
Abstract
This work deals with the synthesis and characterization of the novel 5-((5-chloro-1H-indol-2-yl)methylene)-1,3-diethyl-2-thioxodihydro-pyrimidine-4,6(1H,5H)-dione π-bridge (D–A–D) donor–acceptor–donor compound. Its exo-isomer structure has been proven by XRD-single-crystal analysis for the first time. The IR, UV–Vis., MS, CHN-, 1H and 13C NMR analysis were also carried out. The DFT-optimized structural-parameters were matched with the XRD-crystallographic data. The experimental-XRD-interactions in the lattice were compared to the computed Hirshfeld analysis (HSA), MEP map and Mulliken charge population. The DFT/6-311G(d) calculations like IR/B3LYP, TD-SCF, HOMO–LUMO, GRD and GIAO-NMR have been compared to their corresponding experimental parameters. Non-linear optical (NLO) crystal theoretical-analysis was carried out then compared to urea reference. The compound thermal activity was evaluated in an open-atmosphere by TG/DTG analysis.

Similar content being viewed by others


Background
Barbituric acid and thiobarbituric acid and their derivatives as hypnotic-compounds containing the active methylene are considered being as a good starting material to prepare specific class of heterocyclic molecules via Knoevenagel mild condensation condition [1,2,3,4]. Combination of thiobarbituric acid and different aldehydes via dehydration reactions is a useful synthetic technique to design novel mono-/or poly-substituted thiobarbiturate derivatives [2,3,4,5,6,7,8]. Such compounds recently become highly attractive to pharmaceutical chemists, since it is biological very activity, it used as: anticancer, anti-inflammatory, antioxidant, antibacterial, anti-convulsing, antifungal, antihypnotics and antiangiogenic agents [5,6,7,8,9,10,11,12,13,14]. Moreover, these compounds were broadly used as enzyme inhibitors [15], for example, it was good to inhabit tyrosinase enzyme which contributed to the neurodegeneration associated with Parkinson’s disease [15,16,17]. For such reasons, there is an urgent need to develop novel and active tyrosinase inhibitors; which is considered as a promising breakthrough enzyme-inhibitors compounds.
Many polar-organic crystalline molecules with non-centrosymmetric crystal structures reflected a very high second-order non-linear optical (NLO) properties [18, 19]. Several organic compounds which were prepared through condensation reactions may own NLO-properties; such properties can be enhanced via introducing of π-bridge in between two different functional groups donor–acceptor–donor (D–A–D) in the desired organic compounds [19].
In this study, 5-((5-chloro-1H-indol-2-yl)methylene)-1,3-diethyl-2-thioxodihydro-pyrimidine-4,6 (1H,5H)-dione compound has been prepared through one pot condensation reaction in a good yield, the structure of exo-isomer was confirmed by XRD-single crystal and spectrally characterized. Several experimental spectral measurements were compared with their corresponding theoretical parameters. Initially, exo–endo isomerization reaction was DFT-computed and its T.S was detected under QTS2 level of calculation.
Results and discussion
Synthesis
Linking thiobarbituric acid with suitable aldehydes in order to prepare heterocyclic thiobarbiturate derivatives for structural analysis and pharmaceutical applications become a very broad area of research [1,2,3]. The 5-((5-chloro-1H-indol-2-yl)methylene)-1,3-diethyl-2-thioxodihydro-pyrimidine-4,6(1H,5H)-dione derivative was prepared through one pot Knoevenagel dehydration reaction, as in Scheme 1. Condensation of 1,3-diethyl-2-thioxodihydro-pyrimidine-4,6(1H,5H)-dione with 5-chloro-1H-indole-2-carbaldehyde under reflux conditions revealed the formation of the desired thiobarbiturate in a very good yield. The synthetic methodology here reflected high yield without side products, moreover, short refluxed time was required compared to recent synthetic methodologies [1,2,3,4,5]. Knoevenagel mild condensation condition consider to be fast and easy method of synthesis, therefore, it can be performed in simple lab, moreover, the desired product is attractive to pharmaceutical chemists, several medical applications like: anticancer, antioxidant, anti-inflammatory, antibacterial, antifungal, anti-convulsing and antiangiogenic agents can be evaluated in future work then compared to published applications [5,6,7,8,9,10,11,12]. The structure of exo-isomer was clearly identified by X-ray crystallography together with several physical analyses like: TOF–MS, CHN-EA, UV–visible, IR, 1H and 13C-NMR spectroscopy.

