
Abstract
Background
In view of wide range of biological activities of oxazole, a new series of oxazole analogues was synthesized and its chemical structures were confirmed by spectral data (Proton/Carbon-NMR, IR, MS etc.). The synthesized oxazole derivatives were screened for their antimicrobial and antiproliferative activities.
Results and discussion
The antimicrobial activity was performed against selected fungal and bacterial strains using tube dilution method. The antiproliferative potential was evaluated against human colorectal carcinoma (HCT116) and oestrogen- positive human breast carcinoma (MCF7) cancer cell lines using Sulforhodamine B assay and, results were compared to standard drugs, 5-fluorouracil and tamoxifen, respectively.
Conclusion
The performed antimicrobial activity indicated that compounds 3, 5, 6, 8 and 14 showed promising activity against selected microbial species. Antiproliferative screening found compound 14 to be the most potent compound against HCT116 (IC50 = 71.8 µM), whereas Compound 6 was the most potent against MCF7 (IC50 = 74.1 µM). Further, the molecular docking study has been carried to find out the interaction between active oxazole compounds with CDK8 (HCT116) and ER-α (MCF7) proteins indicated that compound 14 and 6 showed good dock score with better potency within the ATP binding pocket and may be used as a lead for rational drug designing of the anticancer molecule.

Similar content being viewed by others


Background
Multidrug resistance and emergence of new infectious diseases are amongst the major challenges in the treating of microbial infections which necessitates the discovery of newer antimicrobial agents [1]. Cancer is one of the serious health issues and many more novel anticancer agents are needed for effective treatment of cancer [2, 3]. Heterocyclic compounds offer a high degree of structural diversity and have proven to be broadly and economically useful as therapeutic agents like benzoxazole [4, 5], indole [3], Quinoline-Branched Amines [6, 7], pyrimidine analogues [8]. The oxazole moiety is reported to have broad range of biological potential such as anti-inflammatory, analgesic, antibacterial [9], antifungal [10], hypoglycemic [11], antiproliferative [12], antitubercular [13], antiobesity [14], antioxidant [15], antiprogesteronic [16], prostacyclin receptor antagonist [17], T-type calcium channel blocker [18] and transthyretin (TTR) amyloid fibril inhibitory activities [19]. A number of marketed drugs (Fig. 1) are available in which oxazole is the core active moiety such as aleglitazar (antidiabetic) [20], ditazole (platelets aggregation inhibitor) [21], mubritinib (tyrosine kinase inhibitor) [22], and oxaprozin (COX-2 inhibitor) [23].

Marketed drugs containing oxazole
Molecular docking studies provide the most detailed possible view of drug-receptor interaction and have created a new rational approach to drug design. The CDKs (cyclin dependent kinase) is an enzyme family that plays an important role in the regulation of the cell cycle and thus is an especially advantageous target for the development of small inhibitory molecules. Selective inhibitors of the CDKs can be used for treating cancer or other diseases that cause disruptions of cell proliferation [24]. Estrogen receptor alpha (ERα) is the major driver of ~ 75% of all breast cancers. Current therapies for patients with ER+ breast cancer are largely aimed at blocking the ERα signaling pathway. For example, tamoxifen blocks ERα function by competitively inhibiting E2/ERα interactions and fulvestrant promotes ubiquitin-mediated degradation of ERα. Endocrine therapies are estimated to have reduced breast cancer mortality by 25 ± 30% [25].
On the basis of the information obtained from literature survey (Fig. 2), in the present work we hereby report the synthesis, antimicrobial and antiproliferative potentials of oxazole derivatives.

