
Abstract
Background
We investigated strategies to improve foreign protein accumulation in the chloroplasts of the model algae Chlamydomonas reinhardtii and tested the outcome in both standard culture conditions as well as one pertinent to algal biofuel production. The downstream box (DB) of the TetC or NPTII genes, the first 15 codons following the start codon, was N-terminally fused to the coding region of cel6A, an endoglucanase from Thermobifida fusca. We also employed a chimeric regulatory element, consisting of the 16S rRNA promoter and the atpA 5′UTR, previously reported to enhance protein expression, to regulate the expression of the TetC-cel6A gene. We further investigated the accumulation of TetC-Cel6A under N-deplete growth conditions.
Results
Both of the DB fusions improved intracellular accumulation of Cel6A in transplastomic C. reinhardtii strains though the TetC DB was much more effective than the NPTII DB. Furthermore, using the chimeric regulatory element, the TetC-Cel6A protein accumulation displayed a significant increase to 0.3% total soluble protein (TSP), whereas NPTII-Cel6A remained too low to quantify. Comparable levels of TetC- and NPTII-cel6A transcripts were observed, which suggests that factors other than transcript abundance mediate the greater TetC-Cel6A accumulation. The TetC-Cel6A accumulation was stable regardless of the growth stage, and the transplastomic strain growth rate was not altered. When transplastomic cells were suspended in N-deplete medium, cellular levels of TetC-Cel6A increased over time along with TSP, and were greater than those in cells suspended in N-replete medium.
Conclusions
The DB fusion holds great value as a tool to enhance foreign protein accumulation in C. reinhardtii chloroplasts and its influence is related to translation or other post-transcriptional processes. Our results also suggest that transplastomic protein production can be compatible with algal biofuel production strategies. Cells displayed a consistent accumulation of recombinant protein throughout the growth phase and nitrogen starvation, a strategy used to induce lipid production in algae, led to higher cellular heterologous protein content. The latter result is contrary to what might have been expected a priori and is an important result for the development of future algal biofuel systems, which will likely require co-products for economic sustainability.
Similar content being viewed by others

Background
Photosynthetic microalgae are increasingly being used as a platform for chemical production and may in the future become a viable feedstock for renewable fuel. Well-studied genetically tractable species, such as Chlamydomonas reinhardtii, may be used directly for production, e.g. [1] or are used as model systems to develop tools and strategies for ultimate application to other algae species with desired traits. C. reinhardtii has been investigated as a host for high-value heterologous protein expression due to its fast reproduction rate, its ability to grow as a heterotroph, and its well-established genetic transformation techniques [2,3,4]; examples include proteins with value as biopharmaceuticals [5] and in agricultural applications [6].
Chloroplast transformation has several advantages for recombinant protein expression over nuclear transformation. These include precise transgene integration, feasibility to express multiple genes simultaneously, the absence of gene silencing, and the presence of 80–100 identical plastomes per cell resulting in a high transgene copy number [7,8,9]. Transplastomic C. reinhardtii strains do not, however, always accumulate heterologous proteins to detectable levels and strategies to generate strains with consistently high heterologous protein accumulation remain elusive. Many factors can influence plastid transgene expression including host choice [10] and codon usage in the translated region [11]. Modest expression levels have been reported in strains using strong constitutive endogenous promoters (e.g., rbcL, atpA, psbA, psbD, and the rrn operon) paired with their respective UTRs [3]. The choice of the 3′UTR sequence has been shown to have a minor impact on mRNA stability and protein accumulation [12], whereas the 5′UTR was more influential [12, 13].
The 5′UTR sequence is believed to interact directly with the coding region, forming secondary structures which facilitate the function of translational protein factors. High heterologous protein accumulation (≥ 5% TSP) was achieved when utilizing the endogenous psbA promoter and corresponding 5′UTR, but only in a psbA knock-out background which is not capable of photosynthesis [14,15,16]. In photosynthetically competent cells, levels of two proteins, luciferase and 14FN3, were increased when the 16S rRNA gene promoter and the atpA 5′UTR were fused to generate a chimeric regulatory element compared to accumulation in strains in which the endogenous atpA, psbD, or rbcL promoters and 5′ UTRs were used [17]. Fusion of the luciferase gene to the C-terminal of rbcL, a highly abundant Rubisco subunit, also increased foreign protein accumulation by three to fivefold relative to use of the rbcL promoter and UTRs without the coding region [18].
Slight modification of the N-terminus of a transgene can also lead to a dramatic change in plastid expression levels in higher plant chloroplasts [19], but this approach has not been investigated in algae. In tobacco, recombinant protein accumulation improved significantly when the first 10–15 codons, the downstream box (DB), of a highly expressed transgene was fused to the N-terminus of another transgene [20]. The downstream box effect is gene dependent such that the same DB sequence can lead to different yields of specific recombinant proteins [20, 21]. Therefore, empirical optimization is needed to determine the appropriate downstream box sequence that will result in high production of a given recombinant protein.
In this study, we investigated two features of the expression cassette and their impact on the production of Cel6A protein in C. reinhardtii chloroplasts. The objective of this work was to determine whether the downstream box (DB) would also affect protein accumulation in an algal chloroplast and to evaluate the pairing of potential DB improvements with those of the previously reported chimeric regulatory element on the accumulation of Cel6A. Cel6A is an endoglucanase belonging to a group of commercially important enzymes that hydrolyze cellulose [22], and was chosen for the study because it was successfully and variably expressed in tobacco chloroplasts with DB modifications [20]. We also evaluated the impact of growth conditions used to trigger lipid biosynthesis on foreign protein production, given that one impetus for studying protein expression in algae is to increase the value of protein co-products in algal biofuel production systems.
