
Abstract
Masitinib (MST) is an orally administered drug that targets mast cells and macrophages, important cells for immunity, by inhibiting a limited number of tyrosine kinases. It is currently registered in Europe and USA for the treatment of mast cell tumors in dogs. AB Science announced that the European Medicines Agency has accepted a conditional marketing authorization application for MST to treat amyotrophic lateral sclerosis. In our work, we focused on studying in vivo metabolism of MST in Sprague–Dawley rats. Single oral dose of MST (33 mg kg−1) was given to Sprague–Dawley rats (kept in metabolic cages) using oral gavage. Urine was collected and filtered at 0, 6, 12, 18, 24, 48, 72 and 96 h from MST dosing. An equal amount of ACN was added to urine samples. Both organic and aqueous layers were injected into liquid chromatography-tandem mass spectrometry (LC–MS/MS) to detect in vivo phase I and phase II MST metabolites. The current work reports the identification and characterization of twenty in vivo phase I and four in vivo phase II metabolites of MST by LC–MS/MS. Phase I metabolic pathways were reduction, demethylation, hydroxylation, oxidative deamination, oxidation and N-oxide formation. Phase II metabolic pathways were the direct conjugation of MST, N-demethyl metabolites and oxidative metabolites with glucuronic acid. Part of MST dose was excreted unchanged in urine. The literature review showed no previous articles have been made on in vivo metabolism of MST or detailed structural identification of the formed in vivo phase I and phase II metabolites.

Similar content being viewed by others

Introduction
Cancer became a major reason of death [1]. More than four millions new cancer cases reported in developed countries [2, 3]. Molecular targeting strategies were used to treat distributed cancer depending on identifying the tumor suppressors and oncogenes involved in the progress of human cancers [4]. Tyrosine kinase inhibitors (TKIs) (e.g. masitinib) are compounds that target tyrosine kinases enzymes, which are responsible for the activation of numerous proteins in a number of cell signaling pathways. They initiate or stop many functions inside living cells [5]. Blocking the selected activation of these proteins has been shown to have therapeutic benefits in cancer diseases and central nervous system disorders mast cells and macrophages [6, 7]. Tyrosine kinase inhibitors (TKIs) are considered a very important class of targeted therapy [8].
MST (Fig. 1) is new orally administered TKIs. It is already registered in Europe and USA for the treatment of mast cell tumors in dogs [9]. MST is approved under the trade name masivet in Europe and Kinavet in the USA at a dose of 12.5 mg kg−1 per day [10]. Toxicity profile of MST is lower than other TKIs [11]. MST selectively inhibits c-kit tyrosine kinase blocking stem cell factor induced proliferation. It exhibits more activity and selectivity against KIT than imatinib in in vitro studies [11]. In 3 October 2016, AB Science announced that the EMA has accepted a conditional marketing authorization application for MST to treat ALS in human. MST found to be effective for the treatment of severely symptomatic indolent or smouldering systemic mastocytosis [12].

