
Abstract
Background
Septic shock remains a significant cause of death in critically ill patients. During septic shock, some patients will retain microcirculatory disorders despite optimal hemodynamic support (i.e., fluid resuscitation, vasopressors, inotropes). Alterations in the microcirculation are a key pathophysiological factor of organ dysfunction and death in septic shock patients. Ilomedin is a prostacyclin analog with vasodilatory effect and anti-thrombotic properties (i.e., inhibition of platelet aggregation) preferentially at the microcirculatory level. We hypothesize that early utilization of intravenous Ilomedin in septic shock patients with clinical persistence of microperfusion disorders would improve the recovery of organ dysfunction.
Methods
The I-MICRO trial is a multicenter, prospective, randomized, double-blinded, placebo-controlled study. We plan to recruit 236 adult patients with septic shock and persistent microcirculatory disorders (i.e., skin mottling or increased capillary refill time) despite hemodynamic support. Participants will be randomized to receive a 48-h intravenous infusion of either Ilomedin or placebo starting at the earliest 6 h and later 24 h after septic shock. The primary outcome will be the change (delta) of sequential organ failure assessment (SOFA) score between randomization and day 7. Secondary outcomes will include mean SOFA score during the first 7 days after randomization, mortality at day 28 post-randomization, number of ventilation-free survival days in the 28 days post-randomization, number of renal replacement therapy-free survival days in the 28 days post-randomization, number of vasopressor-free survival days in the 28 days post-randomization, and mottling score at day 1 after randomization.
Discussion
The trial aims to provide evidence on the efficacy and safety of Ilomedin in patients with septic shock and persistent microcirculatory disorders.
Trial registration
NCT NCT03788837. Registered on 28 December 2018
Similar content being viewed by others

Background
Septic shock remains a significant cause of death in critically ill patients [1]. Controlling the source of infection, appropriate and early antibiotics administration, and supportive care are the key elements of septic shock treatment [2]. Alterations in microcirculation are a critical pathophysiological factor of organ dysfunction and death in septic shock patients [3, 4]. Several clinical studies have revealed that microcirculatory disorders are associated with poor outcome in septic shock [5,6,7,8]. During septic shock, some patients will retain microcirculatory disorders despite optimal hemodynamic support (fluid resuscitation, vasopressors, inotropes). Preclinical data have furthermore strongly suggested that recruiting the microcirculation could improve organ perfusion and function in sepsis [4]. However, to date, no treatment specifically targeting the microcirculation has been shown to improve the patient’s outcome. Reasons for the microcirculatory defects are the release of local vasoconstrictive agents and intravascular microthrombi. Persistence of mottling, prolonged skin recoloration time, and cyanosis of the extremities are the easily and frequently observed manifestations of these microcirculatory disorders [9]. Ilomedin is a prostacyclin analog with a potent vasodilatory effect together with anti-thrombotic properties (inhibition of platelet aggregation) [10]. Improvement in mesenteric perfusion has, moreover, been observed in experimental sepsis using Ilomedin [11, 12]. Our group has reported that administration of Ilomedin in patients with refractory septic shock was associated with a rapid and sustained improvement in peripheral perfusion [13]. Altogether, Ilomedin may prevent organ dysfunction or improve its recovery in septic shock patients and ultimately improve outcomes. This study aims to assess the effect of Ilomedin infusion on organ function in septic shock patients with persistent peripheral hypoperfusion despite standard of care resuscitation.
Methods/design
Aim, design, and setting of the study
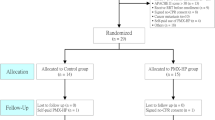
The study protocol is in accordance with the SPIRIT guidelines [14] (Fig. 1). I-MICRO is an academic, investigator-initiated multicenter (~ 25 centers), prospective, parallel-group (two groups), double-blinded, placebo-controlled, randomized trial in adults with septic shock admitted to an intensive care unit. This trial’s aim is whether the administration of Ilomedin could improve organ function compared to placebo. Exploratory secondary outcomes and adjusted analyses. A total of 236 evaluable patients with septic shock and persistence of microcirculation disorders (i.e., skin mottling or increased capillary refill time) will be enrolled in 25 centers with experience in the management of patients with septic shock (list of participating centers is in Appendix 1).