Synthesis of desired thiobarbiturate product
TOF–MS analysis
TOF–MS of the desired compound reflected very good agreement with the C17H16ClN3O2S expected molecular formula; the (MH+) molecular ion peak was detected experimentally to be m/z = 361.3, since the theoretical m/z ion peak found to be 360.2, this seen is consistent with recent result [1, 21].
X-ray crystallographic and optimized structures
Crystals were grown by slowly evaporation of ethanol from compound ethanolic solution. The XRD data was collected on BRUKER APPEX-II CCD diffractometer, using graphite monochromatic Mo Kα radiation, λ = 0.71073 Å, at T = 293(2) K. Details of crystallographic measurement are given in Table 1. Direct methods were utilized to solve the structure using SHELXS-97 program [20]. The light-yellow compound crystal is shown in Fig. 1.

Photograph of compound crystal
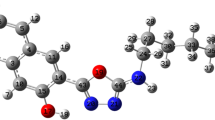
The structure of thiobarbiturate molecule was solved by single-crystal X-ray diffraction and computed by B3LYP/6-311G(d), as in Fig. 2. X-Ray diffraction suitable crystals were grown by recrystallization from ethanol solvent. The compound crystallizes in Triclinic with space group P-1, Z = 4 and cell parameters a = 9.1136 (3) Å, b = 12.7475 (5) Å, c = 15.6198 (6) Å, α = 67.0300 (10)°, β = 81.2960 (10)° and γ = 79.0530 (10)°.

Structure of exo-isomer: I ORTEP and II B3LYP/6-311G(d) optimized structure
In solid state, no solvent molecules were detected in the crystal lattice; the desired thiobarbiturate is composed of thioxodihydro-pyrimidine-4,6(1H,5H)-dione ring bonded to 5-chloro-1H-indole ring via C=C bond, the two ethyl groups which were bonded to the thioxodihydro-pyrimidine-4,6(1H,5H)-dione via the N atoms are in trans positions to each other. The two rings are in one plane which flattens the molecule, XRD structure confirmed such seen since all the atoms in the molecule (except terminal ethyl groups) are with sp2 hybridizations. The structure was solved as dimer, the two molecules connected together via N–H…O strong H-bond in perpendicular planes. The two molecules in the dimer are structurally semi-identical and both solved as exo-isomer stereo-structure (Fig. 2a).
The B3LYP exo-optimized parameters and XRD-structural (bond lengths and angles) are listed in Table 2.
A very good matching between calculated and theoretical structural parameters results were collected, as seen in Fig. 3. The bond length vs. bonds type and the angle value vs. angles type in both experimental and theoretical are very close in their values, as seen in Fig. 3a, c. Excellent graphical correlations between the exp.-XRD and DFT/B3LYP calculated bond distances and angles were found to be 0.9883 and 0.9932, respectively (Fig. 3b, d).

a Bond lengths histogram and b graphical correlation of exp. against DFT, c exp. vs. DFT angles values histogram together with its graphical correlation (d)
Endo/exo DFT-isomerization via sp2–sp2 single flip rotation
Since the thiobarbituric acid is a high symmetrical organic compound, the Knoevenagel condensation reaction with aldehydes thiobarbitone products expected to have no E/Z isomers [21]. Based on the XRD-structure and its energy profile, the exo-isomer is considered to be structurally-favored isomer (exo-isomer steric-less compared to the endo-isomer), as explained in Scheme 2.