Biological profile of oxazole derivatives
Results and discussion
Chemistry
The synthesis of oxazole derivatives (1–15) were accomplished using the synthetic procedure depicted in Scheme 1. At first, 3-acetyl-2H-chromen-2-one (I) was prepared by the reaction of salicylaldehyde and ethyl acetoacetate in the presence of piperidine. Further, the reaction of I with bromine resulted in the formation of 3-(2-bromoacetyl)-2H-chromen-2-one (II). The later was refluxed with urea to synthesize 3-(2-aminooxazol-5-yl)-2H-chromen-2-one (III). The reaction of 3-(2-aminooxazol-5-yl)-2H-chromen-2-one (III) with substituted aldehydes yielded the title compounds 3-(2-(substituted benzylideneamino)oxazol-5-yl)-2H-chromen-2-one derivatives (1–15). The physicochemical and spectral characteristics of the synthesized oxazole derivatives are given in Table 1. Spectral data (FT-IR (KBr, cm−1), 1H/13C–NMR (DMSO-d6, 600 MHz, δ ppm) and Mass spectral) studies helped in determining the molecular structures of the synthesized derivatives (1–15). The IR spectrum indicated that the appearance of bands at 3398–2924 cm−1, 1456–1415 cm−1, 1680–1595 cm−1, 1382–1236 cm−1 and 1724–1693 cm−1 displayed the presence of C–H, C=C, C=N, C–N and C=O groups, respectively in the synthesized compound. The absorption bands around 1292–1130 cm−1 corresponded to C–O–C stretching of oxazole compounds. In case of 1H-NMR spectra the presence of multiplet signals between 6.88 and 8.69 δ ppm reflected the presence of aromatic protons in synthesized derivatives. The compound 14 showed singlet (s) at 6.76 δ ppm because of the presence of OH of Ar–OH. The appearance of singlet (s) at 7.51–8.4 δ ppm and 6.9–7.37 δ ppm is due to the existence of N=CH and C–H of oxazole, respectively. Compound 8 showed multiplet and doublet signals at 3.11 δ ppm and 1.29 δ ppm due to existence of –CH and (CH3)2 groups of –CH(CH3)2 at the para-position. The compounds, 1, 2 and 14 showed singlet at 3.73–3.89 δ ppm due to the existence of OCH3 of Ar–OCH3. The compounds, 3 and 5 showed singlet at 5.08 δ ppm due to the existence of –CH2–O group of (benzyloxy)benzene. The compound 10 displayed doublet signal at 5.59–6.95 δ ppm due to the existence of –CH=CH group of -prop-1-en-1-ylbenzene. The 13C–NMR spectrum indicated that the carbon signals around at 161.1, 128.5 (coumarin), 151.9 (N=CH), 136.1 (oxazole) of the synthesized compounds. Mass of synthesized compounds showed in (M++1).

Synthesis of 3-(2-aminooxazol-5-yl)-2H-chromen-2-one derivatives (1–15)
Antimicrobial activity
The in vitro antimicrobial potential of the prepared oxazole derivatives was determined by tube dilution technique (Table 2, Fig. 3, 4 and 5). The antibacterial screening results revealed that compound 3 was moderately potent against S. aureus with MICsa value of 14.8 µM and compound 8 was moderately active against B. subtilis with MICbs value of 17.5 µM. Compound 3 (MICec = 14.8 µM) was found to be effective against E. coli. Compound 14 (MICpa = 17.3 µM) and compound 6 (MICse= 17.8 µM) exhibited promising activity against P. aeruginosa and S. enterica, respectively. The antifungal activity results indicated that compound 6 (MICan = 17.8 µM) displayed most potent activity against A. niger and compounds 3 and 5 (MICca= 29.6 µM) were found to be moderately potent against C. albicans. The antibacterial screening results are comparable to the standard drug (cefadroxil), whereas antifungal results of compound 6 showed less activity against A. niger and compound 5 showed more against C. albicans than the standard drug (fluconazole) and these compounds may be used as a lead compound to discover novel antimicrobial agents.