Chemical structure of MST
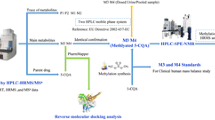
Drug metabolism research is an integral part of the drug discovery process and is very often the factor that determines the success of a given drug to be marketed and clinically used [13]. Drug metabolism research is generally conducted using in vitro and/or in vivo techniques. In vitro techniques involve the incubation of drugs with different types of in vitro preparations (e.g. liver microsomes, hepatocytes) isolated from rats and subsequent sample processing and analysis using spectroscopic techniques [14, 15]. In vivo techniques involve the administration of a single dose of the drug to rat, and the subsequent collection of urine that contain the drugs and their potential metabolites. In this work, we focused in the in vivo phase I metabolites and in vivo phase II MST metabolites identification using LC–MS/MS [16]. All measurements were done using Agilent LC–MS/MS system that consisted of LC (Agilent HPLC 1200) coupled to MS/MS detector (6410 QqQ MS) through an electrospray ionization source (Agilent Technologies, USA) [17].
MST chemical structure contains cyclic tertiary amine. Phase I metabolism of cyclic tertiary amines produces metabolites of oxidative products including N-dealkylation, ring hydroxylation, α-carbonyl formation, N-oxygenation, and ring opening metabolites that can be formed through iminium ion intermediates [18, 19].
Chemicals and methods
Chemicals
All chemicals are listed in Table 1.
In vivo metabolism of MST in Sprague–Dawley Rats
Rat dosing protocol
Male Sprague–Dawley rats (n = 6, average: 340 g, 4 weeks of age) were housed individually in special purpose metabolism cages. Cages are placed in the animal care facility in a 12 h light/dark cycle (7:00–19:00) and were allowed free access to standard animal feed and water that were placed in the special food and water compartments attached to the metabolism cages. Rats were acclimated in metabolism cages for 72 h prior to the start of the study. MST was formulated in (4% DMSO, 30% PEG 300, 5% Tween 80, HPLC H2O) for oral dosing of rats. Doses were individually calculated for each rat such that everyone receives a specific dose. The average dose of MST (Kinavet-CA1) in dogs was 10 mg kg−1. By using the following equations [20,21,22]:
So the dose for each rat was 33.3 mg/kg. All rats except one were given a single dose of MST. All MST doses were administered by oral gavage. Urine draining into the special urine compartments fitted to the metabolism cages were collected prior to drug dosing as blank control reference and at 6, 12, 18, 24, 48, 72 and 96 h following MST dosing. Urine samples taken from all metabolism cages were pooled together, labeled, and stored at (− 20 °C).
Sample preparation
Urine samples were thawed to room temperature and filtered over 0.45 µm syringe filters. Liquid liquid extraction (LLC) was used to extract MST and its related metabolites. Equal volume of ice cold acetonitrile (ACN) was added to each sample then vigorously shaken by vortexing for 1 min. Phase separation [23, 24] between an aqueous sample and a water-miscible solvent (ACN) into two layers achieved by using ice cold ACN that was added to urine and the mixture was stored at 4 °C overnight [25]. Low temperature leads to phase separation of ACN/urine mixture. The pH of urine and the nature of urine matrix which contains high concentration of salt participated in phase separation [26]. As we did not want to miss any MST-related metabolites, both layers were removed and evaporated to dryness under stream of nitrogen. The dried extracts were reconstituted in 1 mL of mobile phase and transferred to 1.5 mL HPLC vials for LC–MS/MS analysis. Control urine samples obtained from rats prior to drug dosing were prepared in the exact way described for each method of sample purification.
LC–MS/MS conditions
The LC–MS/MS parameters optimized for chromatographic separation and identification of rat urine extract components are listed in Table 2.
Identification of in vivo MST metabolites
MST-related metabolites were concentrated in the ACN layer while endogenous urine components and polar metabolites (e.g. glucuronide conjugates) were found in the aqueous layer. Extracted ion chromatograms for the expected metabolites were used to find metabolites in the total ion chromatogram of both organic and aqueous layers. PI studies were for the suspected compounds and results were interpreted and compared with the PI of MST. Mass scan and PI scan modes of the triple quadrupole mass analyzer were used for detection of in vivo phase I and phase II MST metabolites. PI mass spectra were used to propose the metabolite chemical structure by reconstructing the marker daughter ions.
Results and discussion
Identification of in vivo phase I metabolic pathways of MST
The in vivo metabolites of MST underwent fragmentations similar to that of the parent ion that allowed us to identify and determine changes in the metabolite structures. The product ion mass spectra of some metabolites exhibited particular fragmentation pathways that provided more structural information as shown below. Comparison of PI mass spectra between urine extracts with control samples in addition to the comparison of PI of MST and its anticipated metabolites (Table 3) resulted in the detection of twenty in vivo phase I and four phase II metabolites (Fig. 2). Ten in vivo phase I metabolites are reported in the case of in vitro metabolism [27]. We concentrated on the structural identification of the new ten in vivo phase I and the other four in vivo phase II MST metabolites. Metabolic pathways for in vivo phase I metabolites were supposed to be N-demethylation, N-oxide formation, oxidation, oxidative deamination, reduction, oxidative cleavage, benzyl oxidation and hydroxylation while for phase II metabolites were N-conjugation of MST and the N-demethyl metabolite with glucuronic acid and oxidative metabolites glucuronidation.

PI chromatograms: a (MST), b (M1), c (M2–M4), d (M5–M7), e (M8–M10) and f (MO1–MO6)
MST excretion of in rat urine
Part of the MST oral dose was excreted unmetabolized in rat urine. MST parent ion was detected at m/z 499 in full mass scan spectrum. MST of and its major in vivo metabolites (M1 and MO6) excretion in urine was observed after 6 h of dosing. Comparative concentrations of MST, M1 and MO6 were high after 6 h and then began to decline by time until almost vanished after 96 h from dosing as shown in the overlayed PI chromatograms (Check Additional file 1). Peak area ratios of MST and its major metabolite (M1 and MO6) in urine were plotted against time. Peak area ratio of each MST, M1 and MO6 were measured at different collection time considering the biggest peak is 100% (Fig. 3) [28].