Study design. SOFA score, sequential organ failure assessment score; AE, adverse events; SAE, serious adverse events
The primary outcome is the difference between SOFA scores from randomization to day 7 in patients allocated to Ilomedin and in patients allocated to placebo.
The secondary outcomes are as follows:
-
Mortality at day 28
-
Mean SOFA score during the first 7 days after randomization
-
Number of survival days in the 28 days post-randomization
-
Number of ventilation-free survival days in the 28 days post-randomization
-
Number of renal replacement therapy-free survival days in the 28 days post-randomization
-
Number of vasopressor-free survival days in the 28 days post-randomization
-
Mottling score at day 1 after randomization
Characteristics of participants
All participants will be included and randomized by the clinician in charge of the patient. To be included, patients must meet all the following inclusion criteria:
-
1
Patients over 18 years of age with signed informed consent or inclusion under the emergency provisions of the law (Article L1122-1-3 of the PHC/modified by order no. 2016-800 of June 16, 2016 - art. 2)
-
2
Patients in septic shock defined by the third international definition [15]:
-
◦ Suspected or proven infection
-
◦ Organ dysfunction defined by an acute change in total SOFA score ≥ 2
-
◦ And serum lactate level > 2 mmol/L despite standard of care hemodynamic optimization
-
◦ Persistent hypotension requiring vasopressor treatment despite standard of care hemodynamic optimization and mean arterial pressure maintained > 65 mmHg
-
-
3
Persistence of peripheral hypoperfusion (skin mottling and/or finger skin recoloration time > 3 s and/or knee skin recoloration time > 4 s) within 6 to 24 h after norepinephrine onset
Patients will be excluded if any of the following criteria apply:
-
1
Refusal to participate in the study
-
2
Pregnancy and breastfeeding
-
3
Hypersensitivity to Ilomedin or any of the excipients
-
4
Conditions where the hemorrhagic risk may be increased due to the effects of Ilomedin on platelets (i.e., evolving hemorrhage, trauma, intracranial hemorrhage, active gastric ulcer)
-
5
Platelet count < 30,000/mm3
-
6
Unstable angina
-
7
Severe cardiac rhythm disorders since norepinephrine onset (ventricular fibrillation or tachycardia)
-
8
Severe hypoxemia (PaO2/FiO2 < 100)
-
9
Myocardial infarction in the last 6 months
-
10
Lack of social insurance
-
11
Persons deprived of liberty
The patients screened for randomization will present septic shock according to the new Sepsis-3 definition and signs of peripheral hypoperfusion (mottling score ≥ 2 and/or finger skin recoloration time > 3 s, and/or knee skin recoloration time > 4 s) as a surrogate parameter for microcirculatory hypoperfusion. A video has been shown to all investigators (Supplementary files 1 and 2). The study centers will treat patients according to the standard of care [2]. However, the following checklist for hemodynamic optimization concerning the standard of care before randomization:
-
1)
An initial fluid challenge should achieve a minimum of 30 mL/kg of crystalloids (a portion of this may be albumin equivalent).
-
2)
The fluid challenge technique will be applied wherein fluid administration is continued as long as there is a hemodynamic improvement based on dynamic (e.g., change in pulse pressure, stroke volume variation) or static (e.g., arterial pressure, heart rate) variables.
-
3)
Norepinephrine is titrated to target a mean arterial pressure of 65 mmHg in patients without a history of chronic hypertension and 75 mmHg in patients with a history of hypertension if the patient is not currently receiving renal replacement therapy.
-
4)
Dobutamine will be initiated if cardiac dysfunction is diagnosed (i.e., left ventricular ejection fraction < 40% on echocardiography and/or low cardiac index < 2.5 L/min/m2, increased filling pressure-wedge pressure > 18 mmHg on pulmonary artery catheter, and/or ScvO2 < 70% or SvO2 < 60% in a patient unresponsive to fluid challenge).