Antibacterial screening results against Gram positive species

Antibacterial screening results against Gram negative species

Antifungal screening results against fungal species
Anticancer activity
The synthesized derivatives were also screened for their cytotoxic effect using Sulforhodamine B (SRB) assay [26] against two cancer cell lines- human colorectal carcinoma (HCT116) and oestrogen-positive human breast carcinoma (MCF7). In the case of HCT116, compound 14 exhibited good activity with IC50 = 71.8 µM. In the case of MCF7, compound 6 exhibited good activity with IC50 = 74.1 µM. Reference drugs used in the study were 5-flourouracil (for HCT116) and tamoxifen (MCF7). They had yielded IC50 values of 12.7 µM and 4.3 µM, respectively and these compounds may be used as a lead compound to discover novel anticancer agents. Results are displayed in Table 3.
Molecular docking results
The mammalian cyclin-dependent kinase 8 (cdk8) protein which is a component of the RNA polymerase has been one of the proteins responsible for acute lymphoblastic leukaemias. CDK-8 is a heterodimeric kinase protein responsible for regulation of cell cycle progression, transcription and other functions. CDK-8 phosphorylates the carboxyterminal domain of the largest subunit of RNA polymerase II like protein kinases. Therefore, the inhibition of CDK-8 protein may be crucial for controlling cancer [27]. Since compounds were screened through ATP binding pocket so, ATP was used as docking control to compare the binding affinity of compounds within the binding pocket. The synthesized oxazole compounds showed good docking score and were found to interact with important amino acids for the biological function of CDK-8 protein.
Molecular docking were carried out to analyse the binding mode of the most active compound 14 and compound 6 against human colorectal carcinoma HCT116 and oestrogen- positive human breast carcinoma MCF7 cancer cell lines respectively. The molecular docking study was carried out on GLIDE docking program. The compound 14 was docked in the active site of the cyclin dependent kinase cdk8 (PDB: 5FGK) co-crystallized wit 5XG ligand. The results were analysed based on the docking score obtained from GLIDE. Ligand interaction diagram and displayed the binding mode of compound 14 in the active site of cdk8 having co cystallised ligand 5XG and 5-fluorouracil (the standard inhibitor of cancer) is having a different binding mode to that of active compound (Figs. 6 and 7).

Interaction of compound 14 and 5-fluorouracil within the active pocket of cdk-8 protein and interacting amino acid in 2D view

Interaction of 5-fluorouracil within the active pocket of cdk-8 protein and interacting amino acid in 2D view
The compound 6 was docked in the active site of the ER-alpha of MCF-7 (PDB: 3ERT) co-crystallized wit OHT (Tamoxifen) ligand. The results were analysed based on the docking score obtained from GLIDE. Ligand interaction diagram and show the binding mode of compound 6 in the active site of ER apha having co cystallised ligand OHT and Tamoxifen (the standard inhibitor of cancer) is having a different binding mode to that of active compound (Figs. 8 and 9). The docking scores were demonstrated in terms of negative energy; the lower the binding energy, best would be the binding affinity. The results depend on the statistical evaluation function according to which the interaction energy in numerical values as docking scores. The 3D pose of the ligand interaction with receptor can be visualized using different visualization tools [28]. Based on the molecular docking study the selected compounds with good anticancer activity against cancer cell lines (HCT116 and MCF-7) were displayed good interaction with crucial amino acids. Like if we look into the best-fitted compound 14 showed the best dock score (− 7.491) with better potency (71.8 µM) within the ATP binding pocket (Table 4). Compound 6 showed the best dock score (− 6.462) with better potency (74.1 µM) within the ATP binding pocket (Table 5). Thus the docking results suggest that the oxazole derivatives can act as of great interest in successful chemotherapy. CDK-8 may be the target protein of oxazole derivatives for their anticancer activity at lower micromolar concentrations. Based on the docking analysis it is suggested that more structural modifications are required in compounds 6 and 14 to make them more active against cancer cells and to have activity comparable to the standards 5-fluorouracil and tamoxifen.

Interaction of compound 6 and tamoxifen within the active pocket of 3ERT protein and interacting amino acid in 2D view

Interaction of tamoxifen within the active pocket of 3ERT protein and interacting amino acid in 2D view
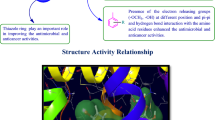
Structure activity relationship
From the antimicrobial and anticancer activities results following structure activity can be derived (Fig. 10):