MST, M1 and MO6 excretion rate
Fragmentation of MST (Fig. 4) was explained in Scheme 1. Comparison of PI of MST with suspected peaks allowed the identification of metabolic changes in the supposed in vivo metabolites.

PI of MST parent ion at m/z 499

Supposed PI of MST
M1 in vivo phase I metabolite
The major metabolic pathway for MST is N-demethyalation. M1 was detected at m/z 485 in mass scan spectrum.
M2, M3 and M4 in vivo phase I metabolite
M2, M3 and M4 were detected at m/z 501 at different retention times in mass scan spectrum of organic urine extract. PI scan for the three metabolites gave different daughter ions. In the case of M2, parent ion at m/z 501 was fragmented to one ion at m/z 401. The daughter ion at m/z 401 supposed that there is no change in the methyl piperazine group. The metabolic pathway for M2 metabolite was supposed to be the reduction of the carbonyl group.
In the case of M3, parent ion at m/z 501 was fragmented to ions at 400.2 and 367.2 (Fig. 5). Metabolic pathways for M3 were supposed to be hydroxylation of pyridine ring and N-demethylation (Scheme 2).

PI mass spectrum of parent ion (M3) at m/z 502

Supposed PIs of M3
In the case of M4, parent ion at m/z 501 was fragmented to two daughter ions at m/z 483 and at m/z 399 (Fig. 6). The daughter ion at m/z 399 supposed that there all metabolic changes occured in the methyl piperazine group. Metabolic pathways for M4 metabolite were hydroxylation and N-demethylation of N-methyl piperazine (Scheme 3).

PI mass spectrum of parent ion (M4) at m/z 501

Supposed PIs of M4
MO1 to MO6 in vivo phase I metabolite
Oxidized MST metabolite (M + O) was detected at m/z 515 in mass scan spectrum at different retention times. Fragmentation of parent ions at m/z 515 gave different daughter ions as shown in the Table 3. The structure of each metabolite was supposed The metabolic pathway for MO metabolites was supposed to be either by hydroxylation or N-oxidation of MST [27].
M5, M6 and M7 in vivo phase I metabolite
M5, M6 and M7 metabolites were detected at m/z 529 in full mass scan spectrum at different retention times. PI scan for parent ions at m/z 529 gave different daughter ions. In the case of M5, parent ion at m/z 529 was fragmented to ions at m/z 511 and at m/z 429 (Fig. 7). The metabolic pathway for M5 was supposed to be benzyl oxidation to carboxylic acid (Scheme 4).

PI mass spectrum of parent ion (M5) at m/z 529

Supposed PIs of M5
In the case of M6, parent ion at m/z 529 was fragmented to ions at 486 and 400 (Fig. 8). The metabolic pathway for M6 was supposed to be hydroxylation and oxidation of methyl piperazine ring (Scheme 5).

PI mass spectrum of parent ion (M6) at m/z 529

Supposed PIs of M6
In the case of M7, parent ion at m/z 529 was fragmented to ions at 511, 399 and 98 (Fig. 9). Metabolic pathways for M7 were supposed to be hydroxylation and oxidation of methyl piperazine ring (Scheme 6).

PI mass spectrum of parent ion (M7) at m/z 529

Supposed PIs of M7
M8, M9 and M10 in vivo phase I metabolite
M8, M9 and M10 metabolites were detected at m/z 531 in full mass scan spectrum at different retention times. PI scan for parent ions at m/z 531 gave different daughter ions. In the case of M8, parent ion at m/z 531 was fragmented to ions at 488, 402 and 123 (Fig. 10). Metabolic pathways for M8 were supposed to be hydroxylation of pyridine and hydroxylation of methyl piperazine ring (Scheme 7).

PI mass spectrum of parent ion (M8) at m/z 531

Supposed PIs of M8
In the case of M9, parent ion at m/z 531 was fragmented to ions at 513, 415, 381 and 123 (Fig. 11). Metabolic pathways for M9 were supposed to be benzyl hydroxylation and hydroxylation of methyl piperazine ring (Scheme 8).