Exo–endo isomerization
The stereo-chemical difference between the exo and endo isomers is controlled by simple flip vertical rotation around highlighted C9sp2–C7sp2 bond; this rotation caused a dramatic change in the C11–C10–C9–C7 dihedral angle from 0° (exo) to 180° (endo). Using this fact, and by neglecting all the expected intermolecular-forces in the both isomers (gaseous state), they were optimized under DFT-B3LYP/6-311++G(d) level of theory, the less global-minimum energies profile of exo-isomer (− 1830.84518316 a.u.) supported to be the more stable isomer (zero reference energy, Eexo = 0.0 kJ) with 1.45 dihedral angle, while for endo-isomer is with − 1830.85592406 a.u., Eendo = 28.2 kJ and 177.82 dihedral angle. The energy of the transition state and its structure were solved using QTS2 method of calculation. T.S energy profile found to be high the exo and endo energy − 1830.83370311 a.u. ET.S = 58.34 kJ, the structure was detected as in between endo/exo isomers structure with 90° dihedral angle C11–C10–C9–C7, that is expected since two rings are in semi-perpendicular to each other. The energies profiles reflected two important seen, first the exo-isomer (less steric) is favored over endo-one (more steric) consisted with the XRD-experimental solved structure, second the rotational isomerization exo⇔endo reaction via 180° sp2–sp2 single bond flip is possible since ΔE is very small, as seen in Fig. 4.

Structures and energy profiles of endo⇔ [T.SQTS2]⇔endo isomerization reaction
Combined crystal interaction, Hirshfeld, MEP and Mulliken charge analysis
The crystal structure of the molecule was solved as dimer form (Fig. 5a); the two molecules which compose the dimer are structural semi-identical, they are nearly flat and linked together via N–H…O H-bond. The short N–H…O hydrogen bonds (2.442 Å) connected the two the molecules reflecting its stability as perpendicular bi-molecule dimer, various intermolecular short interactions such as: C–H…Cl with 2.763 Å, C–H…S with 2.959 Å and π…π with 3.348 Å were detected which stabilized crystal lattice in 3D-network morphology. The nature of these interactions was computed by HSA. The HSA result of the compound is illustrated in Fig. 5b. Since the compound contains number of heteroatoms such as S, N, O and polar H atoms it is expected to have several red-spots on the HSA computed surface [22,23,24]. Sufficient numbers of red-spots were detected on the molecule surface reveals the presence of H-bonds and other short contacts as seen in the dnorm map (Fig. 5c). The main hydrogen bonds with biggest red-point was cited N–H…O intermolecular confirming the connected of two molecules via short interaction in semi-perpendicular plane, the other H-bonds like C–H…Cl and C–H…S were detected as smaller red-points which consisted with their longer distances. Furthermore, the compound was subjected to MEP map analysis, blue-regions on the MEP surface indicated the electrophilic parts [21], for example, proton of the H–N functional group consider to be the strongest electrophile since deep blue color was observed (Fig. 5d). On the other hand, the protons of the phenyl ring are less electrophilie since light blue color was detected. The red or orange colored around S, O and Cl atoms indicated the electronic richness positions (nucleophilic functional groups). For such reason, N–H….O hydrogen bond is highly expected to be formed as main H-bond (red or orange bind blue). Such computational output is consistent with XRD-experimental collected and HSA theoretical results.

a XRD-molecular packing, b HSA dnorm, c HSA H-bond, d MEP map and e Mulliken atomic charges
Exo, endo isomers and their T.S were subjected to DFTB3LYP/6-311G(d) Mulliken charge population analysis as summarized in Fig. 5e and Table 3. The analysis supported the existence of nucleophilic electrons-donor and electrophilic electron-acceptor functional groups in the isomers and the T.S [22]. In general all the hydrogen atoms revealed electrophilic sites in between (~ + 0.16 − 0.26e), proton of amine in exo-isomer was the highest electrophile one with ~ + 0.359e. This seen supported its acidity as well as polarity to form strong H-bonds. The carbonyl oxygen atoms in the exo-isomer showed higher nucleophilicity ~ − 0.418e explaining their roles in formation of several H-bonds in the crystal lattice of the compound. This result is consistent with the XRD packing, MPE and HSA studies.
DFT and experimental 1H NMR
The theoretical and experimental 1H-NMR spectra of the prepared thiobarbiturate are illustrated in Fig. 6. In aliphatic region two broad peaks corresponding to CH3 at 1.3 ppm and CH2 at 4.5 ppm, four peaks corresponding to the C–H aromatic protons in between 7.0–9.0 ppm were recorded, the aldehyde-proton =CH is detected at 9.6 ppm, while the acidic amine-proton (NH) is recorded at 13.1 ppm as in Fig. 6a.