Structure activity relationship of synthesized compounds
-
The different substitution of aldehydes were used to synthesized the final derivatives of 3-(2-aminooxazol-5-yl)-2H-chromen-2-one derivatives played an important role in improving the antimicrobial and anticancer activities. Presence of electron releasing group (–CH(CH3)2) at para position of the substitution part of the synthesized compound 8, increased the antibacterial activity against B. subtilis. Presence of para-(phenoxy-methyl)benzene group (compound 3), enhanced the antibacterial activity against E. coli and S. aureus as well antifungal activity against C. albicans whereas (Compound 5) also improved the antifungal activity against C. albicans.
-
Presence of electron releasing group (OH, OCH3) at meta and para position of the substitution portion of the synthesized compound 14, increased the antibacterial activity against P. aeruginosa and also increased anticancer activity against HCT116 cancer cell line whereas electron withdrawing groups (–Cl) at para-position of the synthesized compound 6, improved the antimicrobial activity against S. enterica and A. niger as well as anticancer activity against MCF7 cancer cell line. These compounds may be used as a lead compound to discover novel antimicrobial and anticancer agents.
Experimental part
The chemicals procured were of analytical grade and were further used without any purification. Melting point (m.p.) was determined in open glass capillaries on a Stuart scientific SMP3 apparatus. Reaction progress of every synthetic step was confirmed by TLC plates on silica gel sheets. 1H and 13C–NMR spectra were determined by Bruker Avance III 600 NMR spectrometer in appropriate deuterated solvents and are expressed in parts per million (δ, ppm) downfield from tetramethylsilane (internal standard). Proton NMR spectra are given as multiplicity (s, singlet; d, doublet; t, triplet; m, multiplet) and number of protons. Infrared (IR, KBr, cm−1) spectra were recorded as KBr pellets on Shimadzu FTIR 8400 spectrometer. Waters Micromass Q-ToF Micro instrument was used for obtaining the Mass spectra.
Synthetic steps of Scheme 1
Step 1: Synthesis of 3-acetyl-2H-chromen-2-one (I) To a solution of salicylaldehyde (0.025 mol) and ethyl acetoacetate (0.025 mol) in methanol (15 mL), 2–3 drops of piperidine was added, shaken with stirring and allowed to stand at room temperature for 30 min. Needle shaped crystals of 3-acetyl-2H-chromen-2-one (I) were obtained which were filtered dried and recrystallized from methanol [29].
Step 2: Synthesis of 3-(2-bromoacetyl)-2H-chromen-2-one (II) To a solution of 3-acetyl-2H-chromen-2-one (0.01 mol) in chloroform (15 mL), bromine (1.7 g) in chloroform (6 mL), was added with intermittent shaking and warming. The mixture was heated on water bath for 15 min to expel most of hydrogen bromide. The solution was cooled, filtered and recrystallized from acetic acid so as to obtain 3-(2-bromoacetyl)-2H-chromen-2-one (II) [29].
Step 3: Synthesis of 3-(2-aminooxazol-5-yl)-2H-chromen-2-one (III) To the methanolic solution of compound II (0.01 mol), urea (0.01 mol) was added. The reaction mixture was refluxed for 12 h, poured on to crushed ice and resultant solid was recrystallized with methanol to obtain III [30].
Step 4: Synthesis of title compounds (1–15) To the solution of compound III (0.01 mol) in methanol (50 mL), different substituted aldehydes (0.01 mol) were added and refluxed for 12 h. The reaction mixture was concentrated to half of its volume after refluxing and poured onto crushed ice. The resulting solution was then evaporated and the residue thus obtained was washed with water and finally recrystallized from methanol to give final compounds (1–15).
In vitro antimicrobial assay
