
Abstract
Background
We report the first case of a missense variant in the APC gene that interrupts splicing by creating a new cryptic acceptor site. The variant, c.289G>A, p.(Gly97Arg), is located in exon 3, and qualitative and semi-quantitative RNA splicing analysis reveal that the variant results in skipping of the last 70 nucleotides of the exon, which leads to the introduction of a frameshift and a premature stop codon.
Case presentation
The variant was detected in two, apparently unrelated, Danish families with an accumulation of colorectal cancers, colonic adenomas and other cancers. The families both have an attenuated familial adenomatous polyposis phenotype, which is consistent with the association of pathogenic variants in the 5′ end of the gene.
One variant-carrier also had Caroli Disease and a Caroli Disease associated hepatic mucinous cystadenocarcinoma. This is the first description of a person with both Caroli Disease and a pathogenic APC variant, and although the APC variant is not known to be connected to the development of the hepatic malformations in Caroli Disease, it remains unclear whether the variant could have contributed to the carcinogenesis of the liver tumour.
Conclusions
Based on functional and co-segregation data we classify the APC c.289G>A, p.(Gly97Arg) variant as pathogenic (class 5). Our findings emphasize the importance of a functional evaluation of missense variants although located far from the exon-intron boundaries.
Similar content being viewed by others

Background
Adenomatous Polyposis Coli (APC) related disorders include familial adenomatous polyposis (FAP), attenuated FAP (AFAP) and Gastric Adenocarcinoma and Proximal Polyposis of the Stomach (GAPPS). Whereas FAP and AFAP can result from intragenic mutations, partial- or whole-gene deletions, GAPPS is only known to be caused by variants in promoter 1B of APC. The conditions are inherited in an autosomal dominant manner with varying degrees of penetrance from almost 100% in classic FAP to 70% at age 80 in AFAP [1,2,3].
Especially FAP, and to a lesser degree AFAP, are in addition to colonic adenomatous polyposis and increased risk of colorectal cancer (CRC) also characterized by an increased risk of extracolonic cancers, especially duodenal, ampullary and thyroid carcinomas, as well as non-malignant conditions and benign neoplasms. The symptoms from FAP to AFAP form a continuum, but FAP is often defined as the presence of > 100 colonic adenomas, early development of colonic adenomas (usually during the second decade) and early onset of CRC with an average age of 40 years at the time of diagnosis. AFAP, on the other hand, is typically characterized by a lower burden of colonic adenomas (most often < 100) with a tendency to right-sided localization and rectal sparing, and higher age at diagnosis [4]. AFAP has in several studies been associated with truncating pathogenic variants in the 5′ and 3′ end of the APC gene as well as exon 9, although the codons involved have varied slightly. A clear genotype-phenotype correlation is still controversial [5,6,7].
Here, we report two families with an extremely rare, pathogenic APC missense variant and AFAP phenotype.
Case presentation
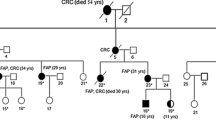
The first family (family 1) was included in a research project aimed at identifying new CRC genes, or genes not previously known to be involved in CRC, in families with an accumulation of CRC, and with no identified pathogenic variant in the DNA mismatch repair (MMR; MLH1, MSH2, MSH6 and PMS2) genes. The family was identified through the Danish HNPCC Register, Clinical Research Centre, Copenhagen University Hospital, Hvidovre. Two siblings (individual II-I and II-II); one with an adenocarcinoma in the transverse colon at age 46 and one with an adenocarcinoma in the duodenum at age 78, were included. They also both had colonic adenomatous polyposis. Another sibling (II-III) was later included in order to perform segregation analysis in the family. She was affected by four primary CRCs and a hepatic mucinous cystadenocarcinoma (Fig. 1; Table 1) and had also been diagnosed with the rare condition Caroli disease (CD). According to the family records, many relatives have suffered from gallstones and have had the gallbladder removed at an early age – as early as the beginning of the third decade - however, gallbladder disease do not segregate with CRC in the family. MLH1, MSH2, MSH6 and APC had previously, in 1999, been tested in the family, but for unknown reasons the APC variant was not detected or interpreted as pathogenic.