1H NMR spectra in DMSO-d6 a experimentally, and theoretically b GIAO, and c ACD-LAB programs
The experimental and theoretical (GIAO and ACD-LAB) in DMSO-d6 were matched as in Fig. 6b and in c. The experimental and calculated protons chemical shifts revealed an excellent correlation, the correlation coefficient determined by GIAO and ACD-LAB against experimental found to be 0.950 and 0.972, respectively.
13C-NMR showed 4C’s with two-signals in the aliphatic region belongs to the CH3–CH2 were detected at 13–44 ppm, 10 C with 10 signals in the aromatic region at 110–147 ppm, 2C’s of C=O are detected at 160.8 ppm and 1C’s of C=S is detected at 173.8 ppm, as seen in Fig. 7. The 13C-NMR chemical shifts are compatible with the expected structure of the desired compound.

Exp. 13C-NMR of the compound in DMSO-d6
FT-IR (DFT and experimental)
The DFT-theoretical and experimental-IR spectra of the thiobarbiturate revealed a number of functional groups conforming its structural formula, as seen in Fig. 8. The characteristic vibrational frequencies revealed several polar functional groups like, N–H, C=O, C=S, N–C and C–Cl and nonpolar like, C–Hph and C–Halphi, C–C and C=C. The main functional groups theoretically and experimentally chemical shifts were explained as: N–H (exp. = 3319 cm−1, DFT = 3635 cm−1), C–HAr (exp. = 3176 cm−1, DFT = 3290 cm−1), C–Haliph (exp. = 2979 cm−1, DFT = 3020 cm−1), C=O (exp. = 1669 cm−1, DFT = 1705 cm−1), C=S (exp. = 1365 cm−1, DFT = 1440 cm−1) and C=C (exp. = 1286 cm−1, DFT = 1325 cm−1), the other functional group vibrations were sited to their positions [1,2,3, 27]. B3LYP theoretical IR frequencies are higher than experimental one, which is expected since the experimental spectra was performed in solid state while the theoretical are in gaseous state. The DFT/Exp. IR correlation coefficient found to be 0.9931 which reflected an excellent agreement between experimental and theoretical IR-analysis as seen in Fig. 8c.

a Exp., b DFT/B3LYP/6-311G(d)-IR and c Exp./DFT correlation coefficient
Electronic, HOMO/LUMO energy and TD-SCF transfer
Experimental UV and theoretical TD-SCF/DFTB3LYP/6-311G(d) spectral analysis of the desired compound were performed in ethanol and chloroform solvents, HOMO and LUMO energy levels for exo-isomer are computed in ethanol as in Fig. 9.

a Experimental UV, b TD-SCF/DFTB3LYP/6-311G(d) electronic spectra and c HOMO/LUMO shape-energy diagram in ethanol
Experimentally, the UV behavior reflected π → π* electronic transition one sharp peak at λmax = 430 nm in both ethanol and chloroform solvents (Fig. 9a). The TD-SCF/DFTB3LYP/6-311G(d) calculations exhibited one abroad band at λmax = 397 nm in both solvents (Fig. 9b). No significant difference in the electronic behaviors (Uv and TD) were detected by changing solvents which reflecting a high degree of harmony between exp. and DFT analysis. The experimental wavelength showed a very good agreement with TD-B3LYP/6-311G(d) an experimentally bathochromic shift with Δλmax = 33 nm were detected. To understand the electron transfer in FMO of the molecule HOMO and LUMO was computed in ethanol [25,26,27], ΔELUMO–HOMO = 0.128 a.u. (3.49 eV). Due to TD-B3LYP/6-311G(d) the main electron transfer at λmax = 397 nm can be attributed to HOMO → LUMO (97%), while λ at 362 nm to HOMO-2 → LUMO (96%) and λ at 445 nm to HOMO-1 → LUMO + 1 (95%).
Global reactivity descriptors (GRD)
GRD quantum parameters can be easily estimated from the energy gap levels using Koopman’s notation as follows:

GRD parameters used by the frontier electron density to explain reactions in conjugated system and predicting the most reactive position in molecule. The conjugated-molecules are detected by a small EHOMO/LUMO, which facilitated the understanding of the structural activities of molecules [22,23,24].
Using the GRD equations above, the electronegativity (χ), electrophilicity (ω), chemical potential (µ), hardness (ƞ) and softness (σ) for exo-isomer were calculated, as shown in Table 4.
The advantage of such quantum parameters have been demonstrated to understand the molecular activities of such compound to be used as metal-coordination ligand or search for other biological applications.
Nonlinear optical (NLO)
The quantum computation of nonlinear optical (NLO) properties of a compound has interesting role for new materials design in optical processing and modern material technology [28]. It is importance in providing optical modulation, frequency shifting, fiber, switching, optical materials laser and optical memory [22,23,24,25,26,27,28]. The NLO quantum calculations like polarizabilities and hyperpolarizabilities became easy and available via the DFT calculation method. Up to our knowledge, no theoretical DFT-computations were reported addressing NLO for thiobarbiturate molecules; therefore, this excited our concern to start such study. To setup the relationship between NLO and structure of the desired molecule the anisotropy polarizability (Δα) the dipole moment (μ), the mean polarizability (α) and the hyperpolarization (β) are calculated using B3LYP/6-31G. Urea has been used as NLO-reference proto-typical; therefore, NLO parameters of the urea were computed in the gaseous phase together with target molecule under the same level of theory, as seen in Table 5. The polarizability, anisotropy polarizability and the hyperpolarizability of title compound were calculated 16.66, 20.234 and 13.872 times better than the urea reference-molecule. It is worth noted that the 5-((5-chloro-1H-indol-2-yl)methylene)-1,3-diethyl-2-thioxodihydro-pyrimidine-4,6(1H,5H)-dione compound has higher NLO properties compared to urea reference, implying of such compounds to be as new NLO promising materials.
Thermal stability
TG/DTG analysis of the desired thiobarbiturate was performed in the temperature range of 0 to 1000 °C in open air atmosphere with heat rate of 5 °C min−1, as seen in Fig. 10. Below 380 °C, the compound displayed good thermal stability, and then it thermally decomposed in one step in the range of 380–520 °C, above 520 °C, the compound was completely decomposed with zero mass residue.

