hyg. 2000;94:575–9.
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28(12):1647–9.
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23(21):2947–8.37.
Hall T. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser. 1999;41:95–8.
Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696–704.
Adaszek L, Winiarczyk S. Molecular characterization of Babesia canis canis isolates from naturally infected dogs in Poland. Vet Parasitol. 2008;152:235–41. doi:10.1016/j.vetpar.2007.12.024.
Beck R, Vojta L, Mrljak V, Marinculić A, Beck A, Živičnjak T, Cacciò SM. Diversity of Babesia and Theileria species in symptomatic and asymptomatic dogs in Croatia. Int J Parasitol. 2009;39:843–8.
Schnittger L, Rodriguez AE, Florin-Christensen M, Morrison DA. Babesia: a world emerging. Infect Genet Evol. 2012;12:1788–809.
Rutkowski R, Krofel M, Giannatos G, Ćirović D, Männil P, Volokh AM, et al. A European concern? Genetic structure and expansion of golden jackals (Canis aureus) in Europe and the Caucasus. PLoS One. 2015;10:e0141236. doi:10.1371/journal.pone.0141236.
Baneth G. Perspectives on canine and feline hepatozoonosis. Vet Parasitol. 2011;181:3–11. doi:10.1016/j.vetpar.2011.04.015.
Patton WS. Preliminary report on a new piroplasm found in the blood of hounds of the Madras-Hunt and subsequently discovered in the blood of a jackal. Bull Soc Pathol Exot. 1910;3:274–80.
Mbaya AW, Aliyu MM, Nwosu CO, Ibrahim UI. Captive wild animals as potential reservoirs of haemo and ectoparasitic infections of man and domestic animals in the arid-region of Northeastern Nigeria. Vet Arh. 2008;78:429–40.
Mihalca AD, Dumitrache MO, Magdaş C, Gherman CM, Domşa C, Mircean V, et al. Synopsis of the hard ticks (Acari: Ixodidae) of Romania with update on host associations and geographical distribution. Exp Appl Acarol. 2012;58:183–206.69.
Dulceanu N, Clipa V, Mardari A. Clinical-therapeutical observations in canine babesiosis. Iaşi: Cercetări Agr în Moldova; 1984. p. 107–8.
Ionita M, Mitrea IL, Pfister K, Hamel D, Buzatu CM, Silaghi C. Canine babesiosis in Romania due to Babesia canis and Babesia vogeli: A molecular approach. Parasitol Res. 2012;110:1659–64.
Imre M, Farkas R, Ilie MS, Imre K, Dărăbuş G. Survey of babesiosis in symptomatic dogs from Romania: Occurrence of Babesia gibsoni associated with breed. Ticks Tick Borne Dis. 2013;4(6):500–2.
Zahler M, Rinder H, Schein E, Gothe R. Detection of a new pathogenic Babesia microti-like species in dogs. Vet Parasitol. 2000;89:241–8.
Lack JB, Reichard MV, Van Den Bussche RA. Phylogeny and evolution of the Piroplasmida as inferred from 18S rRNA sequences. Int J Parasitol. 2012;42:353–63.
Baneth G, Florin-Christensen M, Cardoso L, Schnittger L. Reclassification of Theileria annae as Babesia vulpes sp. nov. Parasit Vectors. 2015;8:1–7.
Harris DJ. Naming no names: Comments on the taxonomy of small piroplasmids in canids. Parasit Vectors. 2016;9:289.
D’Amico G, Dumitrache MO, Matei IA, Ionică AM, Gherman CM, Sándor AD, et al. Ixodid ticks parasitizing wild carnivores in Romania. Exp. App. Acarol. 2017 (in press).
Mircean V, Dumitrache MO, Mircean M, Bolfa P, Györke A, Mihalca AD. Autochthonous canine leishmaniasis in Romania: neglected or (re)emerging? Parasit Vectors. 2014;7:135.
Dumitrache MO, Nachum-biala Y, Gilad M, Mircean V, Cazan CD, Mihalca AD, Baneth G. The quest for canine leishmaniasis in Romania: the presence of an autochthonous focus with subclinical infections in an area where disease occurred. Parasit Vectors. 2016; doi:10.1186/s13071-016-1583-5.
Acknowledgements
The study was conducted under EurNegVec COST Action TD1303 and financially supported by the Ministry of Education, Youth and Sports of the Czech Republic, projects CEITEC 2020 (LQ1601), COST CZ LD14048 and LD14076 and partially funded by IGA UVPS (115/2013/FVL). Samples from Romania were collected as part of UEFISCDI grant TE 299/2015. KH was supported by project LO1218 with financial support from the Ministry of Education, Youth and Sports of the Czech Republic under the NPU I programme. We acknowledge a grant for the development of research organization (RVO: RO0516). We are indebted to all hunters and collaborators involved in sample collection and examination who are not listed as authors, to Cristian Domșa for preparing the maps, and to Markéta Rybářová for her participation in PCR optimization.
Funding
The study was conducted within the frame EurNegVec COST Action TD1303, financially supported by projects from the Ministry of Education, Youth and Sports of the Czech Republic CEITEC 2020 (LQ1601), COST CZ LD14048 and LD14076, partially funded by project LO1218 and by IGA UVPS (115/2013/FVL) and by grant for the development of research organization (RVO: RO0516). Samples from Romania were collected as part of UEFISCDI grant TE 299/2015.
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article. The sequences obtained in this study are avalable in the GenBank database under accesion numbers KX712122–KX712139.
Authors’ contributions
BM, KH, DM and PH wrote the manuscript, GD, IAM, AMI, AAD and CMG performed necropsies. PF, FS, ADM and DM managed the sample collection. PF and BM performed the necropsies. GGD provided the Austrian sample. BM and JV performed the molecular analysis. KH performed the phylogenetic analyses. All authors have read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Mitková, B., Hrazdilová, K., D’Amico, G. et al. Eurasian golden jackal as host of canine vector-borne protists. Parasites Vectors 10, 183 (2017). https://doi.org/10.1186/s13071-017-2110-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-017-2110-z