Tube dilution method [31] was used for evaluating the antimicrobial potential of the compounds and the standard drugs used were cefadroxil (antibacterial) and fluconazole (antifungal). The microbial species used in the study were Gram +ve and Gram −ve bacteria, i.e. MTCC-441 (B. subtilis), MTCC-3160 (S. aureus), MTCC-424 (P. aeruginosa), MTCC 1165 (S. enterica) and MTCC-443 (E. coli). The antifungal potential was assessed against MTCC-227 (C. albicans), and MTCC-281 (A. niger). Double strength nutrient broth I.P. (bacteria) or sabouraud dextrose broth I.P. (fungi) [32] were used for antimicrobial study. Dimethyl sulfoxide was used for preparing the stock solution of the test and reference compounds. Results were noted in MIC after incubating the samples at 37 ± 1 °C (24 h) for bacteria, at 25 ± 1 °C (7 days) for A. niger and at 37 ± 1 °C (48 h) for C. albicans, respectively. The lowest concentration of the tested compound that showed no visible growth of microorganisms in the test tube was noted as MIC.
In vitro anticancer assay
The cytotoxic effect of oxazole derivatives was determined against two different cancer cell lines—human colorectal carcinoma [HCT116] and oestrogen- positive human breast carcinoma (MCF7) using Sulforhodamine-B assay. HCT116 was seeded at 2500 cells/well (96 well plate) whereas MCF7 was seeded at 3000 cells/well (96 well plate). The cells were allowed to attach overnight before being exposed to the respective compounds for 72 h. The highest concentration of each compound tested (100 µg/mL) contained only 0.1% DMSO (non-cytotoxic). Sulforhodamine B (SRB) assay [26] was then performed. Trichloroacetic acid was used for fixing the cells and then staining was performed for 30 min with 0.4% (w/v) sulforhodamine B mixed with 1% acetic acid. After five washes of 1% acetic acid solution, protein-bound dye was extracted with 10 mM tris base solution. Optical density was read at 570 nm and IC50 (i.e. concentration required to inhibit 50% of the cells) of each compound was determined. Data was presented as mean IC50 of at least triplicates.
Molecular docking study
The protein target for oxazole derivatives was identified through the literature. Since the oxazole nucleus has vast medicinal properties, so the targets enzymes/receptors were found targeted with anticancer effect of oxazole compounds were collected for selection [33]. Cyclin-dependent kinase-8 (PDB Id: 5FGK) co-crystallized wit 5XG ligand and ER-alpha of MCF-7 (PDB: 3ERT) co-crystallized wit OHT (Tamoxifen) ligand excellent target against cancer [34], was retrieved from Protein Data Bank (http://www.rcsb.org/pdb/home/home.do) for docking of potent synthesized oxazole compounds. Docking score obtained from GLIDE and ATP binding site was targeted and the grid was created. The active site grid covered the important amino acids interacting with ATP [35].
Conclusion
A series of oxazole derivatives was designed, synthesized and evaluated for its antimicrobial and antiproliferative activities. The biological screening results indicated that the compounds 3, 5, 6, 8 and 14 had the best antimicrobial activity and had MIC values comparable to the standard drugs whereas in the case of anticancer activity, compound 14 was found to be moderate activity against HCT116 while compounds 6 was moderate activity against MCF7. Further molecular docking study indicated that compound 14 showed the best dock score (− 7.491) with better potency (71.8 µM) within the ATP binding pocket. Compound 6 showed the best dock score (− 6.462) with better potency (74.1 µM) within the ATP binding pocket. Hence these compounds may be taken as lead compound for further development of novel antimicrobial and anticancer agents.
References
Keche AP, Hatnapure GD, Tale RH, Rodge AH, Birajdar SS, Kamble VM (2012) Novel pyrimidine derivatives with aryl urea, thiourea and sulfonamide moieties: synthesis, anti-inflammatory and antimicrobial evaluation. Bioorg Med Chem Lett 22(10):3445–3448
Zijian L, Yu W, Lin L, Dazhuang Z, Lihui W, Yanfang Z, Ping G (2014) Design, synthesis and biological evaluation of novel thieno[3,2-d]pyrimidine derivatives containing diaryl urea moiety as potent antitumor agents. Eur J Med Chem 85:215–227
Nagesh N, Raju G, Srinivas R, Ramesh P, Reddy MD, Reddy CR (2015) A dihydroindolizino indole derivative selectively stabilizes G-quadruplex DNA and down-regulates c-MYC expression in human cancer cells. Biochem Biophys Acta 1850:129–140