-
5)
Transfusion threshold will be a hemoglobin target of 7–8 g/dL
Randomization process
Patients in the trial treatment are allocated to treatment with Ilomedin or placebo in a ratio of 1:1. The randomization list will be performed at the CRU “Lariboisière-St Louis,” block-free, and stratified by center. The randomization list will be developed by a different biostatistician than the biostatistician who will conduct the final analysis within the CRU “Lariboisière- St Louis.”
Randomization is carried out by assigning treatment boxes in ascending order of the numbers available on site. Each center receives four treatment boxes with a randomization number on each box. Two new boxes will be sent to centers when they have recruited two participants. To randomize a patient and allocate treatment, the investigator takes the box with the smallest randomization number.
Ilomedin
The intervention is the intravenous administration of Ilomedin or to placebo (0.9% saline) via a central venous catheter. The perfusion will start at the first dose of 0.5 ng/kg/min with increments every 30 min up to a maximum of 1.5 ng/kg/min. The study drug or placebo is administered over 48 h, at the earliest 6 h after the diagnostic of septic shock (i.e., norepinephrine onset) and at last 24 h after the diagnostic.
Ilomedin is supplied in 50 μg/0.5 mL vials. A trained unblinded person reconstitutes the syringe with 50 mL of 0.9% saline to provide a ready-to-use solution. The final concentration of the drug substance Ilomedin is 1 μg/mL. Eight syringes are used for each patient (syringes are changed every 6 h, even if not empty).
The doses for each patient are calculated automatically according to the individual’s ideal body weight, using information from the electronic case report form (eCRF). The calculation results in an individual infusion rate for each level (i.e., 0.5, 1, and 1.5 ng/kg/min) for each patient. Patients, investigation site staff, site research coordinators, persons performing the assessments, the sponsor, central mottling pictures assessor, the staff in charge of treating the patients, and data managers will be blinded to the treatment allocation except for the independent, unblinded statistician approving the randomization scheme. The identification of treatment will be concealed by using a matching placebo to the study product that will be identical in packaging, labeling, and appearance. Unblinding will be requested for any reason considered essential by the investigating physician.
In systemic mean arterial pressure decreases below 65 mmHg after initiation of the treatment, we propose the following algorithm:
-
1.
The need for fluid loading will be assessed using dynamic parameters (i.e., pulse pressure or stroke volume variations when applicable, end-expiratory occlusion test) or fluid challenge.
-
2.
An increase of 25% in norepinephrine of baseline dose will be allowed. If systemic arterial hypotension persists, the infusion rate will be decreased to the previous dose (e.g., 1.5 to 1 ng/kg/min or 0.5 ng/kg/min to zero) every 15 min
In the case of profound hypotension (mean arterial pressure below 50 mmHg) for more than 5 min without identified etiology (e.g., sedation bolus), the treatment will be interrupted.
If hypoxemia occurs after introducing the treatment (or placebo), FiO2 can be increased to reach a SpO2 > 88–92%; if profound hypoxemia occurs (PaO2/FiO2 < 100) after increasing the infusion rate, the treatment will be decreased to the previous dose (e.g., 1.5 to 1 ng/kg/min or 0.5 ng/kg/min to zero) every 15 min.
In case of severe bleeding after randomization (active hemorrhage with hemodynamic instability), the treatment will be discontinued and not resumed.
If the patient requires emergency surgery, the treatment will be interrupted before transfer to the operating room. The treatment can be resumed 6 h after the end of surgery if peripheral hypoperfusion persists for a total duration of treatment (including the preoperative phase) of 48 h.
The administration period is followed by a clinical follow-up period of 28 days after randomization (Fig. 1).
No treatment will be prohibited concerning the research. However, due to the platelet anti-aggregate effect of Ilomedin, some concomitant treatments must be associated with precaution, especially anti-coagulants and platelet anti-aggregate treatments.
Data collection
Data on all patients will be collected by trained study nurses or physicians using a web-based e-CRF and stored in a secured server (Telemedicine Technologies, Boulogne-Billancourt, France). Data collected and time points are presented Table 1. Monitoring is performed by the clinical research organization and the sponsor.
All information required by the protocol will be provided in the case report form and an accompanying explanation given by the investigator for each missing data.