PI mass spectrum of parent ion (M9) at m/z 531

Supposed PIs of M9
In the case of M10, parent ion at m/z 531 was fragmented to ions at 501 and 401 (Fig. 12). Metabolic pathways for M10 were supposed to be oxidative cleavage of N-methyl piperazine ring to carboxylic acid (Scheme 9).

PI mass spectrum of parent ion (M10) at m/z 531

Supposed PIs of M10
M11 in vivo phase I metabolite
M11 was detected at m/z 547 in mass scan spectrum of the urine organic extract. PI chromatogram of urine organic extract at m/z 547 showed one peak at 30.72 min. PI scan for M11 at m/z 547 gave daughter ions at m/z 511. Metabolic reactions for M11 metabolite were supposed to be hydroxylation of benzylic carbon, oxidation of pyridine nitrogen and oxidation of piperazine nitrogen.
In vivo phase I oxidative deamination metabolic pathway (MA1, MA2 and MA3)
The loss of the piperazine moiety by oxidative deamination and rapid further oxidation of the intermediate aldehyde to a carboxylic acid metabolite were observed for MA1, MA2 and MA3 in the aqueous layer of the urine/ACN mixture. Fragmentation of parent ions at m/z 431 and at m/z 447 gave different daughter ions. The structure of each metabolite was supposed.
MA1 was detected at m/z 431 in mass scan spectrum of the aqueous layer urine extract. PI chromatogram of urine aqueous extract at m/z 431 showed one peak at 10.2 min. PI scan for MA1 at m/z 431 gave daughter ions at m/z 255 (Fig. 13). The daughter ion at m/z 255 supposed the loss of the piperazine moiety by oxidative deamination and rapid further oxidation of the intermediate aldehyde to a carboxylic acid (Scheme 10).

PI mass spectrum of parent ion (MA1) at m/z 431

Supposed PIs of MA1
MA2 and MA3 were detected at m/z 447 in mass scan spectrum of the aqueous layer urine extract. PI chromatogram of urine aqueous extract at m/z 447 showed two peaks at 18.6 and 19.5 min. PI scan for MA2 and MA3 at m/z 447 gave different daughter ions at two different retention times (Figs. 14 and 15).

PI mass spectrum of parent ion (MA2) at m/z 447

PI mass spectrum of parent ion (MA3) at m/z 447
In the case of MA2, the daughter ion at m/z 271 supposed the loss of the piperazine moiety by oxidative deamination and rapid further oxidation of the intermediate aldehyde to a carboxylic acid in addition to phenyl hydroxylation (Scheme 11).

Supposed PIs of MA2
In the case of MA3, the daughter ion at m/z 271 supposed the loss of the piperazine moiety by oxidative deamination and rapid further oxidation of the intermediate aldehyde to a carboxylic acid. The other daughter ion at m/z 285 supposed benzyl hydroxylation (Scheme 12).

Supposed PIs of MA3
Identification of in vivo phase II metabolic pathways of MST
Phase II metabolic pathways were supposed to be N-conjugation of MST and the N-demethyl metabolite with glucuronic acid, and glucuronidation of oxidative metabolites (Table 4). Phase II metabolites were found in the aqueous layer of the rat urine extract in a very small concentration compared to in vivo phase I metabolites. Excretion of all in vivo phase II metabolites in urine was observed after 12 h of rat dosing and disappeared rapidly after 48 h of rat dosing.
MG1 in vivo phase II metabolite
MG1 was detected at m/z 675 in mass scan spectrum of the aqueous layer urine extract. PI chromatogram of urine aqueous extract at m/z 675 showed one peak at 18.9 min. PI scan for MG1 at m/z 675 gave daughter ions at m/z 499 and 399 (Fig. 16). The daughter ion at m/z 399 supposed that direct N-conjugation of MST with glucuronic. The other daughter ion at 499 refers to the aglycone (MST) formed in the triple quadrupole by the loss of anhydroglucuronic acid (Scheme 13).

PI mass spectrum of parent ion (MG1) at m/z 675

Supposed PIs of MG1
MG2 in vivo phase II metabolite
MG2 was detected at m/z 661 in mass scan spectrum of the aqueous layer urine extract. PI chromatogram of urine aqueous extract at m/z 661 showed one peak at 18.7 min. PI scan for MG2 at m/z 661 gave daughter ions at m/z 485 (Fig. 17). The daughter ion at 485 refers to the aglycone (N-demethyl MST) formed in the triple quadrupole by the loss of anhydroglucuronic acid (Scheme 14).