TG/DTG curves
Conclusions
Novel 5-((5-chloro-1H-indol-2-yl)methylene)-1,3-diethyl-2-thioxodihydro-pyrimidine-4,6(1H,5H)-dione via Knoevenagel mild condensation condition. Exo–endo isomerization reaction in the desired molecule was computed, T.S structure and energy level was detected under QTS2 level of calculation. The exo-structure was proven by XRD-analysis measurement, several physical analyses like: CHN-EA, MS, IR, UV–Vis., 1H and 13C NMR consisted with such seen. The DFT/B3LYP/6-311G(d) structural optimized data were agreed with the XRD-parameters. The exp. XRD-lattice interactions were computed by HSA, MEP map and Mulliken charge, several H-bonds and π–π stacked short interactions were detected. The DFT/6-311G(d) calculations like B3LYP-IR, TD-SCF, HOMO–LUMO, GRD and GIAO NMR reflected a high agreement with their corresponding experimental parameters. NLO-theoretical calculation showed excellent optical properties of the compound, it is even ~ 20 better than urea-reference. The compound TG/DTG analysis revealed a high thermal stability with one step decomposition reaction.
Experimental
General
The XRD-data was collected on a Bruker APEX-II D8 diffractometer. The NMR spectra were run in DMSO-d6 using Jeol-400 spectrometer. All the chemicals were purchased from Sigma.
Synthesis of 1,3-dimethyl-5-(thien-2-ylmethylene)-pyrimidine-2,4,6-(1H,3H,5H)-trione
A mixture of 1,3-diethyl-2-thioxodihydro-pyrimidine-4,6(1H,5H)-dione (1.0 mmol) and 5-chloro-1H-indole-2-carbaldehyde (1.0 mmol) in 50 mL of distilled water was refluxed and stirred for 5 h until a yellow product was precipitated. Water was decanted, and the yellow product was washed with water then left under an open atmosphere for drying (yield, 85%).
The yellow powder product, with a m.p = 360 °C, was collected; molecular formula C17H16ClN3O2S; (Calcd. C, 56.43; H, 4.46 and N, 11.69. Found: C, 56.28; H, 4.41 and N, 11.53). [M+] m/z = 361.3 (360.2, theoretical). 1H NMR (400 MHz, DMSO-d6): (ppm) 1.2 (m, 6H, 2CH3), 4.5 (b, 4H, 2CH2), 7.0–9.0 (4 m, 14H, Ar’s), 9.2 (s, 2H, –HC=N–), 13.1 (s, 1H, –HN–). 13C-NMR (100 MHz, DMSO-d6): (ppm) 13.5 (2C, CH3CH2), 43.1 (2C, CH3CH2), 110.8, 116.1, 117.7, 121.8, 123.4, 126.1, 131.5, 128.8, 130.7, 139.8, 145.6, 146.9 (10 signals, 10C, Ar’s), 160.8 (2C, C=O), 173.8 (1C, C=S). FT-IR main vibrations, VN–H = 3319 cm−1, VC–HAr = 3176 cm−1, VC–H aliph = 2979 cm−1, VC=O = 1669 cm−1, VC=S = 1365 cm−1, VC=C = 1286 cm−1.
Computational details
Hirshfeld surface analysis (HSA) been performed using the CRYSTAL EXPLORER 3.1 program [29]. All Computational calculations of the desired compound were performed by Gaussian 09 software [30]. The molecule optimization geometries, IR vibrations, HOMO/LUMO, TD-SCF, NLO, GRD analysis were carried on DFT/B3LYP level of theory using 6-311G(d, p) base set, NMR chemical shifts were performed at DFT/B3LYP/level of theory and 6-311++G(d,p) base set via adopting GIAO method [31].
References
Barakat A, Soliman SM, Ghabbour HA, Ali M, Al-Majid AM, Zarrouk A, Warad I (2017) Intermolecular interactions in crystal structure, Hirshfeld surface, characterization, DFT and thermal analysis of 5-((5-bromo-1H-indol-3-yl) methylene)-1, 3-dimethylpyrimidine-2, 4, 6 (1H, 3H, 5H)-trione indole. J Mol Struct 1137:354–361
Serrano JL, Cavalheiro E, Barroso S, Romão MJ, Silvestre S, Almeida P (2017) A synthetic route to novel 3-substituted-2, 1-benzisoxazoles from 5-(2-nitrobenzylidene)(thio) barbiturates. C R Chim 20(11–12):990–995
Ziarani GM, Aleali F, Lashgari N (2016) Recent applications of barbituric acid in multicomponent reactions. RSC Adv 6(56):50895–50922