The data will be transferred in the case report forms as and when they are obtained, whether clinical, labs, or imaging. Erroneous data tracked on case report forms will be replaced by a declared investigator, which ensures the confidentiality of the data and authenticates the interventions.
Daily assessments (e.g., SOFA score) until day 7 and secondary outcomes will be performed by the respective center investigators. All data and other information generated will be held in strict confidence. The patients will be identifiable only by their initials and number. The Standard Protocol Items: Recommendation for Interventional Trials (SPIRIT) reporting guidelines are applied [14] (SPIRIT checklist is provided as supplementary File 3). Data from the trial will be made available to the reviewers upon request by the editor of the journal.
For ancillary studies, blood will be collected (15 mL) at the same time as the sample routinely collected within the 12 first hours after randomization. The aliquots previously labeled and stowed in the specific boxes for the study will be stored at − 80 °C, under the responsibility of the principal investigator of each center. Then, every 6 months, the aliquots will be shipped to the “Centre de Ressources Biologiques” (CRB) of the Lariboisière Hospital.
Statistical analysis
This trial’s primary aim is to demonstrate superiority in the intent-to-treat analysis of Ilomedin versus placebo on delta SOFAJ0–J7. The null hypothesis is that there are no differences in delta SOFAJ0–J7 between the two treatment groups.
Delta SOFAJ0–J7 will be compared between the two groups using the non-parametric Mann-Whitney test. The efficacy of Ilomedin will be considered if the null hypothesis for the primary endpoint is rejected and if the treatment difference is in favor of Ilomedin in the sense of a shift to higher delta SOFAJ0–J7.
We need to include N = 117 patients per group to have an 80% power to detect a difference between the two groups corresponding to a 1 point difference in delta SOFA between the two groups. Continuous variables will be summarized using the number of observations; mean; standard deviation; minimum; maximum; 25%, 50%, and 75% quartiles; and the two-sided 95% confidence intervals. Means, medians, minimum, maximum, and standard deviations will be presented to one further decimal place.
Categorical variables will be expressed as absolute and relative frequencies (percentages). Percentages will be rounded to one decimal place, and there may be occasions where the total of the percentages does not exactly equal 100%. Missing values will be imputed by multiple imputation techniques.
Data monitoring and interim analysis
A steering committee provides scientific direction for the trial and meets periodically to assess its operational progress every 9 months. It also provides scientific input and addresses policy issues regarding the protocol. The committee chair is responsible for communicating with the Data Safety and Monitoring Committee (DSMC) when appropriate. The Steering Committee is comprised of the coordinating investigator, Dr. François Dépret; scientific officer, Pr Matthieu Legrand; the statistician, Pr Eric Vicaut; a representative of the sponsor (AP-HP DRCI), Elodie Lemadre; and members selected on the basis of their outstanding expertise in the field and contribution to the study, Pr Daniel De Backer, Pr Bruno Levy, Pr Marc Leone, Pr Jacques Duranteau, and Pr Hafid Ait Oufella.
An independent DSMC monitors the quality of the trial and has access to the trial outcome and accumulated safety data, including serious adverse events (SAEs), suspected unexpected severe adverse reactions, and mortality. The composition of the DSMC is presented in Appendix 2. Besides, the DSMC will review the safety data from a clinical and safety point of view on an on-going basis.
An interim safety analysis will be scheduled once the first 50 patients have been included and have completed their 28-day clinical observation phase. The data analyst for the primary analyses will be blinded. An interim analysis will be made after evaluating 50% of patients by an unblinded statistician and reviewed by the DSMC based on clean data on the primary and secondary target variables and the latest status of safety data. This analysis will allow three possible recommendations of the DSMC to the sponsor:
-
Early stop of the study if the p value for the main criterion is less than p = 0.0035 (according to O’Brien-Fleming boundaries)
-
Sample size reassessment (only increase in sample size should be accepted (calculated from the ADDPLAN software))
-
Stopping for futility if the predictive power is less than 50%
The nominal alpha value for the interim analysis of the main criterion will be equal to 0.0035 and to 0.049 for the final analysis of this criterion (according to O’Brien-Fleming boundaries). All other tests will be two-sided at a 5% significance level.