Pedigree of family 1 and 2, and segregation analyses of the APC variant c.289G>A, p.(Gly97Arg). Abbreviations: CRC: Colorectal cancer, adn: Colonic adenomas, DC: Duodenal cancer, LC: Hepatic cancer, MC: Breast cancer, EC: Endometrial cancer, PC: Pancreatic cancer, GC: Gastric cancer, n/a: Not available. Numbers in () refer to age at diagnosis (or death, if the person did not develop cancer). +: APC variant c.289G>A carrier, ?: APC variant c.289G>A carrier status unknown
The second family (family 2) was referred to Department of Clinical Genetics due to an accumulation of gastrointestinal cancers and adenomas. The proband (individual III-I) had a total colectomy at age 62 due to multiple (> 100) colonic adenomas, and the pathological evaluation revealed adenocarcinoma in two of the adenomas. A colonoscopy on her 64-year-old brother (III-II) revealed that he had approximately 20 adenomas. The family history included at least three additional cases of gastrointestinal cancers (Fig. 1; Table 1).
All living individuals received written and oral information as well as genetic counselling, and a written informed consent was obtained. As for the research project, ethical approval was obtained from the Danish Committee on Health Research Ethics (reference: H-4-2014-050).
In individual II-I and II-II, family 1, we analysed a research gene panel consisting of 34 defined or candidate genes for colorectal cancer. In individual III-I, family 2, we analysed a clinical CRC gene panel consisting of 17 CRC/polyposis predisposing genes. Both panels included APC (supplementary table S1 provides a complete list of the included genes). The analyses were next-generation sequencing based (Illumina, San Diego, California, USA) and performed on germline DNA obtained from peripheral blood.
In all three individuals we identified the APC variant c.289G>A, p.(Gly97Arg). Subsequent segregation analyses showed that a third sibling (II-III) in family 1 also carried the variant, as well as two individuals in family 2 (II-I and III-II); pedigrees can be seen in Fig. 1. Several healthy individuals from family 1 have also been tested, none of whom had inherited the variant; they were all following a surveillance program with regular colonoscopies without evidence of polyposis.
The APC c.289G>A variant has to the best of our knowledge only been published once in a Chinese patient with mild FAP [8], but has not been reported in any population allele frequency database (dbSNP, ExAC or gnomAD) including in 2000 exomes from Danish patients with diabetes [9]. In silico splice prediction software (SpliceSiteFinder-like, MaxEntScan and NNSPLICE; all integrated in the Alamut visual software (v.2.11)) all predicted that a strong cryptic acceptor splice site was introduced 70 base pairs into exon 3, which could lead to aberrant splicing and a lack of 70 nucleotides in the mRNA and probably nonsense mediated mRNA decay.
To confirm this hypothesis, we analysed RNA isolated from whole blood samples from individual III-I (family 2) by qualitative RT-PCR and semi-quantitative capillary electrophoresis (CE) using primers located in exon 1 and exon 5, respectively (primer sequences are available upon request).
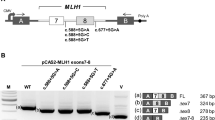
The RT-PCR analysis detected two major transcripts in the APC c.289G>A carrier, while only one transcript was identified in the two control samples (Fig. 2a). Sanger sequencing revealed that the upper transcript corresponded to the reference full-length (572 bp), while the lower band represented a transcript lacking 70 bp of exon 3 (502 bp (3p-70), Fig. 2b, c). Semi-quantitative CE supports the RT-PCR data and showed the control samples only expressed the full-length transcript, while the carrier expressed the full-length and a transcript corresponding to exon 3 lacking 70 nucleotides (3p(− 70)). The data moreover indicated that the 3p(− 70) transcript contributed to ~ 50% of the total transcript expression in the carrier (Fig. 2d). To further determine the contribution of the c.289G>A variant allele to APC reference transcript expression we Sanger sequenced an RT-PCR product using primers located in the skipped region of exon 3 and in exon 4 (primer sequences are available upon request) from the carrier (Fig. 2e). The data showed a monoallelic contribution of the wt allele, supporting that only the 3p(− 70) transcript is produced from the variant allele.