Methods
Chlamydomonas strain and growth conditions
Chlamydomonas reinhardtii wild-type CC-125 strain and the engineered transformants were maintained in minimal media (20 mM Tris, 0.68 mM K2HPO4, 7.26 mM KH2PO4, and 7.5 mM NH4Cl) [23], with the addition of 150 µg/mL spectinomycin for the transplastomic strains. Experiments were performed in 50 mL batch cultures of either minimal medium or TAP medium (20 mM Tris, 17 mM Acetate, 0.68 mM K2HPO4, 7.26 mM KH2PO4, and 7.5 mM NH4Cl) [23], with trace metal additions prepared as described previously [24]. All experimental cultures were incubated at 25 °C with constant shaking (110 rpm), open to the atmosphere and exposed to continuous fluorescent white light (80 µmol/m2 s) unless otherwise stated. Cellular growth was monitored by measuring the auto-fluorescence of chlorophyll using a 10-AU Fluorometer (Turner Designs, San Jose, CA), equipped with a Daylight White Lamp F4T5D (EX: 340–500 nm, EM: > 665 nm). Cell counts were made using a Neubauer hemacytometer (Spencer Bright Line®) at 40× magnification with an optical microscope (model 1864, Southern Precision Instruments, San Antonio, TX, USA).
Cloning and plasmid construction
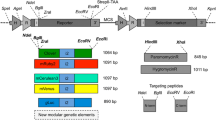
Expression cassettes assembled as illustrated in Fig. 1a were cloned at the EcoRI and SalI restriction sites of the pUC19 vector. The regulatory elements were amplified from the C. reinhardtii chloroplast genome. The chimeric construct of the 16S rRNA gene promoter and the atpA 5′UTR was engineered as described previously [17]. The coding sequence of the Thermobifida fusca cel6A (lacking the signal peptide), TetC-cel6A, and NPTII-cel6A genes were amplified from the pGG86, pTetCCel6A, and the pNPTIICel6A plasmids, respectively [20]. The aadA gene, a selectable marker conferring resistance to spectinomycin and streptomycin [25], was amplified from the pPTDNA-aadA plasmid [20]. DNA sequences from a non-coding region between the psbA and 5S rRNA genes were used to flank the expression cassettes (Fig. 1a), which allow integration at the unique BamHI site [12] via homologous recombination [26]. DNA fragments were amplified via PCR using a high fidelity Phusion polymerase (Finnzymes, Thermo Fisher Scientific, Waltham, MA), and assembled by restriction digestion and ligation reactions (NEB, Ipswich, MA). Primers used for cloning are listed in Additional file 1: Table S1.

Engineered constructs and site of integration of recombinant genes into the C. reinhardtii chloroplast genome. a Schematic diagram of the transformation vectors used to integrate the recombinant genes into the inverted repeat of the chloroplast. The cel6A coding region is regulated by the endogenous atpA promoter, 5′UTR, and terminator in pCHR23, pCHR24, and pCHR25 plasmids; and by the 16S promoter and atpA 5′UTR chimeric construct and the atpA terminator in pCHR72, pCHR73, and pCHR74 plasmids. The cel6A gene is cloned with no added downstream box (DB) in pCHR23 and pCHR72, with the NPTII DB in pCHR24 and pCHR73, and the TetC DB in pCHR25 and pCHR74 plasmids. Primers used to screen for accurate gene insertion and homoplasmicity are indicated by arrows. b DNA agarose gels of PCR products generated using the C. reinhardtii cell lysate and gene-specific primers. The Fwd1 and Rev1 primers were designed around the insertion site. PCR reaction conditions using the Fwd1 and Rev1 primers were chosen to generate amplicons < 500 bp. Of all the tested strains, only the parent wild-type template gave a product, which confirms the homoplasmic state of the engineered strains (top gel). Fwd2 and Rev2 primers were designed within the cel6A sequence and used to confirm transgene integration (second gel from the top). NPTII-Fwd and TetC-Fwd designed within the respective DB sequence and were used in pairs with Rev2 primer to confirm the transgene identity (bottom two gels)
Chloroplast transformation
C. reinhardtii chloroplasts were transformed via particle bombardment with a biolistic device (BioRad, Hercules, CA) [27]. Briefly, C. reinhardtii wild-type cells were grown to a density of 1 × 106 cell/mL. Cells were harvested at 2700×g for 5 min and resuspended in minimal media to a concentration of 5 × 107 cell/mL. Concentrated cultures were spread in the center (3 cm in diameter) of minimal media plates supplemented with 150 µg/mL spectinomycin, and bombarded with 1-µm gold particles (BioRad, Hercules, CA) coated with the appropriate plasmid DNA. Following bombardment, plates were incubated under dim light until colonies appeared. Several rounds of selection on minimal media with 150 µg/mL spectinomycin were conducted to obtain a homoplasmic cell line.