The investigator will assess the severity of each adverse event and record all serious and non-serious adverse events in the case report form (Appendix 3). The investigator will also assess the potential causal relationship between the serious adverse events and the drug. Serious adverse events require a notification without delay by the investigator to the sponsor.
The sponsor will notify all the investigators any information that could adversely affect the safety of the participants. Assistance Publique Hôpitaux de Paris (AP-HP) is the sponsor of this study and has delegated power to its Clinical Research and Development Department (DRCD) to conduct the study under Article L.1121-1 of the French Public Health Code. AP-HP reserves the right to terminate the study at any time for medical or administrative reasons. In this case, the investigator will be informed accordingly.
In the case of lost to follow-up, the investigator will do his or her best to contact him or her in order to know whether he is alive. Mailing address, phone number, and phone number of at least one relative will be collected at inclusion to contact the patient. The contact details of the patient and the trusted person are recorded in the patient’s medical record and the eCRF. Furthermore, all patients are recorded in the French social security system and can be followed. If an individual leaves the research prematurely, participant data can be used unless an objection was recorded when the patient signed the consent form. If the consent was withdrawn, no data about the individual could be used unless stated otherwise. In practice, the participant is excluded from the research (Fig. 2).

CONSORT flow chart of the study
It should be noted that all amendments will be validated by the institutional review board. Once validated, the content of the amendments will be communicated to all investigators through contact emails, newsletters (on a three-month basis), and investigators’ meeting.
In accordance with good clinical practices, the sponsor is responsible for obtaining the permission of all parties involved in the research to guarantee direct access to all locations where the research will be carried out, to the source data, and to the source documents and the reports, with the goal of quality control and audit by the sponsor. Furthermore, the investigators will make available to those in charge of data monitoring, quality control, and audit relating to the biomedical research, the documents, and personal data strictly necessary for these controls, under the legislative and regulatory provisions in force (Articles L.1121-3 and R.5121-13 of the French Public Health Code).
Discussion
Septic shock with persistent peripheral perfusion disorders is associated with a very poor prognosis with no specific pharmaceutical therapies. The administration of Ilomedin has the potential to improve clinical outcomes through the prevention of organ failure. The I-MICRO trial aims to detect a beneficial effect of Ilomedin on organ function in patients with septic shock and persistent peripheral perfusion disorders or to provide the basis for an additional pivotal trial. Other important clinical outcomes, including survival will be explored as secondary endpoints. The design of this study lies in the identification of patients who are most likely to benefit from adjunctive treatment recruiting the microcirculation, i.e., remaining with altered perfusion despite standard of care. Mottling and increase capillary refilling time have previously been associated with alteration of the microcirculation in septic shock. Their observation is furthermore associated with poor outcomes. The ease of measuring these clinical symptoms without additional test or device makes them generalizable criteria to decide rescue strategies in sepsis. The results of this trial should provide essential and critical information on the feasibility, safety, and efficacy for preventing organ failure or for recovery from organ failure in this sub-population of septic shock with a very high risk of death. This trial will also provide essential data for designing a future larger clinical trial with mortality as the primary endpoint in this population.
Trial status
The current protocol is version 3.0, dated December 25, 2019. Recruitment began on July 3, 2019. The approximate completion date for recruitment is in July 2022. This trial was prospectively registered before the recruitment began. The study protocol was approved by the institutional review board (IRB) of Sud-Est V on October 30, 2018 (approval number 2018-001709-10) and from the Agence Nationale de Sécurité du Medicament et des Produits de santé (MEDAECNAT-2018-07-00015) and was registered in the clinical trial on December 28, 2018 (NCT03788837) and in EudraCT (N° 2018-001709-10) (Supplementary files 4 and 5). The Scientific director will be the first author, the principal investigator last author, and the methodologist the penultimate one. Each site principal investigator will be listed as an author from the second author based on the number of patients included on each site (if at least one patient is included from the site). All other investigators will be listed as collaborators.
Availability of data and materials
Data from the trial will be made available to the reviewers upon request by the editor of the journal.