Kakkar S, Tahlan S, Lim SM, Ramasamy K, Mani V, Shah SAA, Narasimhan B (2018) Benzoxazole derivatives: design, synthesis and biological evaluation. Chem Cent J 12(92):1–16
Kakkar S, Kumar S, Narasimhan B, Lim SM, Ramasamy K, Mani V, Shah SAA (2018) Design, synthesis and biological potential of heterocyclic benzoxazole scaffolds as promising antimicrobial and anticancer agents. Chem Cent J 12(96):1–11
Reddy MD, Fronczek FR, Watkins EB (2016) Rh-Catalyzed, Regioselective, C−H Bond functionalization: access to quinoline-branched amines and dimers. Org Lett 18:5620–5623
Motati DR, Uredi D, Watkins EB (2018) A general method for the metal-free, regioselective, remote C–H halogenation of 8-substituted quinolines. Chem Sci 9:1782–1788
Raju G, Srinivas R, Reddy MD, Reddy ChR, Nagesh N (2014) Studies on non-covalent interaction of coumarin attached pyrimidine and 1-methyl indole 1,2,3 triazole analogues with intermolecular telomeric G-quadruplex DNA using ESI-MS and spectroscopy. Nucleosides, Nucleotides Nucleic Acids 33(7):489–506
Tanitame A, Oyamada Y, Ofuji K, Fujimoto M, Suzuki K, Ueda T, Terauchi H, Kawasaki M, Nagai K, Wachi M, Yamagishi J (2004) Synthesis and antibacterial activity of novel and potent DNA gyrase inhibitors with azole ring. Bioorgan Med Chem 12(21):5515–5524
Ryu CK, Lee RY, Kim NY, Kim YH, Song AL (2009) Synthesis and antifungal activity of benzo[d]oxazole-4,7-diones. Bioorgan Med Chem Lett 19(20):5924–5926
Kumar A, Ahmad P, Maurya RA, Singh AB, Srivastava AK (2009) Novel 2-aryl-naphtho [1,2-d]oxazole derivatives as potential PTP-1B inhibitors showing antihyperglycemic activities. Eur J Med Chem 44(1):109–116
Kumar D, Kumar NM, Sundaree S, Johnson EO, Shah K (2010) An expeditious synthesis and anticancer activity of novel 4-(3´-indolyl)oxazole. Eur J Med Chem 45(3):1244–1249
Moraski GC, Markley LD, Chang M, Cho S, Franzblau SG, Hwang CH, Boshoff H, Miller MJ (2012) Generation and exploration of new classes of antitubercular agents: the optimization of oxazolines, oxazoles, thiazolines, thiazoles to imidazo[1,2-a]pyridines and isomeric 5,6-fused scaffolds. Bioorgan Med Chem 20(7):2214–2220
Jadhav RD, Kadam KS, Kandre S, Guha T, Reddy MMK, Brahma MK, Deshmukh NJ, Dixit A, Doshi L, Potdar N, Enose AA, Vishwakarma RA, Sivaramakrishnan H, Srinivasan S, Nemmani KVS, Gupte A, Gangopadhyay AK, Sharma R (2012) Synthesis and biological evaluation of isoxazole, oxazole and oxadiazole containing heteroaryl analogs of biarylureas as DGAT1 inhibitors. Eur J Med Chem 54:324–342
Parveen M, Ali A, Ahmed S, Malla AM, Alam M, Silva PSP, Silva MR, Lee DU (2013) Synthesis, bioassay, crystal structure and ab initio studies of Erlenmeyer azlactones. Spectrochim Acta A Mol Biomol Spectrosc 104:538–545
Jin C, Manikumar G, Kepler JA, Cook CE, Allan GF, Kiddoe M, Bhattacharjee S, Linton O, Lundeen SG, Sui Z (2007) Synthesis and identification of novel 11β-aryl-4´,5´-dihydrospiro[estra-4,9-diene-17β,4´-oxazole] analogs with dissociated antiprogesterone activities. Bioorgan Med Chem Lett 17:5754–5757
Brescia MR, Rokosz LL, Cole AG, Stauffer TM, Lehrach JM, Auld DS, Henderson I, Webb ML (2007) Discovery and preliminary evaluation of 5-(4-phenylbenzyl)oxazole4-carboxamides as prostacyclin receptor antagonists. Bioorgan Med Chem Lett 17(5):1211–1215
Lee JE, Koh HY, Seo SH, Baek YY, Rhim H, Cho YS, Choo H, Pae AN (2010) Synthesis and biological evaluation of oxazole derivatives as T-type calcium channel blockers. Bioorgan Med Chem Lett 20(14):4219–4222
Razavi H, Powers ET, Purkey HE, Adamski-Werner SL, Chiang KP, Dendle MTA, Kelly JW (2005) Design, synthesis, and evaluation of oxazole transthyretin amyloidogenesis inhibitors. Bioorgan Med Chem Lett 15(4):1075–1078
Davis A, Ward SE (2015) The handbook of medicinal chemistry: principles and practice. Royal Soc Chem 1:642
Mussoni L, Poggi A, De Gaetano G, Donati MB (1978) Effect of ditazole, an inhibitor of platelet aggregation, on a metastasizing tumour in mice. Br J Cancer 37(1):126–129