PCR screening for transformants
To confirm the accurate transgene placement and to assess the homoplasmy of the engineered transplastomic strains, PCR reactions were performed using sets of primers listed in Additional file 1: Table S1. Wild-type and transplastomic cells were treated with 10 mM Tris–EDTA lysis buffer and boiled for 10 min in preparation for PCR tests [28]. Taq polymerase (NEB, Ipswich, MA) was used in all PCR reactions for 35 cycles following the manufacturer’s instructions. Gene-specific primers were used to confirm the proper transgene insertion. To confirm the homoplasmy of the transformants, Fwd1 and Rev1 primers (Additional file 1: Table S1) were designed to amplify a sequence at the insertion site that is disrupted if the homologous recombination is successful.
SDS-PAGE and immunoblotting
The wild-type and transplastomic C. reinhardtii cultures were grown in TAP or minimal media and harvested by centrifugation (5000×g for 15 min) at the growth phase indicated for each experiment. Pellets were resuspended in 2 mL of 10 mM sodium phosphate buffer pH 6.8 with 1 mM PMSF [29]. Resuspended pellets were frozen-thawed for three cycles for a minimum of 1.5 h for each freezing cycle. Cells were then subjected to three rounds of probe sonication with 20/40 s on/off duty cycle on ice. The cell debris was separated from the supernatant by micro-centrifugation at 13,000 rpm for 10 min, and the soluble fraction was stored at − 20 °C until used. The total soluble protein (TSP) was quantified by Bradford assays (BioRad, Hercules, CA).
TSP samples were denatured by the addition of 2× Laemmli SDS-loading buffer (BioRad, Hercules, CA) and boiling for 5 min, then electrophoresed in 12% polyacrylamide pre-cast gels (BioRad, Hercules, CA), and transferred to PVDF membranes (BioRad, Hercules, CA). Membranes were blocked by incubation with 5% (w/v) milk in TBST (0.1 M Tris, pH 7.4, 0.15 M NaCl, 0.1% Tween 20) and then probed with a polyclonal anti-Cel6A primary antibody (kindly donated by David Wilson, Cornell University, Ithaca, NY) for 1 h. The anti-Cel6A primary antibody was diluted 1:10,000 in Antibody Signal Enhancer solution (Amresco, Solon, OH). The secondary antibody, ECL peroxidase-labeled anti-rabbit antibody (GE Healthcare Life Sciences, Marlborough, MA), was diluted 1:20,000 in Antibody Signal Enhancer solution. Membranes were incubated with Clarity Western ECL Substrate solution (BioRad, Hercules, CA) for 1–2 min and visualized on CL-Xposure film (Thermo Fisher Scientific). Exposure time varied depending on the signal intensity. Purified Cel6A protein was provided by David Wilson (Cornell University, Ithaca, NY). Immunoreactive bands were quantified using ImageJ 1.47 software and the Cel6A concentration in the algal extracts was determined by comparison to the relative blot density of known amounts of a purified Cel6A control in the same gel.
RNA blotting
Total RNA was extracted from C. reinhardtii wild-type and transplastomic strains using TriZol reagent according to manufacturer’s instructions (Invitrogen, Carlsbad, CA). RNA concentration was quantified via spectrophotometric absorption at 260 nm, and RNA samples (20 µg per strain) were electrophoresed on a 1.2% agarose formaldehyde gel in MOPS buffer (20 mM MOPS, 5 mM Na Acetate, and 1 mM EDTA, pH 8.0). After electrophoresis, RNA was transferred to a Hybond N+ membrane (Amersham Biosciences, Piscataway, NJ) and UV crosslinked. RNA was detected by hybridization with 32P-labeled cel6A gene-specific probe prepared by a Random Labeling kit (Ambion, Thermo Fisher Scientific, Waltham, MA). The membrane was exposed to a PhosphorImager screen for detection (Molecular Dynamics, Amersham Biosciences, Piscataway, NJ).
Nitrogen starvation time course experiment
The TetC-cel6A-expressing strain was cultured in minimal media supplemented with 150 µg/mL spectinomycin until late logarithmic phase (3 × 106 cell/mL). Cultures were then centrifuged at 5000×g for 15 min, and pellets were resuspended in minimal media with or without added nitrogen (7.5 mM NH4Cl). Biological duplicates of each growth medium for three different incubation times (5, 24 or 48 h) were prepared. Cell counts were measured immediately after resuspension (t = 0) and then at the end of each incubation. Cell counts were normalized to that measured at t = 0 to determine the fold change in cell density. Cultures were collected and treated as described above for foreign protein quantification.
Results
Recombinant constructs and chloroplast transformation
Expression cassettes were designed to generate transplastomic C. reinhardtii strains that would express the native T. fusca cel6A gene with no codon optimization because the chloroplast and bacterial genomes share a similar codon bias and the native sequence was successfully employed for plastid expression in tobacco [20]. In some cassettes, the downstream box (DB) of either the TetC (the fragment C from tetanus toxin) or NPTII (the neomycin phosphotransferase reporter enzyme) genes was inserted immediately after the start codon of cel6A (Fig. 1a). The expression of the cel6A variants was regulated by one of two endogenous promoters, the atpA promoter or the 16S rRNA promoter (16SP), cloned upstream of the atpA 5′UTR sequence (Fig. 1a). Plasmids containing the expression cassettes were transformed into the C. reinhardtii chloroplasts by biolistic particle bombardment and the transgenes were integrated at the BamHI site within the intergenic region between the psbA and 5S rRNA genes within the C. reinhardtii chloroplast genome (Fig. 1a). Transformants were selected on minimal media plates supplemented with spectinomycin, and multiple rounds of streaking were conducted to isolate homoplasmic cell lines. PCR screening was used to verify that the transgene was inserted in the correct location and that the transplastomic strains were homoplasmic (Fig. 1b).