Abbreviations
- SOFA:
-
Sequential organ failure assessment
- PaO2 :
-
Partial arterial oxygen pressure
- FiO2 :
-
Inspired fraction of oxygen
- eCRF:
-
Electronic case report form
- CRB:
-
Center de Ressources Biologiques
- DSMC:
-
Data and Safety Monitoring Committee
- APHP:
-
Assistance Publique Hôpitaux de Paris
- DRCD:
-
Clinical Research and Development Department
- SAE:
-
Serious adverse events
- IRB:
-
Institutional review board
References
Rudd KE, Johnson SC, Agesa KM, Shackelford KA, Tsoi D, Kievlan DR, et al. Global, regional, and national sepsis incidence and mortality, 1990–2017: analysis for the global burden of disease study. Lancet. 2020;395:200–11.
Levy MM, Evans LE, Rhodes A. The surviving sepsis campaign bundle: 2018 update. Intensive Care Med. 2018;44:925–8.
Legrand M, Ait-Oufella H, Ince C. Could resuscitation be based on microcirculation data? Yes. Intensive Care Med. 2018;44:944–6.
Legrand M, De Backer D, Dépret F, Ait-Oufella H. Recruiting the microcirculation in septic shock. Ann Intensive Care. 2019;9 Available from: https://annalsofintensivecare.springeropen.com/articles/10.1186/s13613-019-0577-9. Cited 2019 Dec 5.
Lima A, Jansen TC, van Bommel J, Ince C, Bakker J. The prognostic value of the subjective assessment of peripheral perfusion in critically ill patients. Crit Care Med. 2009;37:934–8.
Donati A, Damiani E, Domizi R, Scorcella C, Carsetti A, Tondi S, et al. Near-infrared spectroscopy for assessing tissue oxygenation and microvascular reactivity in critically ill patients: a prospective observational study. Crit Care. 2016;20:311.
Ait-Oufella H, Lemoinne S, Boelle PY, Galbois A, Baudel JL, Lemant J, et al. Mottling score predicts survival in septic shock. Intensive Care Med. 2011;37:801–7.
De Backer D, Donadello K. Assessment of microperfusion in sepsis. Minerva Anestesiol. 2015;81:533–40.
De Backer D, Ospina-Tascon G, Salgado D, Favory R, Creteur J, Vincent J-L. Monitoring the microcirculation in the critically ill patient: current methods and future approaches. Intensive Care Med. 2010;36:1813–25.
De Backer D. Is there a place for prostacyclin in the treatment of septic shock? Intensive Care Med. 2001;27:1110–2.
Rasmussen I, Arvidsson D, Zak A, Haglund U. Splanchnic and total body oxygen consumption in experimental fecal peritonitis in pigs: effects of dextran and iloprost. Circ Shock. 1992;36:299–306.
Manasia A, Kang H, Hannon E, Lu Y, Oropello J, Leibowitz A, et al. Effects of the stable prostacyclin analogue iloprost on mesenteric blood flow in porcine endotoxic shock. Crit Care Med. 1997;25:1222–7.
Dépret F, Sitbon A, Soussi S, De Tymowski C, Blet A, Fratani A, et al. Intravenous iloprost to recruit the microcirculation in septic shock patients? Intensive Care Med. 2018;44:121–2.
Chan A-W, Tetzlaff JM, Altman DG, Laupacis A, Gøtzsche PC, Krleža-Jerić K, et al. SPIRIT 2013 Statement: defining standard protocol items for clinical trials. Ann Intern Med. 2013;158:200.
Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (sepsis-3). JAMA. 2016;315:801.
Acknowledgements
To Morgane Gilg and Benedicte Rossignol from the INI-CRCT Network for their logistic support.
Collaborators: Antoine Kimmoun, MD, PhD; Alexandre Mebazaa, MD, PhD; Samuel Gaugain, MD; Jules Audard, MD; Jean-Yves Lefrant, MD, PhD; Bruno Megarbane, MD, PhD; Julien Pottecher, MD, PhD; Philippe Montravers, MD, PhD; Alexy Tran Dinh, MD, PhD; Romain Sonneville, MD, PhD; Thomas Rimmele, MD, PhD; Muriel Fartoukh, MD, PhD; Vincent Labbé, MD; Hervé Mentec, MD; Gaëtan Plantefeve MD; Anne-Laure Fedou, MD; Damien Barraud, MD; Hélène Nougue, MD, PhD; Stéphane Gaudry, MD, PhD; Carole Ichai, MD, PhD; Cécile Aubron, MD, PhD; Arnaud Mari, MD; Jean-Michel Constantin, MD, PhD
Funding
The study is funded by a grant from the Programme Hospitalier de Recherche Clinique - PHRC National 2017 from the French Minister of Health. The funding body has no role in the design of the study; collection, analysis, and interpretation of the data; and writing of the manuscript.