RNA and allele-specific analyses of the APC c.289G>A variant. a Gel electrophoresis analysis of RT-PCR products from an APC c.289G>A carrier and two controls (C1 and C2). b Sanger sequencing of the upper RT-PCR product. c Sanger sequencing of lower RT-PCR product. d Capillary electrophoresis analysis of the RT-PCR products from an APC c.289G>A carrier and two controls (C1 and C2). FL: Reference full-length transcript, 3p(− 70): Transcript lacking 70 bp of exon 3. e Sanger sequence of an RT-PCR product using primers designed to target the skipped region in exon 3 and to amplify the heterozygote c.289G>A variant in the carrier. The Sanger sequence show a monoallelic contribution of the wt allele
Discussion and conclusions
We report two Danish families with a segregating APC missense variant. The variant is absent from all allele frequency databases, but was recently reported in a Chinese patient reported to have mild FAP, however no functional analyses were performed. Here we show that the variant creates a cryptic splice site resulting in a frameshift. We have data from four generations in both families, and although the families appear to be unrelated, they could have a common ancestor; however, since the variant has been detected in another patient from China, with a similar phenotype, it is unlikely to represent a Danish founder variant not related to AFAP. Although most published pathogenic APC mutations are truncating, disease-causing missense variants have been reported, but they are typically located in the exon-intron boundaries and result in splicing defects. The identified APC c.289G>A variant is located one third into exon 3 and creates a cryptic splice site, and several findings suggest that this variant is indeed pathogenic. Firstly, the phenotype and inheritance patterns in the families are consistent with APC-associated polyposis. Secondly, the variant segregates with disease in both families. Thirdly, our functional data confirm that the APC c.289G>A variant introduces a cryptic splice acceptor site that results in a major transcript isoform lacking 70 bp of exon 3, which is predicted to encode a truncated APC protein. Based on both the functional and clinical data we therefore classify the APC c.289G>A, p.(Gly97Arg) variant as pathogenic (class 5) in accordance with the ACMG-AMP guidelines for interpretation of sequence variants [10].
In family 1, individual II-III was – in addition to four colorectal cancers – also diagnosed with CD and an intrahepatic mucinous cystadenocarcinoma. CD is a rare congenital condition with cystic dilatations of the intrahepatic bile ducts. The condition is progressive, and the individuals typically develop symptoms later in life. CD can, among other symptoms, be characterized by recurrent cholangitis, cholelithiasis and gallbladder stones and is associated with an increased risk of cholangiocarcinoma, which affects up to 14% of the individuals. Different inheritance patterns and genes have been suggested to be involved in CD, i.e. autosomal recessive inheritance due to PKHD1 mutations [11, 12]. In family 1 several family members suffered from cholelithiasis; the family might also have an autosomal dominant genetic predisposition to gall bladder disease, but we have no reason to believe that CD and the APC variant should be linked in any way, which is substantiated by the fact that the APC variant and gall bladder disease did not segregate in the family. On the other hand, it cannot be excluded that the APC variant could have contributed to the carcinogenesis of the CD-associated intrahepatic mucinous cystadenocarcinoma, however due to lack of tumour tissue, it has not been possible to perform APC loss of heterozygosity (LOH) analysis in the tumour.
The variant carriers in this study all have phenotypes consistent with AFAP; most had polyp count < 100 and onset of cancer at age 46–88, and although individual II-II in family 1 and individual III-I in family 2 both had large numbers of adenomas, they both developed cancer at a relatively high age (62-years-old and 78-years-old, respectively). This is in accordance with the association of a less severe phenotype and pathogenic variants in APC codons 1–177 [5,6,7]. On the other hand, an increasing amount of evidence suggests that modifier variants in other genes, low risk single nucleotide polymorphisms (SNPs) and non-genetic factors such as lifestyle influence the cancer risk and possibly also account for some of the intrafamilial variation [13]. Further research will help to elucidate the possible clinical implications of both the genotype and the role of modifying variants.
Availability of data and materials
The data obtained during the current study are available from the corresponding author on reasonable request.
Abbreviations
- APC :
-
Adenomatous Polyposis Coli
- AFAP:
-
Attenuated Familial Adenomatous Polyposis
- FAP:
-
Familial Adenomatous Polyposis
- GAPPS:
-
Gastric Adenocarcinoma and Proximal Polyposis of the Stomach
- CRC:
-
Colorectal Cancer
- DNA:
-
Deoxy Ribonucleic Acid
- MMR:
-
Mismatch Repair
- MLH1 :
-
MutL homolog 1
- MSH2 :
-
MutS homolog 2
- MSH6 :
-
MutS homolog 6
- PMS2 :
-
PostMeiotic Segregation increased 2
- Gly:
-
Glycine
- Arg:
-
Arginine
- mRNA:
-
Messenger Ribonucleic Acid
- RNA:
-
Ribonucleic Acid
- RT-PCR:
-
Reverse Transcription Polymerase Chain Reaction
- CE:
-
Capillary Electrophoresis
- Bp:
-
Base pairs
- ACMG-AMP:
-
American College of Medical Genetics and Genomics and the Association for Molecular Pathology
- CD:
-
Caroli Disease
- PKHD1 :
-
Polycystic Kidney and Hepatic Disease 1
- SNP:
-
Single Nucleotide Polymorphism
- AC:
-
Adenocarcinoma
- MCAC:
-
Mucinous cystadenocarcinoma
- IDC:
-
Invasive ductal carcinoma
- EEA:
-
Endometrioid adenocarcinoma
- n/a:
-
Not available
- Adn:
-
Colonic adenomas
- Duo:
-
Duodenal cancer
- LC:
-
Hepatic cancer
- MC:
-
Breast cancer
- EC:
-
Endometrial cancer
- PC:
-
Pancreatic cancer
- GC:
-
Gastric cancer
- Wt:
-
Wild type
References
Jasperson KW, Tuohy TM, Neklason DW, Burt RW. Hereditary and familial Colon Cancer. Gastroenterology. 2010;138(6):2044–58.