Quantification of Cel6A accumulation in C. reinhardtii transplastomic strains
Six transplastomic strains engineered to express the cel6A gene under the control of different expression element combinations were tested for Cel6A accumulation. When the coding region was prefaced by the atpA promoter and 5′UTR, immunoreactive bands of the recombinant protein were visible on blots only after extended exposure time. Notably, TetC-Cel6A and NPTII-Cel6A accumulation was higher than Cel6A but levels were very low and only one transformant of the two tested exhibited detectable protein (Fig. 2, top blot). Examples of different accumulation levels for the same recombinant protein from independent homoplasmic lines have been documented [16].

Effect of various regulatory elements on Cel6A accumulation in C. reinhardtii transplastomic strains. Immunoblots showing the Cel6A protein accumulation in C. reinhardtii strains expressing the cel6A, NPTII-cel6A, and TetC-cel6A genes under the regulation of the atpA promoter and 5′UTR (top blot) or the 16S promoter and atpA 5′UTR (bottom blot) constructs. Extended exposure time to X-ray film, up to several hours, was used for the top blot, while short exposure time, 1–2 min, was used to observe the immunoreactive bands in the bottom blot. All lanes contained 50 µg total soluble protein (TSP) extracted from the wild-type and transplastomic C. reinhardtii strains. Lanes A and B contain protein extracted from two independent transplastomic single colony isolates. Control lane contained 0.1 µg purified Cel6A protein. Quantification of the Cel6A concentration in the algal extracts was done by comparing the relative blot density of bands to those of the purified Cel6A control
Previously, the strong endogenous promoter 16SP was reported to boost the plastid expression of the luciferase and 14FN3 genes in C. reinhardtii when coupled to the atpA 5′UTR, in comparison to the atpA promoter and 5′UTR combination [17]. In our experiments, the 16SP and atpA 5′UTR chimeric regulatory construct did not improve the native Cel6A or the NPTII-Cel6A production to levels detectable on immunoblots (Fig. 2, bottom blot). However, the chimeric regulatory construct in combination with the TetC DB significantly increased the amount of TetC-Cel6A protein (to ~ 0.3% of total soluble protein, TSP) in both homoplasmic lines (Fig. 2). This result is consistent with earlier work which showed that TetC-Cel6A accumulation was higher than that of NPTII-Cel6A in tobacco [20]. Crude cell extracts of TetC-Cel6A-containing algae were tested for enzymatic activity using Carboxymethyl Cellulose (CMC) as a substrate and showed greater activity than wild-type cell extracts demonstrating that transgenic protein was active (Additional file 2: Figure S1).
Characterization of cel6A mRNA in C. reinhardtii transplastomic strains
To evaluate the cel6A mRNA expression level when regulated by the 16SP and atpA 5′UTR chimeric construct and to investigate a possible correlation between the protein yield observed in Fig. 2 and the transcription of the corresponding transgene, RNA blots were performed (Fig. 3). The stained gel provides visual confirmation that equal amounts of RNA were loaded in each lane (Fig. 3, top gel). Hybridization with a cel6A-specific probe revealed similar accumulation of cel6A mRNA in all of the transplastomic strains at the predicted size (1.3 kb) (Fig. 3, bottom gel). Consistent with this result, RT-qPCR analysis showed comparable expression levels of the cel6A, NPTII-cel6A, and TetC-cel6A genes when controlled by the 16SP and atpA 5′UTR in transplastomic cell-wall mutant strains (Additional file 3: Figure S2). Taken together, the differences in Cel6A accumulation observed on protein blots (Fig. 2, bottom gel and Additional file 3: Figure S2b) are not due to differences in transcript accumulation between strains.

RNA blotting of the wild-type and transplastomic C. reinhardtii strains containing the cel6A genes regulated by the 16S promoter and atpA 5′UTR chimeric construct. The RNA bands from the ethidium-bromide-stained agarose gel are shown in the top gel. Total RNA was hybridized with a radiolabeled cel6A probe to evaluate the accumulation of cel6A transcripts in cel6A, NPTII-cel6A, and TetC-cel6A transplastomic strains as well as its absence in the wild-type RNA sample (bottom gel)
Transplastomic strain growth and recombinant protein accumulation
To further characterize the strains expressing the DB-cel6A variants, growth of the transplastomic strains expressing the cel6A, NPTII-cel6A, or TetC-cel6A under the control of the 16SP and atpA 5′UTR chimeric construct, in addition to the wild-type strain, was monitored. Algal growth in TAP medium was quantified by measuring the auto-fluorescence of chlorophyll. All transplastomic strains displayed an average growth rate of 1.75 ± 0.08/day, which was indistinguishable from wild type, and reached a similar maximum cell density (Fig. 4a). Assessment of recombinant TetC-Cel6A protein accumulation throughout the entire cellular growth phase including after 2 days of senescence reveals fairly constant levels, with a modest peak during late exponential phase (t = 84 h, Fig. 4b).