Author information
Authors and Affiliations
Consortia
Contributions
ML initiated the trial, contributed to the trial design, reviewed the manuscript, and is the scientific responsible and drafted the manuscript. HAO, DDB, JD, ML, BL, and PR are principal investigators at the highest recruiting sites. EV contributed to the design of the trial, drafted the statistical analysis concept of the trial, and contributed to the drafting of the manuscript. FD contributed to the design of the trial, reviewed the manuscript, is the principal investigator, and drafted the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This trial is conducted in patients who are potentially unable to consent. Therefore, patients can be included under the emergency provisions of the law (Article L1122-1-3 of the PHC/Order N° 2016-800 of June 16, 2016 - art. 2). Obviously, given the inclusion and non-inclusion criteria, the consent of patients or relatives, if present, cannot be collected at baseline. Thus, given the extreme emergency not allowing collecting the prior consent of the person or relative, the protocol provides that their consent is not necessary. In case of successful resuscitation, delayed consent will be asked to the patient for further participation. The participant shall be informed regarding the study during his or her hospitalization as soon as his or her condition allows. Thereafter, if the patient agrees, he or she signs the delayed consent form to pursue his or her participation in the research, and a “no objection to the use of his/her data” form for the possible continuation of this research will be completed.
According to the law (art. L1122-2 and L1121-8 of the Public Health Code), if the patient has a legal representative (guardian designated by law), it belongs to the patient’s guardian to give his or her consent for further participation to the research and the collection and the exploitation of the data.
The study protocol was approved by the institutional review board (IRB) of Sud-Est V on October 30, 2018 (approval number 2018-001709-10) and from the Agence Nationale de Sécurité du Medicament et des Produits de santé (MEDAECNAT-2018-07-00015) and was registered in the clinical trial on December 28, 2018 (NCT03788837) and in EudraCT (N° 2018-001709-10).
Consent for publication
As a clinical image or other clinical and personal data are not shown, consent for publication is not applicable.
Competing interests
ML reports consulting fees from Novartis, lecture fees from Baxter and Fresenius, and research support from Shingotec.
HAO declares no conflict of interest
DDB is a consultant to and material for studies by Edwards Lifesciences.
ML declares COIs with Amomed (consulting, lecture), Aguettant (consulting), MSD (consulting, lecture), Pfizer (lecture), Aspen (lecture), Orion (lecture), and Octapharma (lecture).
BL is a member of an advisory board working for Orion Pharma and has received honoraria from the company for his participation in the board and for giving invited lectures at industry symposia.
PR is a co-founder of CardioRenal and declares personal fees (consulting) from Novartis, NovoNordisk, Relypsa, AstraZeneca, Grünenthal, Idorsia, Stealth Peptides, Fresenius, Vifor Fresenius Medical Care Renal Pharma, Vifor, and Clinical Trials Mobile Application, and lecture fees from Bayer and CVRx.
EV declares no conflict of interest.
FD received lecture fees from Sedana medical, a research grant from the French ministry of health.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1:. Supplementary video 1.
Additional file 2:. Supplementary video 2.
Additional file 3:.
SPIRIT 2013 Checklist: Recommended items to address in a clinical trial protocol and related documents.
Additional file 4:.
Clinical trial authorization for medicinal products for human use
Additional file 5:.
Protection of persons committee.