Lednicer VD, Mitscher LA (1977) The organic chemistry of drug synthesis. Wiley, New York, p 1
Greenblatt DJ, Matlis R, Scavone JM, Blyden GT, Harmatz JS, Shader RT (1985) Oxaprozin pharmacokinetics in the elderly. Br J Clin Pharmacol 19:373–378
Kumar S, Lim SM, Ramasamy K, Vasudevan M, Shah SAA, Selvaraj M, Narasimhan Balasubramanian (2017) Synthesis, molecular docking and biological evaluation of bis-pyrimidine Schiff base derivatives. Chem Cent J 11(89):1–16
Horibata S, Rice EJ, Mukai C, Marks BA, Sams K, Zheng H, Anguish LJ, Coonrod SA, Danko Charles G (2018) ER-positive breast cancer cells are poised for RET-mediated endocrine resistance. PLoS ONE 13(4):1–21
Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, Warren JT, Bokesch H, Kenney S, Boyd MR (1990) New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst 82(13):1107–1112
Peyressatre M, Prével C, Pellerano M, Morris MC (2015) Targeting cyclin-dependent kinases in human cancers: from small molecules to peptide inhibitors. Cancer 7:179–237
Bassyouni F, El Hefnawi M, El Rashed A, Rehim MA (2017) Molecular modeling and biological activities of new potent antimicrobial, anti-inflammatory and anti-nociceptive of 5-nitro indoline-2-one derivatives. Drug Des 6(2):1–6
Sahu SK, Mishra A, Behera RK (1996) Synthesis of thiazole benzothiazole, oxadiazole, thiadiazole, triazole and thiazolidinone incorporated coumarins. Indian J Heterocy Ch. 6:91–94
Singh I, Kaur H, Kumar S, Kumar A, Lata S, Kumar A (2010) Synthesis of new coumarin derivatives as antibacterial agents. Int J Chem Tech Res 2(3):1745–1752
Cappuccino JC, Sherman N (1999) Microbiology-a laboratory manual. Addison Wesley, California, p 263
Pharmacopoeia of India (2007) Controller of publication, ministry of health department, vol 1. Govt. of India, New Delhi, p 37
Kaur R, Kaur P, Sharma S, Singh G, Mehndiratta S, Bedi PM, Nepali K (2015) Anti-cancer pyrimidines in diverse scaffolds: a review of patent literature. Recent Pat Anti-Canc 10(1):23–71
Amin KM, Awadalla FM, Eissa AAM, Abou- Seri AM, Hassan GS (2011) Design, synthesis and vasorelaxant evaluation of novel coumarin-pyrimidine hybrids. Bioorg Med Chem 19:6087–6097
Singh J, Kumar M, Mansuri R, Sahoo GC, Deep A (2016) Inhibitor designing, virtual screening and docking studies for methyltrans-ferase: a potential target against dengue virus. J Pharm Bioallied Sci 8(3):188–194
Zhang W, Liu W, Jiang X, Jiang F, Zhuang H, Fu L (2011) Design, synthesis and antimicrobial activity of chiral 2-(substituted-hydroxyl)-3-(benzo[d]oxazol-5-yl)propanoic acid derivatives. Eur J Med Chem 46(9):3639–3650
Singh I, Kaur H, Kumar S, Lata S, Kumar A, Kumar A (2010) Synthesis and antibacterial activity of 3-chloro 4-(substitutedphenyl) azetidinonyl/thiazolidinonyl-4-(3-acetanilido) oxa/thiazoles. Int J Pharm Sci Res 1(2):148–168
Ohnmacht SA, Micco M, Petrucci V, Todd AK, Reszka AP, Gunaratnam M, Carvalho MA, Zloh M, Neidle S (2012) Sequences in the HSP90 promoter form G-quadruplex structures with selectivity for disubstituted phenyl bis-oxazole derivatives. Bioorg Med Chem Lett 22(18):5930–5935
Authors’ contributions
Authors BN, SK and SK have designed synthesized and carried out the antimicrobial activity and SML, KR, VM and SAAS have carried out the spectral analysis, interpretation and cytotoxicity study of synthesized compounds. All authors read and approved the final manuscript.
Acknowledgements
The authors are thankful to Head, Department of Pharmaceutical Sciences, Maharshi Dayanand University, Rohtak, for providing necessary facilities to carry out this research work.
Competing interests
The authors declare that they have no competing interests.
Ethics approval and consent to participate
Not applicable.
Funding
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Kakkar, S., Kumar, S., Lim, S.M. et al. Design, synthesis and biological evaluation of 3-(2-aminooxazol-5-yl)-2H-chromen-2-one derivatives. Chemistry Central Journal 12, 130 (2018). https://doi.org/10.1186/s13065-018-0499-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13065-018-0499-x