TetC-Cel6A accumulation during growth of transplastomic C. reinhardtii strains. a Growth curve of C. reinhardtii wild-type and transformed strains harboring the cel6A, NPTII-cel6A, and TetC-cel6A genes regulated by the 16SP and atpA 5′UTR, grown in TAP media. A representative of triplicate growth curves for each cell line is shown. b Evaluation of the TetC-Cel6A protein accumulation throughout the cellular growth phases. The wild-type cell line was used as a negative control. All lanes contained 50 µg total soluble protein (TSP) extracted from either the wild-type or the TetC-cel6A strain
Effect of nitrogen starvation on transplastomic Cel6A accumulation
Algae are a potential feedstock for biofuel production and nitrogen deprivation promotes starch and triacylglycerol (TAG) biosynthesis [30]. N-starvation also triggers other changes in cell physiology, including the down regulation of most genes involved in photosynthesis and a transition to gametogenesis in some species, including C. reinhardtii [31]. To determine the effect of nitrogen starvation on the accumulation of recombinant TetC-Cel6A, cells expressing the TetC-cel6A gene regulated by the 16SP and atpA 5′UTR chimeric construct were cultivated in minimal media until late logarithmic growth, then pelleted, and resuspended in N-free or N-replete minimal media for 5, 24, and 48 h. The TetC-Cel6A accumulation was evaluated at each time point using immunoblots (Fig. 5a).

Effect of nitrogen starvation on the accumulation of TetC-Cel6A. a Immunoblots showing the TetC-Cel6A protein accumulation after 5, 24, and 48 h of resuspension in N-replete (top blot) or N-deplete (bottom blot) minimal media. Exposure time was 2 min for both blots. All lanes contained 10 µg total soluble protein. Control lane contained 0.01 µg purified Cel6A protein. The TetC-Cel6A protein accumulation was quantified in two biological replicates by comparing the relative blot density of bands to those of a purified Cel6A control. b–d Several parameters monitored in the TetC-Cel6A expressing cells grown under N-replete or N-deplete media for 5, 24, and 48 h. Data are the average of two biological replicates per treatment and time point. Error bars are the standard error of the mean. Statistical analysis were performed by Student’s t test, *p < 0.05 and ns = not significant
Following resuspension, the transplastomic strain maintained cell division at a similar rate in both media for the first 5 h (fold change in cell counts from t = 0 is shown in Fig. 5b). After 48 h, cell numbers were significantly lower in the N-deplete cultures, presumably because of gametogenesis triggered by N-limitation; similar observations were reported previously [1]. In contrast, the plateau in cell density in the N-replete medium was likely caused by carbon limitation or shading.
Distinct changes in cellular protein content, both TSP and TetC-Cel6A, were observed in the two media. After just 5 h, N-deplete cells had twice the TSP compared to N-replete cells (Fig. 5c), perhaps due to changes in cellular metabolism causing a shift of N resources from storage into protein synthesis. Subsequently, the protein content doubled in N-deplete cells and then leveled off after 24 and 48 h, respectively, whereas N-replete cells did not exhibit significant changes in protein accumulation through the same time period (Fig. 5c). The TetC-Cel6A content of cells (ng/cell) mirrored extractable protein to a large extent, increasing gradually in N-replete cells and increasing more rapidly in N-deplete cells, with the highest cellular levels recorded after 24 h of N-starvation (Fig. 5d). We do not believe that changes in cellular protein content are due to differences in extraction efficiency; at least one other study noted no change in this parameter even as cell wall thickness increased with N-depletion [32].
Discussion
The development of economical and renewable sources of fuel is critical to a sustainable future. One key to the economic success of algae-derived biofuel is the development of protein co-products that may also be of value for biofuel production or sold as animal feed [33, 34]. In this work, we developed transplastomic C. reinhardtii strains that can produce Cel6A, an endoglucanase from T. fusca, which has been previously shown to accumulate variably in tobacco chloroplasts. The high conservation of chloroplasts across microalgae species makes it likely that regulatory strategies successful in C. reinhardtii will be transferrable to related algae with better lipid accumulation traits, such as Chlorella C596 [35].
It was previously shown that the expression of two heterologous proteins could be enhanced when the strong 16S rRNA gene promoter was used with the atpA 5′UTR in place of the endogenous atpA promoter [17]. In this study, the 16SP and atpA 5′UTR chimeric regulatory construct improved protein accumulation in only one of the three engineered constructs, the one in which the TetC downstream box (DB) was N-terminally fused to the cel6A gene immediately after the start codon (Fig. 2). In the strain containing this construct, TetC-Cel6A accumulated to 0.3% TSP. In tobacco, the DB of both the TetC and NPTII genes were utilized to enhance plastid production of cel6A, resulting in 10.7 and 0.9% TSP for the TetC-Cel6A and NPTII-Cel6A proteins, respectively, compared to 0.1% when using yet another unique DB from GFP [20]. In the tobacco study, the DB sequence was postulated to increase the stability of the mRNA transcript. We have now shown that the TetC DB is more effective than the NPTII DB in conjunction with the cel6A ORF in C. reinhardtii chloroplasts as well (Fig. 2).