Appendices
Appendix 1
Appendix 2
I-MICRO trial Data and Safety Monitoring Committee
Pr Charle Edouard Luyt, Medical Intensivist, Paris, France
Pr Etienne Gayat, Statistician, Paris, France
Pr Atul Pathak, Pharmacologist, Toulouse, France
Pr Olivier Sitbon, Pneumologist, Le Kremlin-Bicêtre, France
Appendix 3
List of events and serious adverse events
The investigator must document the serious adverse event as thoroughly as possible and provide the medical diagnosis, if possible. The investigator assesses the severity of the adverse events by using more general terms:
-
Mild: tolerated by the patient, does not interfere with daily activities
-
Moderate: sufficiently uncomfortable to affect daily activities
-
Serious: preventing daily activities
The investigator must assess the causal relationship between the serious adverse events and the investigational medicinal product(s).
The method used by the investigator is based on the WHO Uppsala Monitoring Center method and uses the following causality terms:
-
Certain
-
Probable/likely
-
Possible
-
Unlikely (not ruled out)
These terms are defined as follows (extracted from the WHO-UMC causality categories, version dated 17 April 2012).
Serious adverse events that require a notification without delay by the investigator to the sponsor
As per article R.1123-49 of the French Public Health Code (PHC), the investigator must notify the sponsor without delay on the day when the investigator becomes aware of any serious adverse event which occurs during a trial as described in Article L.1121-1(1) PHC, except those which are listed in the protocol (see section 10.1.2.2.2) and, if applicable, in the investigator’s brochure as not requiring a notification without delay.
A serious adverse event is any untoward medical occurrence that:
1- Results in death 2- Is life-threatening 3- Requires inpatient hospitalization or prolongation of existing hospitalization 4- Results in persistent or significant disability/incapacity 5- Is a congenital anomaly/birth defect |
Specific features of the protocol
Other events that require the investigator to notify the sponsor without delay
The expected serious adverse events associated with no specific research procedures or exams. The investigator must notify the sponsor of the following events immediately if they occur within 48 h of treatment.
-
Cardiovascular collapse defined as a drop of mean arterial pressure of 25% or more baseline value within 15 min after initiating treatment.
-
Cardiac arrest.
-
Ventricular tachycardia or ventricular fibrillation.
-
Acute myocardial infarction (ST+).
-
Severe hypoxemia will be defined as PaO2/FiO2 < 100 mmHhg.
-
Hypotension will be defined as a drop of mean arterial pressure of 25% or more from baseline value within 15 min after introducing the treatment.
-
A major bleeding episode that will be defined as:
-
◦ Epistaxis if it lasts for more than 5 min, if it is repetitive (i.e., two or more episodes of true bleeding, i.e., no spots on a handkerchief, within 24 h), or if it leads to an intervention (packing, electrocautery, etc.).
-
◦ Gingival bleeding if it occurs spontaneously (i.e., unrelated to tooth brushing or eating) or if it lasts for more than 5 min.
-
◦ Hematuria if it is macroscopic and if either spontaneous or lasts for more than 24 h after instrumentation (e.g., catheter placement or surgery) of the urogenital tract.
-
◦ Macroscopic gastrointestinal hemorrhage: at least 1 episode of melena or hematemesis, if clinically apparent.
-
◦ Rectal blood loss, if more than a few spots.
-
◦ Hemoptysis, if more than a few speckles in the sputum, or intramuscular hematoma.
-
◦ Subcutaneous hematoma if the size is larger than 25 cm2 or larger than 100 cm2 if provoked
-
◦ Multiple sources of bleeding events
-
Monitoring of such events is part of the standard of care in ICU patients (e.g., arterial pressure monitoring, hemoglobin level monitoring, blood gas monitoring, etc.).
Serious adverse events that do not require the investigator to notify the sponsor immediately
These serious adverse events are only recorded in the “adverse event” section of the case report form (e-CRF)
Adverse events that are “not serious” but which are significant for the safety of participants
Expected non-serious adverse events are:
-
Transient and rapidly reversed arterial hypotension
-
Mild hypoxemia
-
Not severe bleeding
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Legrand, M., Oufella, H.A., De Backer, D. et al. The I-MICRO trial, Ilomedin for treatment of septic shock with persistent microperfusion defects: a double-blind, randomized controlled trial—study protocol for a randomized controlled trial. Trials 21, 601 (2020). https://doi.org/10.1186/s13063-020-04549-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13063-020-04549-y