Mahmudov KT, Kopylovich MN, Maharramov AM, Kurbanova MM, Gurbanov AV, Pombeiro AJ (2014) Barbituric acids as a useful tool for the construction of coordination and supramolecular compounds. Coord Chem Rev 265:1–37
Nikoofar K, Khademi Z (2017) Barbituric acids in organic transformations, an outlook to the reaction media. Mini Rev Org Chem 14(2):143–173
Laxmi SV, Reddy YT, Kuarm BS, Reddy PN, Crooks PA, Rajitha B (2011) Synthesis and evaluation of chromenyl barbiturates and thiobarbiturates as potential antitubercular agents. Bioorg Med Chem Lett 21(14):4329–4331
Kenchappa R, Bodke YD, Asha B, Telkar S, Sindhe MA (2014) Synthesis, antimicrobial, and antioxidant activity of benzofuran barbitone and benzofuran thiobarbitone derivatives. Med Chem Res 23(6):3065–3081
Biradar JS, Sasidhar BS, Parveen R (2010) Synthesis, antioxidant and DNA cleavage activities of novel indole derivatives. Eur J Med Chem 45(9):4074–4078
Dhorajiya BD, Bhakhar BS, Dholakiya BZ (2012) Synthesis, characterization, solvatochromic properties, and antimicrobial evaluation of 5-acetyl-2-thioxo-dihydro-pyrimidine-4, 6-dione-based chalcones. Med Chem Res 22(9):4075–4086
Singh P, Kaur M, Design Verma P (2009) synthesis and anticancer activities of hybrids of indole and barbituric acids—identification of highly promising leads. Bioorg Med Chem Lett 19(11):3054–3058
Radwan MA, Ragab EA, Sabry NM, El-Shenawy SM (2007) Synthesis and biological evaluation of new 3-substituted indole derivatives as potential anti-inflammatory and analgesic agents. Bioorg Med Chem 15(11):3832–3841
Johnson SR, Zheng W (2006) Recent progress in the computational prediction of aqueous solubility and absorption. AAPS J 8(1):E27–E40
Ulrich CM, Bigler J, Potter JD (2006) Non-steroidal anti-inflammatory drugs for cancer prevention: promise, perils and pharmacogenetics. Nat Rev Cancer 6(2):130–140
Penthala NR, Ponugoti PR, Kasam V, Crooks PA (2013) 5-((1-Aroyl-1H-indol-3-yl) methylene)-2-thioxodihydro-pyrimidine-4, 6 (1H, 5H)-diones as potential anticancer agents with anti-inflammatory properties. Bioorg Med Chem Lett 23(5):1442–1446
Figueiredo J, Serrano JL, Cavalheiro E, Keurulainen L, Yli-Kauhaluoma J, Moreira VM, Ferreira S, Domingues FC, Silvestre S, Almeida P (2018) Trisubstituted barbiturates and thiobarbiturates: synthesis and biological evaluation as xanthine oxidase inhibitors, antioxidants, antibacterial and anti-proliferative agents. Eur J Med Chem 143:829–842
Harriman GC, Brewer M, Bennett R, Kuhn C, Bazin M, Larosa G, Skerker P, Cochran N, Gallant D, Baxter D, Picarella D (2008) Selective cell adhesion inhibitors: barbituric acid based α4β7—MAdCAM inhibitors. Bioorg Med Chem Lett 18(7):2509–2512
Wang J, Radomski MW, Medina C, Gilmer JF (2013) MMP inhibition by barbiturate homodimers. Bioorg Med Chem Lett 23(2):444–447
Di Bella S, Fragalà I, Ledoux I, Diaz-Garcia MA, Marks TJ (1997) Synthesis, characterization, optical spectroscopic, electronic structure, and second-order nonlinear optical (NLO) properties of a novel class of donor–acceptor bis (salicylaldiminato) nickel (II) Schiff base NLO chromophores. J Am Chem Soc 119(40):9550–9557
Lacroix PG (2001) Second-order optical nonlinearities in coordination chemistry: the case of bis (salicylaldiminato) metal Schiff base complexes. Eur J Inorg Chem 2:339–348
Spek AL (2009) Structure validation in chemical crystallography. Acta Cryst 65:148–155