Protein accumulation from chloroplast transgenes is likely influenced by several factors including transcription and translation regulators, RNA turnover, and/or protein turnover. Analyses of RNA blots included herein revealed comparable monocistronic cel6A transcript accumulation regardless of the presence or identity of the DB (Fig. 3), which leads us to conclude factors other than transcript abundance are mediating recombinant Cel6A accumulation. Coragliotti et al. also showed that changes in mRNA levels of plastid-expressed green fluorescent protein (GFP) and bacterial luciferase did not result in concomitant changes in protein accumulation [36]. They concluded that the main factor limiting recombinant protein accumulation in their study was poor mRNA translation. The authors proposed that the coding region of the transgene might interact with the 5′UTR forming a stable and defined secondary structure which in turn facilitates binding to translation activators [12, 36]. As noted above, the TetC DB improved Cel6A accumulation in both tobacco [20] and in C. reinhardtii (Fig. 2); however, the 5′UTRs utilized in these constructs were different. The T7g10 and atpA 5′UTR sequences were used in tobacco and C. reinhardtii, respectively. Therefore, it is plausible to speculate that the TetC DB along with the cel6A coding sequence is actually the critical region for stabilization or successful initiation of translation. This assertion is further supported by the earlier finding in tobacco that while using the same T7g10 5′UTR, the NPTII DB was more effective than the TetC DB when coupled to the coding region of bglC [21]. Only when the mechanism whereby the DB enhances the production of recombinant proteins is known will we be able to design a priori an effective DB for a given gene.
With respect to the longer term goal of using algae to produce proteins commercially as a co-product in biofuel production, we have shown that this expression cassette is largely compatible with that goal. In particular, the 16SP and atpA 5′UTR regulation resulted in stable TetC-Cel6A accumulation at different growth stages (Fig. 4b) making it possible to grow such algae in continuous or batch cultures with less attention to precise timing of protein harvest. Braun-Galleani et al. reported that cultivation conditions should be optimized for different recombinant proteins, as conditions leading to high accumulation of a given protein would not necessarily be optimal for another [10]. We did observe that TetC-Cel6A accumulation was roughly 60% lower when using photoautotrophic cultivation (0.12% TSP in minimal media) compared to mixotrophic growth (0.3% TSP in TAP media) but we did not attempt any optimization with respect to medium composition.
In production systems, nitrogen starvation is commonly used to induce lipid production. C. reinhardtii begins to accumulate starch after 1 day of N-starvation and then TAGs after 2–3 more days [37]. During this same interval, vegetative C. reinhardtii cells also differentiate into haploid gametes (mt+ and mt−), to prepare for sexual reproduction [38]. Our experiments reveal that accumulation of foreign protein is not compromised when cells are initially shifted to N-limitation; in fact during the first 24 h of N-limitation, recombinant TetC-Cel6A accumulation is double that measured in N-replete cells (Fig. 5d). Numerous physiological transitions occur during N-depletion and therefore active protein production machinery is likely required in the chloroplasts. We do not know whether the rapid increase in cellular Cel6A levels is due to a particular regulatory element in our construct or is simply due to a general upregulation of all protein synthesis.
Conclusions
Our work demonstrates that a foreign downstream box (DB) fusion can significantly enhance the accumulation of a heterologous endoglucanase in C. reinhardtii chloroplasts, but a better understanding of the post-transcriptional mechanism involved is still needed to determine how best to increase accumulation of other proteins. We show that when the chimeric regulatory construct of 16SP and 5′UTR was used, the TetC-Cel6A production was unaltered by the cellular growth, though was influenced by medium composition and N-limitation. Perhaps contrary to expectations, our experiment reveals a substantial increase in cellular foreign protein accumulation associated with a short period of N-deprivation. Further work is needed to determine if this benefit can be realized in conjunction with systems designed to generate lipid-rich biomass. In summary, the downstream box used in conjunction with the right regulatory elements holds great promise for production of recombinant protein production in green algal chloroplasts.
Change history
10 December 2019
In the original version of the article [1], a calculation error resulted in a 3-order of magnitude mistake for the y-axis of the data reported in Fig. 5c and d.
Abbreviations
- DB:
-
downstream box
- 16SP:
-
16S rRNA gene promoter
- 5′UTR:
-
5′ untranslated region
- 3′UTR:
-
3′ untranslated region
- NPTII:
-
the neomycin phosphotransferase reporter enzyme
- TetC:
-
the fragment C from tetanus toxin gene
- T7g10:
-
gene 10 of the bacteriophage T7
- Cel6A:
-
an endoglucanase
- aadA :
-
aminoglycoside adenine transferase
- atpA :
-
ATP synthase subunit A
- rbcL :
-
Rubisco large subunit
- TSP:
-
total soluble protein
- PCR:
-
polymerase chain reaction
References
Iwai M, Ikeda K, Shimojima M, Ohta H. Enhancement of extraplastidic oil synthesis in Chlamydomonas reinhardtii using a type-2 diacylglycerol acyltransferase with a phosphorus starvation-inducible promoter. Plant Biotechnol J. 2014;12(6):808–19.
Maul JE, Lilly JW, Cui LY, dePamphilis CW, Miller W, Harris EH, Stern DB. The Chlamydomonas reinhardtii plastid chromosome: islands of genes in a sea of repeats. Plant Cell. 2002;14(11):2659–79.
Specht E, Miyake-Stoner S, Mayfield S. Micro-algae come of age as a platform for recombinant protein production. Biotechnol Lett. 2010;32(10):1373–83.
Doron L, Segal N, Shapira M. Transgene expression in microalgae-from tools to applications. Front Plant Sci. 2016;7:505.
Gong YM, Hu HH, Gao Y, Xu XD, Gao H. Microalgae as platforms for production of recombinant proteins and valuable compounds: progress and prospects. J Ind Microbiol Biotech. 2011;38(12):1879–90.
Erpel F, Restovic F, Arce-Johnson P. Development of phytase-expressing Chlamydomonas reinhardtii for monogastric animal nutrition. BMC Biotechnol. 2016;16:29.
Bock R. Engineering plastid genomes: methods, tools, and applications in basic research and biotechnology. Annu Rev Plant Biol. 2015;66:211–41.