PI mass spectrum of parent ion (MG2) at m/z 661

Supposed PIs of MG2
MG3 and MG4 in vivo Phase II metabolites
MG3 and MG4 were detected at m/z 691 in mass scan spectrum of the aqueous layer urine extract. PI chromatogram of urine aqueous extract at m/z 691 showed two peaks at 18.6 and 19.5 min. PI scan for MG3 and MG4 at m/z 691 gave different daughter ions at two different retention times (Figs. 18, 19).

PI mass spectrum of parent ion (MG3) at m/z 691

PI mass spectrum of parent ion (MG4) at m/z 691
In the case of MG3, the daughter ion at m/z 515 supposed that direct O-glucuronidation of hydroxy MST. The daughter ion at 515 refers to the aglycone (hydroxy MST) formed in the triple quadrupole by the loss of anhydroglucuronic acid. (Scheme 15). Hydroxylation was supposed to be in the N-methyl piperazine ring. In the case of MG4, the daughter ion at m/z 515 supposed that direct O-glucuronidation of hydroxy MST. The daughter ion at 515 refers to the aglycone (hydroxy MST) formed in the triple quadrupole by the loss of anhydroglucuronic acid (Scheme 16). The other daughter at m/z 415 supposed that the hydroxylation of benzyl carbon.

Supposed PIs of MG3

Supposed PIs of MG4
Conclusions
MST was excreted partially unchanged in rat urine. Twenty in vivo phase I metabolites were formed by oral dosing of MST to Sprague–Dawley rats through six metabolic pathways: N-demethylation, N-oxidation, oxidation, reduction, hydroxylation and oxidative deamination. Four in vivo phase II glucuronide conjugates were found in the aqueous layer of rat urine extract (Fig. 20).