Li J, Woods SL, Healey S, Beesley J, Chen X, Lee JS, et al. Point mutations in exon 1B of APC reveal gastric adenocarcinoma and proximal polyposis of the stomach as a familial adenomatous polyposis variant. Am J Hum Genet. 2016;98(5):830–42.
Burt RW, Leppert MF, Slattery ML, Samowitz WS, Spirio LN, Kerber RA, et al. Genetic testing and phenotype in a large kindred with attenuated familial adenomatous polyposis. Gastroenterology. 2004;127(2):444–51.
Knudsen AL, Bisgaard ML, Bülow S. Attenuated familial adenomatous polyposis (AFAP). A review of the literature. Familial Cancer. 2003;2(1):43–55.
Sieber OM, Segditsas S, Knudsen AL, Zhang J, Luz J, Rowan AJ, et al. Disease severity and genetic pathways in attenuated familial adenomatous polyposis vary greatly but depend on the site of the germline mutation. Gut. 2006;55(10):1440–8.
Nieuwenhuis MH, Vasen HFA. Correlations between mutation site in APC and phenotype of familial adenomatous polyposis (FAP): a review of the literature. Crit Rev Oncol Hematol. 2007;61(2):153–61.
Newton KF, Mallinson EKL, Bowen J, Lalloo F, Clancy T, Hill J, et al. Genotype-phenotype correlation in colorectal polyposis. Clin Genet. 2012;81(6):521–31.
Wang D, Zhang Z, Li Y, Xu C, Yu Y, Li M, et al. Adenomatous polyposis coli gene mutations in 22 Chinese pedigrees with familial adenomatous polyposis. Med Sci Monit. 2019;25:3796–803.
Lohmueller KE, Sparsø T, Li Q, Andersson E, Korneliussen T, Albrechtsen A, et al. Whole-exome sequencing of 2,000 Danish individuals and the role of rare coding variants in type 2 diabetes. Am J Hum Genet. 2013;93(6):1072–86.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology sue. Genet Med. 2015;17(5):405–24.
Yonem O, Bayraktar Y. Clinical characteristics of Caroli’s disease. World J Gastroenterol. 2007;13(13):1930–3.
Fabris L, Fiorotto R, Spirli C, Cadamuro M, Mariotti V, Perugorria MJ, et al. Pathobiology of inherited biliary diseases: a roadmap to understand acquired liver diseases. Nat Rev Gastroenterol Hepatol. 2019;16(8):497–511.
Talseth-Palmer BA. The genetic basis of colonic adenomatous polyposis syndromes. Hered Cancer Clin Pract. 2017;15(1):1–7.
Acknowledgements
We thank the families included in this study. Moreover, Anne Lauridsen is thanked for technical assistance with the splicing analysis.
Funding
This study was funded by Danish Cancer Society, Rigshospitalet, Anna og Hans Steffensens Fond til støtte for Dansk Kræftforskning, Aase og Ejnar Danielsens Fond og A.P. Møller Fonden til Lægevidenskabens Fremme.
Author information
Authors and Affiliations
Contributions
MD, KW and AMG included the patients in the research project and analysed and interpreted the research gene panel data. KW handled family 2 clinically. MD was a major contributor in writing the manuscript. TvOH and JHF analysed and interpreted the splicing analyses. MBM and LAB were responsible of the laboratory gene panel analyses and variant interpretation. JPH and GLW performed the histological examination of the tissue. All authors read and approved the final manuscript.
Authors’ information
Not applicable.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All living individuals received written and oral information as well as genetic counselling, and a written informed consent was obtained. As for the research project, ethical approval was obtained from the Danish Committee on Health Research Ethics (reference: H-4-2014-050).
Consent for publication
All living patients have given consent for publication.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Supplementary Table S1.
Genes included in our gene panels. A) The research CRC gene panel. B) The clinical CRC gene panel.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Djursby, M., Wadt, K., Frederiksen, J.H. et al. A rare missense variant in APC interrupts splicing and causes AFAP in two Danish families. Hered Cancer Clin Pract 18, 8 (2020). https://doi.org/10.1186/s13053-020-00140-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13053-020-00140-3