Olejniczak SA, Lojewska E, Kowalczyk T, Sakowicz T. Chloroplasts: state of research and practical applications of plastome sequencing. Planta. 2016;244(3):517–27.
Hanson MR, Gray BN, Ahner BA. Chloroplast transformation for engineering of photosynthesis. J Exp Bot. 2013;64(3):731–42.
Braun-Galleani S, Baganz F, Purton S. Improving recombinant protein production in the Chlamydomonas reinhardtii chloroplast using vivid verde fluorescent protein as a reporter. Biotechnol J. 2015;10(8):1289–97.
Mayfield SP, Schultz J. Development of a luciferase reporter gene, luxCt, for Chlamydomonas reinhardtii chloroplast. Plant J. 2004;37(3):449–58.
Barnes D, Franklin S, Schultz J, Henry R, Brown E, Coragliotti A, Mayfield SP. Contribution of 5′- and 3′-untranslated regions of plastid mRNAs to the expression of Chlamydomonas reinhardtii chloroplast genes. Mol Genet Genom. 2005;274(6):625–36.
Eberhard S, Drapier D, Wollman FA. Searching limiting steps in the expression of chloroplast-encoded proteins: relations between gene copy number, transcription, transcript abundance and translation rate in the chloroplast of Chlamydomonas reinhardtii. Plant J. 2002;31(2):149–60.
Manuell AL, Beligni MV, Elder JH, Siefker DT, Tran M, Weber A, McDonald TL, Mayfield SP. Robust expression of a bioactive mammalian protein in Chlamydomonas chloroplast. Plant Biotechnol J. 2007;5(3):402–12.
Rasala BA, Muto M, Lee PA, Jager M, Cardoso RM, Behnke CA, Kirk P, Hokanson CA, Crea R, Mendez M, Mayfield SP. Production of therapeutic proteins in algae, analysis of expression of seven human proteins in the chloroplast of Chlamydomonas reinhardtii. Plant Biotechnol J. 2010;8(6):719–33.
Surzycki R, Greenham K, Kitayama K, Dibal F, Wagner R, Rochaix JD, Ajam T, Surzycki S. Factors effecting expression of vaccines in microalgae. Biologicals. 2009;37(3):133–8.
Rasala BA, Muto M, Sullivan J, Mayfield SP. Improved heterologous protein expression in the chloroplast of Chlamydomonas reinhardtii through promoter and 5′ untranslated region optimization. Plant Biotechnol J. 2011;9(6):674–83.
Muto M, Henry RE, Mayfield SP. Accumulation and processing of a recombinant protein designed as a cleavable fusion to the endogenous Rubisco LSU protein in Chlamydomonas chloroplast. BMC Biotechnol. 2009;9(1):26.
Kuroda H, Maliga P. Sequences downstream of the translation initiation codon are important determinants of translation efficiency in chloroplasts. Plant Physiol. 2001;125(1):430–6.
Gray BN, Ahner BA, Hanson MR. High-level bacterial cellulase accumulation in chloroplast-transformed tobacco mediated by downstream box fusions. Biotechnol Bioeng. 2008;102(4):1045–54.
Gray BN, Yang HJ, Ahner BA, Hanson MR. An efficient downstream box fusion allows high-level accumulation of active bacterial beta-glucosidase in tobacco chloroplasts. Plant Mol Biol. 2011;76(3–5):345–55.
Paulova L, Patakova P, Branska B, Rychtera M, Melzoch K. Lignocellulosic ethanol: technology design and its impact on process efficiency. Biotechnol Adv. 2015;33(6):1091–107.
Togasaki RK, Brunke K, Kitayama M, Griffith M. Isolation of intact chloroplasts from Chlamydomonas reinhardtii with Beckman centrifugal elutriation system. In: Biggins J, editor. Progress in photosynthesis research, vol. 3. Dordrecht: Springer; 1987. p. 499–502.
Hunter SH. Organic growth essentials of the aerobic nonsulfur photosynthetic bacteria. J Bacteriol. 1946;52(2):213–21.
Cerutti H, Johnson AM, Gillham NW, Boynton JE. A eubacterial gene conferring spectinomycin resistance on Chlamydomonas reinhardtii: integration into the nuclear genome and gene expression. Genetics. 1997;145(1):97–110.
Ramesh VM, Bingham SE, Webber AN. A simple method for chloroplast transformation in Chlamydomonas reinhardtii. Methods Mol Biol. 2004;274:301–7.
Guzman-Zapata D, Macedo-Osorio KS, Almaraz-Delgado AL, Duran-Figueroa N, Badillo-Corona JA. Production of recombinant proteins in the chloroplast of the green alga Chlamydomonas reinhardtii. Methods Mol Biol. 2016;1385:69–85.
Noor-Mohammadi S, Pourmir A, Johannes TW. Method to assemble and integrate biochemical pathways into the chloroplast genome of Chlamydomonas reinhardtii. Biotechnol Bioeng. 2012;109(11):2896–903.
Diaz-Troya S, Florencio FJ, Crespo JL. Target of rapamycin and LST8 proteins associate with membranes from the endoplasmic reticulum in the unicellular green alga Chlamydomonas reinhardtii. Eukaryot Cell. 2008;7(2):212–22.