Chemical structure of MST and identified metabolic pathways in Rat. The main metabolic pathway was the N-demethylation
References
Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T et al (2008) Cancer statistics, 2008. CA Cancer J Clin 58(2):71–96
Sinha R, El-Bayoumy K (2004) Apoptosis is a critical cellular event in cancer chemoprevention and chemotherapy by selenium compounds. Curr Cancer Drug Targets 4(1):13–28
Cozzi P, Mongelli N, Suarato A (2004) Recent anticancer cytotoxic agents. Curr Med Chem Anti Cancer Agents 4(2):93–121
Barinaga M (1997) From bench top to bedside. Science 278(5340):1036–1039
Schlessinger J (2000) Cell signaling by receptor tyrosine kinases. Cell 103(2):211–225
Özvegy-Laczka C, Cserepes J, Elkind NB, Sarkadi B (2005) Tyrosine kinase inhibitor resistance in cancer: role of ABC multidrug transporters. Drug Resist Updates 8(1):15–26
Steeghs N, Nortier JW, Gelderblom H (2007) Small molecule tyrosine kinase inhibitors in the treatment of solid tumors: an update of recent developments. Ann Surg Oncol 14(2):942–953
Natoli C, Perrucci B, Perrotti F, Falchi L, Iacobelli S (2010) Tyrosine kinase inhibitors. Curr Cancer Drug Targets 10(5):462–483
Daly M, Sheppard S, Cohen N, Nabity M, Moussy A, Hermine O et al (2011) Safety of masitinib mesylate in healthy cats. J Vet Intern Med 25(2):297–302
Marech I, Patruno R, Zizzo N, Gadaleta C, Introna M, Zito AF et al (2014) Masitinib (AB1010), from canine tumor model to human clinical development: where we are? Crit Rev Oncol Hematol 91(1):98–111
Dubreuil P, Letard S, Ciufolini M, Gros L, Humbert M, Castéran N et al (2009) Masitinib (AB1010), a potent and selective tyrosine kinase inhibitor targeting KIT. PLoS ONE 4(9):e7258
Lortholary O, Chandesris MO, Livideanu CB, Paul C, Guillet G, Jassem E et al (2017) Masitinib for treatment of severely symptomatic indolent systemic mastocytosis: a randomised, placebo-controlled, phase 3 study. Lancet 389(10069):612–620
Kumar GN, Surapaneni S (2001) Role of drug metabolism in drug discovery and development. Med Res Rev 21(5):397–411
Kadi AA, Al-Shakliah NS, Yin W, Rahman AM (2017) In vitro investigation of metabolic profiling of newly developed topoisomerase inhibitors (ethyl fluorescein hydrazones, EtFLHs) in RLMs by LC–MS/MS. J Chromatogr B 1054:93–104
Rahman AM, Al-Shakliah NS, Yin W, Kadi AA (2017) In vitro Investigation of Metabolic Profiling of a Potent Topoisomerase Inhibitors Fluorescein Hydrazones (FLHs) in RLMs by LC-MS/MS. J Chromatogr B 1054:27–35
Attwa MW, Kadi AA, Darwish HW, Alrabiah H (2018) LC-MS/MS reveals the formation of reactive ortho-quinone and iminium intermediates in saracatinib metabolism: phase I metabolic profiling. Clin Chim Acta 482:84–94
Attwa MW, Kadi AA, Darwish HW, Amer SM, Alrabiah H (2018) A reliable and stable method for the determination of foretinib in human plasma by LC-MS/MS: application to metabolic stability investigation and excretion rate. Eur J Mass Spectrom 1469066718768327. https://doi.org/10.1177/1469066718768327
Masic LP (2011) Role of cyclic tertiary amine bioactivation to reactive iminium species: structure toxicity relationship. Curr Drug Metab 12(1):35–50
Kadi AA, Attwa M, Darwish HW (2018) LC-ESI-MS/MS reveals the formation of reactive intermediates in brigatinib metabolism: elucidation of bioactivation pathways. RSC Adv 8(3):1182–1190
Shin J-W, Seol I-C, Son C-G (2010) Interpretation of animal dose and human equivalent dose for drug development. J Korean Med 31(3):1–7
Nair AB, Jacob S (2016) A simple practice guide for dose conversion between animals and human. J Basic Clin Pharm 7(2):27–31
Reagan-Shaw S, Nihal M, Ahmad N (2008) Dose translation from animal to human studies revisited. FASEB J Off Publ Fed Am Soc Exp Biol 22(3):659–661 (Epub 2007/10/19)
Varaprath S, Salyers KL, Plotzke KP, Nanavati S (1999) Identification of metabolites of octamethylcyclotetrasiloxane (D(4)) in rat urine. Drug Metab Dispos Biol Fate Chem 27(11):1267–1273
Bylda C, Thiele R, Kobold U, Volmer DA (2014) Recent advances in sample preparation techniques to overcome difficulties encountered during quantitative analysis of small molecules from biofluids using LC–MS/MS. Analyst 139(10):2265–2276
Yoshida M, Akane A (1999) Subzero-temperature liquid − liquid extraction of benzodiazepines for high-performance liquid chromatography. Anal Chem 71(9):1918–1921
Valente IM, Gonçalves LM, Rodrigues JA (2013) Another glimpse over the salting-out assisted liquid–liquid extraction in acetonitrile/water mixtures. J Chromatogr A 1308:58–62
Amer S, Kadi AA, Darwish HW, Attwa MW (2017) Identification and characterization of in vitro phase I and reactive metabolites of masitinib using a LC-MS/MS method: bioactivation pathway elucidation. RSC Adv 7(8):4479–4491
Amer SM, Kadi AA, Darwish HW, Attwa MW (2017) LC–MS/MS method for the quantification of masitinib in RLMs matrix and rat urine: application to metabolic stability and excretion rate. Chem Cent J 11(1):136
Authors’ contributions
AK, SA, HD and MA established the experiment design. Practical work was performed by MA. Data were analyzed by AK, HD, SA and MA. HD and MA wrote the first draft of the manuscript. AK and SA contributed in editing the manuscript. AK, SA and HD supervised the research work. All authors read and approved the final manuscript.
Acknowledgements
The authors would like to extend their sincere appreciation to the Deanship of Scientific Research at the King Saud University for funding this work through the Research Group Project No. RGP-322.
Competing interests
The authors declare that they have no competing interests.
Data availability
All data supporting the results in this article can be found in the manuscript or the Additional file.
Ethics approval and consent to participate
Animal Care Center guidelines of Pharmacy College at King Saud Univesity were applied for Rats’ maintenance. The Local Animal Care and Use Committee at KSU approved these guidelines.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional file
Additional file 1.
Additional figures.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Kadi, A.A., Amer, S.M., Darwish, H.W. et al. Characterization of in vivo metabolites in rat urine following an oral dose of masitinib by liquid chromatography tandem mass spectrometry. Chemistry Central Journal 12, 61 (2018). https://doi.org/10.1186/s13065-018-0429-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13065-018-0429-y