Barakat A, Islam MS, Al-Majid AM, Ghabbour HA, Atef S, Zarrouk A, Warad I (2018) Quantum chemical insight into the molecular structure of l-chemosensor 1, 3-dimethyl-5-(thien-2-ylmethylene)-pyrimidine-2, 4, 6-(1 H, 3 H, 5 H)-trione: naked-eye colorimetric detection of copper (II) anions. J Theor Comp Chem 17(01):1850005
Warad I, Barakat A (2017) Synthesis, physicochemical analysis of two new hemilabile ether-phosphine ligands and their first stable bis-ether-phosphine/cobalt (II) tetrahedral complexes. J Mol Struct 1134:17–24
Asadi Z, Esrafili MD, Vessally E, Asnaashariisfahani M, Yahyaei S, Khani A (2017) A structural study of fentanyl by DFT calculations, NMR and IR spectroscopy. J Mol Struct 1128:552–562
Tarı GÖ, Ceylan Ü, Uzun S, Ağar E, Büyükgüngör O (2018) Synthesis, spectroscopic (FT-IR, UV–Vis), experimental (X-Ray) and theoretical (HF/DFT) study of:(E)-2-Chloro-N-((4-nitrocyclopenta-1, 3-dienyl) methylene) benzenamine. J Mol Struct 1156:74–82
Ahmed NH, Yasin Kh, Ayub Kh, Mahmood T, Tahir NM, Khan AB, Hafeez M, Ahmed M, Ul-Haq I (2016) Click one pot synthesis, spectral analyses, crystal structures, DFT studies and brine shrimp cytotoxicity assay of two newly synthesized, 4,5-trisubstituted 1,2,3-triazoles. J Mol Struct 1106:430–439
Sherzaman S, Rehman S, Ahmed M, Khan B, Mahmood T, Ayub Kh, Tahir M (2017) Thiobiuret based Ni(II) and Co(III) complexes: synthesis, molecular structures and DFT studies. J Mol Struct 1148:388–396
Ahmed M, Yasin Kh, Khan R, Mahmood T, Ayub Kh, Malik D, Hafeez M, Khan A, Tahir M (2017) Synthesis, crystal structure, spectral analysis, DFT studies and antimicrobial activity of ethyl 6-(4-(ethoxycarbonyl)-1H-1,2,3-triazol-1-yl)pyridine-3-carboxylate. J Chem Soc Pak 39:640–649
Thorat KG, Sekar N (2017) Pyrrole-thiazole based push-pull chromophores: an experimental and theoretical approach to structural, spectroscopic and NLO properties of the novel styryl dyes. J Photochem Photobiol A Chem 333:1–7
Wolff SK, Grimwood DJ, McKinnon JJ, Jayatilaka D, Spackman MA (2007) Crystal explorer 2.1. University of Western Australia, Perth
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA et al (2010) Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford
Mahmood T, Kosar N, Ayub K (2017) DFT study of acceleration of electrocyclization in photochromes under radical cationic conditions: comparison with recent experimental data. Tetrahedron 73:3521–3528
Authors’ contributions
AB and IW conceived and designed the experiments; MSA performed the experiments; AMA analyzed the data; AB contributed reagents/materials/analysis tools; HAG solved the chemical structure by X-ray single crystal technique; AZ and IW carried out the computational studies; AB and IW wrote the paper. All authors read and approved the final manuscript.
Acknowledgements
The authors would like to extend their sincere appreciation to the Deanship of Scientific Research at King Saud University for its funding this Research group NO (RGP-257).
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
All data and materials are fully available without restriction.
Ethics approval and consent to participate
Not applicable.
Funding
This study was funded by the Deanship of Scientific Research at King Saud University (NO. RGP-257).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Altowyan, M.S., Barakat, A., Al-Majid, A.M. et al. Vibrational spectral analysis, XRD-structure, computation, exo⇔endo isomerization and non-linear optical crystal of 5-((5-chloro-1H-indol-2-yl)methylene)-1,3-diethyl-2-thioxodihy-dropyrimidine-4,6 (1H,5H)-dione. BMC Chemistry 13, 11 (2019). https://doi.org/10.1186/s13065-019-0524-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13065-019-0524-8