Rodolfi L, Zittelli GC, Bassi N, Padovani G, Biondi N, Bonini G, Tredici MR. Microalgae for oil: strain selection, induction of lipid synthesis and outdoor mass cultivation in a low-cost photobioreactor. Biotechnol Bioeng. 2009;102(1):100–12.
Miller R, Wu GX, Deshpande RR, Vieler A, Gartner K, Li XB, Moellering ER, Zauner S, Cornish AJ, Liu BS, et al. Changes in transcript abundance in Chlamydomonas reinhardtii following nitrogen deprivation predict diversion of metabolism. Plant Physiol. 2010;154(4):1737–52.
Yap BH, Crawford SA, Dagastine RR, Scales PJ, Martin GJ. Nitrogen deprivation of microalgae: effect on cell size, cell wall thickness, cell strength, and resistance to mechanical disruption. J Ind Microbiol Biotechnol. 2016;43(12):1671–80.
Harun R, Singh M, Forde GM, Danquah MK. Bioprocess engineering of microalgae to produce a variety of consumer products. Renew Sust Energ Rev. 2010;14(3):1037–47.
Hannon M, Gimpel J, Tran M, Rasala B, Mayfield S. Biofuels from algae: challenges and potential. Biofuels. 2010;1(5):763–84.
Mansfeldt CB, Richter LV, Ahner BA, Cochlan WP, Richardson RE. Use of de novo transcriptome libraries to characterize a novel oleaginous marine Chlorella species during the accumulation of triacylglycerols. PLoS ONE. 2016;11(2):e0147527.
Coragliotti AT, Beligni MV, Franklin SE, Mayfield SP. Molecular factors affecting the accumulation of recombinant proteins in the Chlamydomonas reinhardtii chloroplast. Mol Biotechnol. 2011;48(1):60–75.
Siaut M, Cuine S, Cagnon C, Fessler B, Nguyen M, Carrier P, Beyly A, Beisson F, Triantaphylides C, Li-Beisson YH, Peltier G. Oil accumulation in the model green alga Chlamydomonas reinhardtii: characterization, variability between common laboratory strains and relationship with starch reserves. BMC Biotechnol. 2011;11(1):7.
Sager R, Granick S. Nutritional control of sexuality in Chlamydomonas reinhardtii. J Gen Physiol. 1954;37(6):729–42.
Authors’ contributions
LVR conducted the N-starvation experiment and wrote the manuscript. HY engineered the transplastomic strains, verified the homoplasmy state, and evaluated the RNA transcript accumulation. MY engineered the cell-wall mutant lines and conducted the qRT-PCR experiment. LVR, MRH, and BAA analyzed the results. BAA and MRH edited the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We thank Professor David Stern and Dr. Shih-Chi Hsu, Boyce Thompson Institute at Ithaca NY, for advice on methodological techniques; and the late Professor David Wilson, Cornell University at Ithaca NY, for providing the Cel6A purified protein and primary antibody.
Competing interests
The authors declare that they have no competing interests.
Availability of supporting data
All data generated or analyzed during this study are included in this published article and its additional files.
Consent for publication
All authors agree to publication.
Ethics approval and consent to participate
Not applicable.
Funding
This research was supported in part by the Cornell University Agricultural Experiment Station federal formula funds, Project No. NYC-123412 received from Cooperative State Research, Education, and Extension Service, U.S. Department of Agriculture. Any opinions, findings, conclusions, or recommendations expressed in this publication are those of the authors and do not necessarily reflect the view of the U.S. Department of Agriculture.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1: Table S1.
Primers used in this work.
Additional file 2: Figure S1.
The Cel6A enzymatic activities were determined by the Carboxymethyl Cellulose (CMC) assay. Total proteins from wild-type and TetC-cel6A cells (50 µg/sample) were incubated with 0.4 M CMC in a water bath at 50 °C for 1 or 2 h. Reactions were quenched by the addition of DNS buffer, and samples were then heated to 95 °C for 10 min for color development. The optical density was assayed at 540 nm to quantify cellobiose accumulation. Sixty minute assays with purified Cel6A were included as a positive control. The wild-type extracts exhibited a baseline enzymatic activity which could be attributed to one or more endogenous enzymes known to be present in Chlamydomonas reinhardtii, as previously published by Blifernez-Klassen et al. (2012) [1].
Additional file 3: Figure S2.
a RT-qPCR analyses of cel6A expression levels in the chloroplast transformed cell-wall mutants and the wild-type strains. The same cloning strategy described in Fig. 1a (pCHR72, pCHR73, pCHR74) was carried out to generate the cel6A, NPTII-cel6A, and TetC-cel6A cell lines in the cell-wall mutant background (CR4349), via the glass bead method as previously described [2, 3]. The expression of the cel6A gene in the transgenic strains is under the regulation of the 16S promoter and atpA 5’UTR construct. The transcript levels are shown as a fold-change relative to the expression level of rbcL gene (n = 3 ± SD; *: p > 0.0001). b Immunoblot comparison of the Cel6A protein accumulation in the transformed cell-wall mutant strains expressing the cel6A, NPTII-cel6A, and TetC-cel6A genes under the regulation of the 16S promoter and atpA 5’UTR construct, and grown in minimal media. Each lane contains 50 µg total soluble protein.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Richter, L.V., Yang, H., Yazdani, M. et al. A downstream box fusion allows stable accumulation of a bacterial cellulase in Chlamydomonas reinhardtii chloroplasts. Biotechnol Biofuels 11, 133 (2018). https://doi.org/10.1186/s13068-018-1127-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13068-018-1127-7